

Abstract
Digestive diseases play major role in development and complications of other disorders including diabetes. For example, Crohn’s disease (CD) is an inflammatory bowel disease associated with Mycobacterium avium subspecies paratuberculosis. The inflammation is a complex process that involves the activity of both innate and adaptive immune responses. CD lesions are primarily due to T cell response, however; innate immune response has a significant role in initiating its pathogenesis. Toll-like receptors and NOD-like receptors promote the activity of nuclear factor (NF)-κB pathway for cytokines production. This results in the production of high levels of tumor necrosis factor-α, interleukin (IL)-1β and IL-6. Moreover, intestinal inflammation of CD is related to increased activity of NMDA receptors and the release of substance P. Imbalanced magnesium homeostasis in CD is a frequent finding in CD, Diabetes and others. The loss of such a major mineral affects many physiological processes in the body including its role as an immunomodulator. This review aims to (1) describe the significance of hypomagnesemia in the release of pro-inflammatory mediators in CD; (2) demonstrate effects of magnesium on pathways like NF-κB; (3) address the role of hypomagnesemia in the activity of CD; and (4) examine possible future research to establish a standard magnesium supplementation strategy; helping patients with CD or other disorders to maintain a sustained remission.
Keywords: Diabetes, Crohn’s disease, Hypomagnesemia, Inflammatory bowel disease, Mycobacterium paratuberculosis
Core tip: Magnesium is an essential trace mineral, which plays key role as an immunomodulator in many pathways leading to homeostasis. Hypomagnesemia is common in patients with Crohn’s disease (CD) and may be the cause of upregulation of pro-inflammatory factors leading to aggravating symptoms. Therefore, understanding the role of magnesium in maintaining a healthy immune response is important for effective treatment of patients with CD.
INTRODUCTION
Inflammatory bowel disease (IBD) generally describes a group of conditions sharing the characteristic of chronic inflammation of the gastrointestinal tract. The two most common conditions in this category are ulcerative colitis (UC) and Crohn’s disease (CD)[1]. In both conditions, the immune system is mistaken food particles and normal flora for foreign materials[2,3]. This will induce an immune response attracting the leukocytes to infiltrate the intestine. The result is destruction of intestinal mucosal cells leading to a state of chronic inflammation. Its distribution and involvement varies between UC and CD. UC is usually confined to the colon while CD can affect any site throughout the gastrointestinal tract from mouth to anus[4]. As both conditions progress, continuation of lesions in the colon becomes a characteristic for UC[5], whereas skipping some locations in the gastrointestinal tract or regional enteritis becomes a characteristic for CD[6]. Moreover, small bowels and the beginning of the large bowels are commonly affected in CD[1]. This difference in lesion locations contributes to the variations in the clinical presentation of UC and CD, as well as the severity of complications.
According to the Center for Disease Control and Prevention, both sexes are equally susceptible to IBD[7], with a majority of the affected population in between their 10 to 30 years of age[8]. Among the most susceptible are Caucasian and Ashkenazi Jewish origins[9]. As gold standard diagnosing criteria for IBD are lacking and the condition gets frequently misclassified, precise incidence and prevalence rates are limited. However, both conditions are noted to be at its highest rate of new diagnoses in industrialized North America and Europe for CD and UC, respectively[8]. In the United States, an estimated 1.4 million individuals suffer from IBD[3,7], of which 20.2 per 100000, per person years suffer from CD[8]. Although the etiology for IBD has not been well established, genetic components[4,8], diet, and environmental factors such as smoking[3] are associated with an increased risk of pathogenesis. Nevertheless, the impact of IBD in the United States creates a huge burden in the health care system, especially for CD with an estimated cost of $2.29 billion annually[10].
In particular, the high cost in CD can be attributed to therapeutic management, physician visits, and hospital stays because of the chronic nature and recurrence of the disease[10]. A detailed review of factors that can influence the persistence of CD might lead to establishing therapeutic strategies that can maintain remission for relatively longer periods. One of these factors includes nutritional deficiency due to malabsorption of vitamins and minerals such as magnesium, a frequent finding in CD patients particularly during high activity of the disease[11-15]. Magnesium deficiency or hypomagnesemia is very understudied and underestimated especially when it comes to its relation to CD (Figure 1). This finding encourages further research about the role of magnesium in inflammation and the possibility of linking this to the breakouts of the CD.
Figure 1.
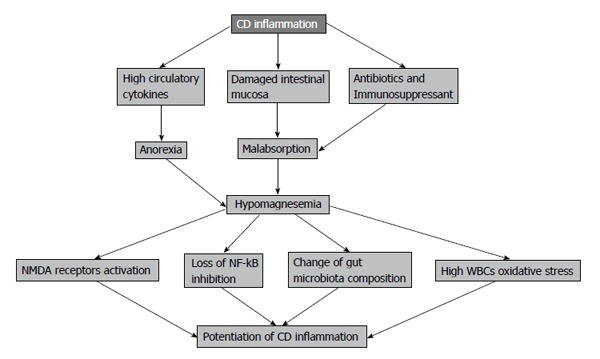
Role of hypomagnesaemia in Crohn’s disease complications.
This review aims to (1) describe the significance of low magnesium levels in the release of pro-inflammatory mediators like interleukin 1 (IL-1), IL-6, tumor necrosis factor (TNF)-α as CRP levels; (2) demonstrate effects of magnesium on inflammatory response pathways like nuclear factor (NF)-κB; (3) address the role of hypomagnesemia in the activity of CD as one of the associated nutritional deficiencies; and (4) examine possible future research to establish a standard magnesium supplementation strategy to CD patients maintaining remission for relatively longer periods.
INFLAMMATION IN CD: INNATE VS ADAPTIVE
CD is primarily a T cell autoimmune disorder[16,17], however; innate immune response has a significant role in its pathogenesis, as we will demonstrate in this paper (Table 1). Traditionally, T helper 1 (Th1) cells were believed to be the main immune cells responsible for most of the intestinal tissue damage in CD[17,18]. Th1-related cytokines like interferon (IFN)γ, which acts as the major inflammatory mediator in CD[19], are released as a response to Th1 stimulation by IL-12 from naïve T cells[19,20]. Therefore, it was plausible to think that it is possible to control CD significantly if antibodies against IL-12 and IFNγ (Fontolizumab) are used as a potential therapeutic option[17]; however, this was eclipsed when the administration of these antibodies (anti-IL-12 and anti-IFNγ) showed a limited improvement in cases with active CD[20]. Recently, it was suggested that CD mucosal lesions are not caused only by Th1 cytokines, instead there is a possibility that other cells and mediators are also involved rather than Th1 cells alone as summarized in Table 1[17,20]. Examining mucosa of a terminal ileum from a CD patient before the appearance of lesions showed a large population of Th1 and macrophages releasing IFNγ and TNF-α, respectively; while samples from well-formed lesions presented a relatively equal response from Th1 and another set of cells, Th17, with dominance of their cytokines, IFNγ and IL-17A[17].
Table 1.
Key immune effectors for Crohn’s disease inflammatory processes
| Role | Ref. | |
| Toll-like receptors | Their overexpression promotes NF-κB pathway leading to immune intolerance to gut normal flora | [17,24,25,28] |
| IL-1 TNF-α | Stimulation of intestinal stromal cells to release matrix metalloproteinases leading to mucosal damage | [17,20] |
| Th1 Th17 cells | Secretion of IFNγ, IL-17 and IL-21 activating macrophages (MØ) and stromal cells to release MMPs | [4,17] |
| NMDA receptors, SP | Intestinal neuronal inflammation | [21,22] |
NF-κB: Nuclear factor-kappa B; IL-1: Interleukin 1; MMPs: Matrix metalloproteinases; TNF: Tumor necrosis factor; IFN: Interferon; Th1: T helper 1; NMDA: N-methyl-D-aspartic acid; SP: Substance P.
As shown in Table 2, this finding suggested a different set of active cells and cytokines presented as the disease progresses from early to late stages. Active lesions in CD are produced after a cascade of steps starting from the antigen presenting cells in the intestine. These cells get activated by luminal antigens triggering the differentiation of naïve T cells into either Th1 cells by IL-12 release or Th-17 cells by IL-6, IL-23, and transforming growth factor beta[4,17,20]. IL-21 from Th-1 and IL-17 from Th-17 will stimulate the release of matrix-degrading proteases from stromal cells[17,20]. Also, IFNγ from Th-1 will further activate macrophages that produce IL-1β and TNF-α, which will further trigger the release of more proteases[20]. Th17 are not stable cells, and as the inflammation continues to progress, they convert to Th-1 releasing more IFNγ during formation of late mucosal lesions[19]. This explains the persistence of high IFNγ levels towards late CD stages[20]. Therefore, it becomes clear that early stages of CD are dominated by Th1 cells, while late stages are mixed in control between Th1 and Th17 (Table 1)[17].
Table 2.
Magnesium and inflammation
| Role | Ref. | |
| NF-κB Pathway | Inhibition of NF-κB p65 phosphorylation and stabilization of IκB protein | [29,39] |
| NMDA receptors, SP | Low magnesium enhances calcium influx through NMDA receptors | [12,38] |
| Neutrophilic oxidative stress | Increased levels of superoxide anions and nitric oxide in magnesium deficiency | [12] |
| Gut microbiota | Low magnesium changes the composition and intestinal permeability | [43] |
NF-κB: Nuclear factor-kappa B; SP: Substance P; NMDA: N-methyl-D-aspartic acid; IκB: Inhibitory kappa B.
Furthermore, the effects of the adaptive immune system extend to the enteric nervous system. Neuronal inflammation and damage is a well-documented problem in IBD generally and in CD specifically[21,22]. A suggested mechanism for this pathology is explained through high activity of NMDA receptors in enteric neurons (Table 2). Leading to elevated levels of intracellular calcium, as a result, substance P (SP) will be released from these cells acting as a pro-inflammatory mediator increasing release of other inflammatory mediators such as TNF-α, IL-1 and IL-6 from macrophages and neutrophils, which adds to the overall exaggerated immune response in CD[21,22].
SP is a tachykinin peptide that has a high affinity for neurokinin-1 (NK-1) receptors on macrophages, neutrophils and mast cells, and the role of SP as a pro-inflammatory agent was proved when a specific antagonist for NK-1 receptors blocked the release of pro-inflammatory cytokines decreasing inflammation and severity of DSS-induced colitis in rats[22]. On the other hand, release of TNF-α and IL-1β has a role in the re-innervation of smooth muscle of intestine damaged in CD; through activation of NF-κB they were able to increase glial cell line derived neurotrophic factor expression in smooth muscles of intestine, making these cytokines neurotrophic and neurotoxic ones at the same time in a sense[21].
In contrast, the innate immune also plays a role in the pathogenesis of CD. Most of the commensal microbes we have in our bodies are located in the intestine, and this intestinal-microbial interface provides a large surface area for innate and adaptive immune response activities[23]. Cells like DC, M cells, and intestinal epithelial cells have the ability to detect and respond to these microbes[23]. This ability is provided by their surface expression of toll-like receptors (TLR), as well as intracellular NOD-like receptors (NLR), which are molecules that sense microbes through recognition of their PAMP[23-26]. PAMP that stimulates TLRs include bacterial lipoproteins, peptidoglycans and most importantly lipopolysaccharides (LPS)[24]. As shown in several studies[24,26,27], overexpression of TLRs (especially TLR2 and TLR4) is frequently observed in patients with IBD, which contributes to the dysfunction of immune tolerance to gut normal flora.
TLR and NLR promote the activity of NF-κB pathway, a major signaling pathway regulating the immune response through cytokine production and cell survival[24,25,28]. NF-κB molecules are transcription factors stimulating a group of genes responsible for immune, inflammatory and apoptosis processes[24-26]. NF-κB induces the expression of its repressor IκB that binds to NF-κB molecules preventing their nuclear translocation[25]. Once IκB is phosphorylated, NF-κB becomes free to translocate into nucleus inducing the expression of pro-inflammatory cytokines[25,29]. Significance of this pathway in the inflammatory process of CD was established by the use of triptolide, a potent anti-inflammatory and immunosuppressant extracted from Chinese herb Tripterygium wilfordii Hook F[24]. The study showed that triptolide has down regulated TLR2 and TLR4 expression as well as NF-κB nuclear translocation, resulting in reduction in levels of pro-inflammatory cytokines in CD[24]. This finding suggested triptolide as a possible immunomodulator option for CD, and at the same time showed the strong link between TLR/NF-κB/pro-inflammatory cytokines in CD dysregulated immune response[24].
NUTRITIONAL LOSS IN CD
The inflammation caused by both innate and adaptive immune system leads to progression of CD, causing the loss of a significant functional intestinal area due to villous atrophy, fistulae formation and bacterial overgrowth[30]. These changes in the intestinal structure lead to loss of a variety of proteins, lipids, sugars, as well as vitamins and minerals, which locates the body in a negative nutritional status[13,15]. Other factors contributing to this negative balance in CD include: anorexia, abdominal pain, fasting for different tests, and medications like sulfasalazine used to control the disease or surgeries leading to short bowel syndrome[14,31]. For the same mentioned reasons, magnesium loss is a frequent finding in patients with CD as a result of the imbalanced magnesium homeostasis[13].
MAGNESIUM HOMEOSTASIS
This is maintained by the cooperation between three organs: intestine, kidneys and bones[32]. In the intestine, distal parts of jejunum and ileum are the most common sites for magnesium uptake[12,32]. Approximately 80%-90% of dietary magnesium absorption is achieved via paracellular transport, which depends on the permeability of tight junctions[32]. In addition, low expression of claudin 1, 3, 4, 5 and 8 proteins in the jejenum and ileum enables the passage of magnesium ions[33]. This mechanism is passive, allowing a majority of magnesium absorption without energy cost[12,32]. The rest of dietary magnesium is absorbed by the active transcellular transport via TRPM6 and TRPM7[12,32]. The latter mechanism allows magnesium to be transported into the blood from intestine through cell membrane[12,32]. Once absorbed, magnesium is stored mainly in bone tissue but traces can be found in muscles, where it acts as a natural calcium antagonist to control muscle contraction[12,32]. Lastly, most of the magnesium excreted from the body is processed by the kidneys, where 90%-95% of filtered magnesium daily gets retrieved via passive and active transport mechanisms[12,32].
MAGNESIUM IS NO TRACE ELEMENT: IT IS AN ESSENTIAL GIANT MINERAL
Magnesium is an important mineral in the human body like calcium, potassium and sodium[34,35]. When it comes to physiology, it is truly a ‘‘chronic regulator’’ and a ‘‘forgotten electrolyte’’[34]. Magnesium is a cofactor for over 300 enzymes catalyzing phosphorylation reactions; it creates the proper conformational changes on their active sites so they fit their specific substrates, which regulates about 30% of total body proteins functions[34]. Regulation of cell cycle and apoptosis is achieved through many magnesium-dependent kinases, adding more weight on the significance of this mineral[34]. Production of the most common second messengers like c-AMP and c-GMP for different signal transduction pathways have magnesium involved in their regulation as well[34]. It is involved in the transport of many other electrolytes including calcium, potassium and sodium through its role in sodium/potassium ATPase activity, which explains the ‘’refractory’’ nature of their disturbances to conventional treatment if the level of magnesium is low[34,36].
ROLE OF MAGNESIUM IN INFLAMMATION
Several studies have shown the importance of magnesium in inflammation that linked its low levels to many medical conditions such as diabetes type 2[37] (Barbagallo, 2007 #168), obesity, metabolic syndrome, osteoporosis, and cardiovascular diseases (Table 2)[12,38]. Levels of many pro-inflammatory cytokines varies depending on magnesium balance in the body, and among these cytokines, TNF-α IL-1 and IL-6 have the strongest relation[12,29,38]. Also, levels of CRP, a well-studied inflammatory indicator of low-grade and chronic inflammation synthesized by the liver, vary with magnesium status changes as well[12,38]. Effects of magnesium on inflammatory responses and mediators are widely distributed; therefore, it will be discussed separately as follows: (1) Magnesium as an anti-proinflammatory cytokine. Inhibition of NF-κB activity and increasing levels of IκBα are the backbone for this function (Table 2)[29]. NF-κB pathway is stimulated widely in the human body to regulate inflammation, cancer fighting, and cell survival[29]. Expression of cytokines IL-6 and TNF-α is induced during inflammatory responses triggered by TLR and NLR, which stimulates a downstream pathway to translocate NF-κB into nucleus for pro-inflammatory cytokines production[29,39]. IκBα is unstable due to its amino acids composition that is rich in proline, threonine and serine, explaining the constitutive breakdown rate affecting it[29,40,41]. IκBα level in monocytes were tested before and after magnesium sulfate supplementation following stimulation of TLR by LPS and it showed that level of IκBα is higher in presence of higher intracellular magnesium[29]. The reason was not related to increased IκBα expression and protein synthesis, rather it was increased stability of IκBα, as proved by the use of protein synthesis inhibitor before and after TLR stimulation[29]. On the other hand, phosphorylation of NF-κB p65 is essential for its translocation, as well as its transcriptional effect to induce cytokines production; it was shown to decreased following magnesium supplement in LPS-TLR stimulated monocytes[29]. At the same time, expression of IκBα was decreased in the presence of increased intracellular magnesium sulfate supporting the finding of decreased activation of NF-κB in high levels of cellular magnesium sulfate[29,39]. As a result of inhibition of NF-κB translocation and transcriptional effect and the decreased phosphorylation of NF-κB, levels of TNF-α and IL-6 were significantly lower in LPS-TLR stimulated cells in the presence of high cellular magnesium levels[29,39]. Also, magnesium has preserved and stabilized more IκBα, which led to more suppression in NF-κB related cytokine production following LPS stimulation. In another study, it was shown that IL-8 expression is decreased following the same steps mentioned for LPS stimulation of cytokine production[39]; (2) Oxidative stress and magnesium. In an experiment conducted on magnesium deficient rats, there was a 40% increase in the level of superoxide anions and nitric oxide levels (Table 2)[12]. Also, there were increased levels of neutrophilic basal superoxide anions, as well as prostacyclin, prostaglandin E2, and thromboxane A2[12]. Red blood cells glutathione levels were decreased in the same experiment showing declining body antioxidant potentials in increased oxidative stress as a result of low cellular magnesium levels[12]; (3) Magnesium effect on NMDA receptors. Magnesium is a natural calcium antagonist and this was discussed in role of magnesium in muscle contraction[32]. From another perspective, NMDA receptors have a threshold of activation and it is lowered in states of decreased extracellular magnesium levels[38]. This will lead to an increase in calcium influx into the cell through NMDA receptors, resulting in increased production of pro-inflammatory prostaglandin E2, which was decreased upon blocking NMDA receptors[12,38]. Also, as calcium levels increases intracellular, the level of SP increases as a result stimulating NK-1 receptors leading to production of inflammatory mediators from macrophages, monocytes and neutrophils[38]. It is noteworthy to mention that the increase in NK-1 and substance P are well-known findings in IBD[12]. In addition to that, magnesium binds to the regulatory gates of calcium channels limiting calcium influx into the cell, and low extracellular magnesium levels will enhance the calcium influx triggering a greater inflammatory response[38]; (4) Magnesium, gut microbiota and intestinal permeability. It has been established before that gut microbiota [mainly bifidobacteria; a gram positive, non-motile anaerobic bacteria (Table 2)[42]] are decreased in endotoxemia, high fat mass index and glucose utilization disturbances[43,44]. Similarly, in another experiment, cecal content of bifidobacteria and lactobacilli were decreased in short-term (four days) magnesium deficient rats[43]. On the other hand, prolonged magnesium deficiency (21 d) has actually increased the cecal content of the mentioned bacteria, suggesting an adaptive response by the bacteria and an established demand for magnesium[43]. Bifidobacteria are microorganisms known for their ability to lower intestinal LPS content and thus enhance the mucosal barrier performance[43,45]. As the drop of magnesium levels decreases the cecal bacterial content, it also causes change in intestinal mucosal barrier, where mRNA of two of the junction proteins (ZO-1 and Occ) were noticed to decrease in ileum and proximal colon resulting in increased intestinal permeability for bacterial products and especially LPS to be increased systemically[43]. Accordingly, it was noticed that expression of CD14 receptors that bind LPS was elevated in gut in magnesium deficient mice, as well as increased expression of CD68 supporting the infiltration of monocytes in proximal colon[43]. The overall content of mRNA of TNF-α and IL-6 in proximal colon was increased in magnesium deficient mice[43]. These findings showed the effect of low magnesium on cellular inflammatory stress, which seemed to be limited to proximal colon rather than ileum[43]. Prolonged magnesium deficiency has an impact on the composition of gut microbiota as more bifidobacteria and lactobacilli will be present, and less bacteroids in the intestine[43]; and (5) Magnesium and C-reactive protein. As several studies investigated effects of dietary modification on inflammatory processes, CRP was among the most common inflammation indicators used for evaluation[46]. High levels of CRP were linked to obesity, metabolic syndrome, cardiovascular diseases and IBD[47]. They all share having an inflammatory component in their etiology and CRP was the tested variable in many studies[12]. As levels of IL-1β, IL-6 and TNF-α increase in the plasma, liver will respond by increasing production of CRP[38]. More specifically, serum high sensitive CRP (hs-CRP) has been used frequently due to its stability and easy detection, which has a normal level in plasma of < 3.0 mg/L[48]. Different conditions with low-grade or chronic inflammation states shared the sign of having elevated hs-CRP, indicating the strong inflammatory component they have[46]. CD activity has been strongly correlated to hs-CRP level[49], which is considered to be one of the main laboratory values that increase in relapses. Back to our mineral, magnesium is a significant immunomodulator that affects many inflammatory responses, and therefore its homeostasis is crucial for the overall body homeostasis. Low magnesium levels (< 1.2 mg/dL) were correlated to elevated levels of TNF-α, IL-1β, IL-6 and hs-CRP in plasma[12,38,46]. A study conducted on 5007 children (1999-2002) showed a significant increase in risk of having high CRP (1.94 times more) in children taking less than 50% of magnesium RDI[38]. One of the most interesting findings about magnesium and hs-CRP is that it was developed at University of South Carolina, showing that magnesium is the highest dietary factor in a 42-item dietary anti-inflammatory index they made for the study[46,48].
MAGNESIUM LOSS IN CD
With more than 32% of American people not meeting the daily requirement of magnesium dietary intake (4.5 mg/kg per day for adults), hypomagnesemia became a real concern for many practitioners[14]. Therefore, IBD adds a major cause for developing hypomagnesemia at different rates ranging from 13% to 88% of patients[14]. This deficiency is caused by many factors in CD including anorexia, food avoidance, intestinal surface loss due to diarrhea, fistulae or surgery as well as malabsorption[14]. Intestinal uptake of magnesium is defected dramatically as inflammatory processes of CD result in villus atrophy and fistulae formation, on top of increased bowel movement not allowing the time for magnesium absorption[32]. As the majority of magnesium absorption occurs passively, there will be no sufficient concentration gradient for magnesium uptake in intestine, as well as destructed enterocytes, losing the active transport component of magnesium absorption[32].
Nutritional loss in patients with CD is variable based on the disease activity status. Usually during remission of the disease, the body demand for macronutrient is covered by diet. However, micronutrient loss is frequent and supplementation is usually required even during remission of the disease[15]. Due to the chronic and extensive damage of intestinal mucosal cells, oral magnesium supplement is not recommended and parenteral forms are encouraged since the bioavailability will not be a concern in this case[13].
As CD result in malabsorption and loss of many vitamins, vitamin D in particular has a direct influence on magnesium and its intestinal absorption[32]. Claudin proteins involved in paracellular mechanism of magnesium absorption (the major mechanism) are regulated by active vitamin D, thus in CD, loss of fat soluble vitamins including vitamin D will lead to decreased magnesium absorption and hypomagnesemia[32]. As 75% of CD patients will require surgery at some point due to intestinal disease complications, short bowel syndrome will be a major cause of malabsorption affecting the levels of many nutrients including magnesium as well[14].
Also, magnesium absorption in the intestine is subjected to the amount of protein in diet and this is decreased in CD due to anorexia produced by circulating cytokines and food avoidance by the patients because of abdominal pain[32]. For all of those factors, magnesium will be in negative balance in CD patients (Figure 1).
EFFECTS OF MAGNESIUM ON CROHN’S PATHOGENESIS
By looking back at magnesium significance in the human body, it is obvious that the major effect magnesium can have on CD is from the immunity and inflammation point of views. However, other fields for magnesium influence on CD can be calcium disturbances, intestinal nerve supply and gut microbiota composition.
As established before in this paper, magnesium has the potential to be an effective cytokine antagonist. In different studies magnesium showed immunomodulation capabilities through controlling expression of pro-inflammatory cytokines, oxidative stress and neuronal damage. Through its effect on NF-κB, intracellular magnesium was able to limit NF-κB nuclear translocation as well as p65 phosphorylation activation step. On the other hand, intracellular magnesium preserved and stabilized IκBα limiting its degradation and applying more inhibitory effect on NF-κB pathways. As CD progresses and intestinal lesions develop, exposure of TLR and NLR to LPS will be more likely (Table 2). LPS is a potent TLR stimulator, which will activate NF-κB subsequently leading to production of IL-1, IL-6 and TNF-α. Hypomagnesemia is a frequent finding in CD, which means that the inhibitory effect on NF-κB pathway will be absent allowing more production of IL-6 and TNF-α that will trigger more mucosal damage via activation of the release of matrix-degrading proteases from intestinal stromal cells[20].
Among the structural changes that occur to intestinal mucosa due to CD, gut microbiota composition changes are also related to magnesium deprivation[43]. Short-term and long-term magnesium deficiency in mice showed significant changes in intestinal permeability and bacterial adaptation[43]. Low magnesium levels decreased bifidobacteria and increased risk of LPS endotoxemia since bifidobacteria can lower intestinal content of LPS[43]. Down-regulation of junction proteins ZO-1 and Occ mRNA as a result of magnesium deprivation caused an increase in intestinal permeability and this finding alone can describe the significant effect of hypomagnesemia in CD patients[43]. Role of magnesium in alleviating LPS immune response is essential for immune tolerance to gut commensal bacteria.
Among the inflammatory mediators associated with CD, CRP is one of the most sensitive markers of CD activity and relapsing status[50]. Its short half-life made it superior to other markers like ESR and fibrinogen which have longer half-lives and interference with other agents[50]. As demonstrated in many studies[12,38], serum CRP levels are elevated in diabetes type 2, obesity, metabolic syndrome, atherosclerosis, osteoporosis and alcoholism indicating the inflammatory component that links them together in etiology. At the same time, all of these conditions were also associated by hypomagnesemia suggesting a strong relationship between magnesium and CRP and its possible application on CD where levels of magnesium are reduced[12,38].
Calcium influx into neurons is largely regulated by extracellular magnesium[12,38]. Low magnesium levels due to CD will lower NMDA receptors activation threshold allowing more calcium entry into neurons[12,38]. Also, gated calcium channels will lose magnesium regulation over them and will allow more calcium influx, which adds to the overall free intracellular calcium[12,38]. Elevated levels of intracellular calcium are able to increase the release of inflammatory SP that trigger more production of IL-1, IL-6 and TNF-α resulting in increase in the oxidative stress affecting intestinal sensory innervation causing symptoms like tenesmus (feeling of incomplete defecation) and frequent bowel movements[12].
Hypocalcaemia in CD is characteristic and has more than one cause resulting loss of many calcium functions throughout the body. First, as more vitamin D is lost in the frequent diarrhea associated with CD, calcium absorption at the intestine will be impaired[32]. On the other hand, low magnesium levels associated with CD results in increased calcium influx shifting most calcium into cells and causing calcium levels in plasma to drop even more[12,38].
Role of magnesium in muscle contraction as a calcium antagonist is essential for intestinal smooth muscle function in creating efficient peristalsis. It allows for periods of relaxation following contraction cycles caused by calcium. Also, SP is a regulator of smooth muscle contractility and hypomagnesemia elevates its levels leading to abnormal intestinal smooth muscle function. This function is essential to control bowel movement frequency in CD which is increased as a result of magnesium deficiency[32].
As levels of antioxidant vitamins like vitamin A, vitamin C and vitamin E are decreased in CD due to intestinal loss, oxidative stress effect on different cells increases including intestinal cells as well. Low magnesium levels showed an association with increased oxidative stress in individuals with no CD as levels of lipid peroxidation increase and production of free radicals like superoxide anion and nitric oxide is promoted[12]. These changes in CD with addition of hypomagnesemia augment the load of oxidative stress all over the body.
TARGETS FOR FUTURE DIRECTIONS
The integration of magnesium functions throughout the body is enormous and fields of future studies of that are numerous. However, when it comes to CD association with hypomagnesemia, some targets are very promising for possible maintenance of remission or even a cure of CD.
Most of drugs used to control CD have a long list of side effects and a possible toxicity with chronic use. Magnesium has shown great potentials on affecting the same pathways involved in CD inflammation as many therapeutic agents. On the other hand, magnesium is not expected to be cytotoxic and this hypothesis is very promising if magnesium is tested on specific regimens to block NF-κB signaling pathway in CD patients.
Role of magnesium in controlling calcium entry in neurons is significant in intestinal sensory innervation[12]. Hypomagnesemia leads to more nerve damage following SP activation and this effect carries a potential of modifying intestinal smooth muscle function and innervation in CD if magnesium supplement are tested for that.
Another area that is poorly understood is the role of magnesium in CD as a cofactor for most kinases and the possible changes of this rule during the disease. It is unclear whether this function affects CD activity and if so, in what way this is applied in cases of hypomagnesemia associated with CD.
Possible changes in gut microbiota composition following magnesium level alterations represent a big opportunity to further explore the role of normal flora in developing CD. Other minerals could play a similar role for the short term or long term changes in their levels. As data suggested, certain strains of microbiota like bifidobacteria turned out to have a role in lowering LPS and contributing to the intestinal mucosal barrier function and tolerance. Could other strains have different functions involved in immune responses and tolerance? A hypothesis can be based on the most common commensal strains and their possible role in local and systemic immune responses and possible implications in autoimmune diseases.
Magnesium supplementation for CD patients is strongly suggested by several research data. Maintaining magnesium homeostasis throughout the course of the disease is expected to minimize the inflammatory damage of CD improving the condition of many patients. However, conventional magnesium supplementation itself causes diarrhea which is the main reason magnesium is lost in CD. A therapeutic strategy for magnesium administration is strongly recommended.
CONCLUSION
CD is primarily an innate immunity dysfunction, and this disturbance develops to trigger an adaptive immune response resulting in mucosal intestinal surface damage. Among the numerous functions magnesium has throughout the body, immunomodulation is by the far the most involved function in CD activity and development. Chronic diarrhea among other problems in CD results in long term loss of magnesium, which makes hypomagnesemia a frequent finding in most CD patients. As the data showed, restoration of magnesium levels in CD patients can limit the activity of NF-κB, which is responsible for production of pro-inflammatory cytokines involved in CD inflammation. Additionally, many studies have suggested the disturbances in gut normal flora composition following short and long term hypomagnesemia. These have resulted in loss of immune tolerance to normal flora at the intestinal interface. Involvement of hypomagnesemia in NMDA receptors and SP release creates a direct effect on neuronal function of intestine and smooth muscle activity as well.
This review highlights some of the well-known functions of magnesium and their potential rule in shaping CD activity. It was strongly suggested by data that magnesium could play a significant rule in controlling CD. This review demonstrates a possible mutual effect of CD on magnesium level as well as hypomagnesemia on CD inflammatory processes. Future nutritional studies as well as medical research are expected to focus more efforts to better understand effects of magnesium and CD on each other. Knowledge of these effects would create a strong basis for development of a potential therapeutic strategy to modulate the vast inflammatory effects CD has on its patients.
Footnotes
P- Reviewer: Izawa KP, Zhao JB S- Editor: Wen LL L- Editor: A E- Editor: Liu SQ
References
- 1.Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet. 2012;380:1590–1605. doi: 10.1016/S0140-6736(12)60026-9. [DOI] [PubMed] [Google Scholar]
- 2.Hou JK, Abraham B, El-Serag H. Dietary intake and risk of developing inflammatory bowel disease: a systematic review of the literature. Am J Gastroenterol. 2011;106:563–573. doi: 10.1038/ajg.2011.44. [DOI] [PubMed] [Google Scholar]
- 3.Loftus EV. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology. 2004;126:1504–1517. doi: 10.1053/j.gastro.2004.01.063. [DOI] [PubMed] [Google Scholar]
- 4.Naser SA, Arce M, Khaja A, Fernandez M, Naser N, Elwasila S, Thanigachalam S. Role of ATG16L, NOD2 and IL23R in Crohn’s disease pathogenesis. World J Gastroenterol. 2012;18:412–424. doi: 10.3748/wjg.v18.i5.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ordás I, Eckmann L, Talamini M, Baumgart DC, Sandborn WJ. Ulcerative colitis. Lancet. 2012;380:1606–1619. doi: 10.1016/S0140-6736(12)60150-0. [DOI] [PubMed] [Google Scholar]
- 6.Lockhart-Mummery HE, Morson BC. Crohn’s disease (regional enteritis) of the large intestine and its distinction from ulcerative colitis. Gut. 1960;1:87–105. doi: 10.1136/gut.1.2.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Centers for Disease Control and Prevention. Inflammatory Bowel Disease (IBD) 2014. Available from: http: //www.cdc.gov/ibd/ [Google Scholar]
- 8.Ponder A, Long MD. A clinical review of recent findings in the epidemiology of inflammatory bowel disease. Clin Epidemiol. 2013;5:237–247. doi: 10.2147/CLEP.S33961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanauer SB. Inflammatory bowel disease: epidemiology, pathogenesis, and therapeutic opportunities. Inflamm Bowel Dis. 2006;12 Suppl 1:S3–S9. doi: 10.1097/01.mib.0000195385.19268.68. [DOI] [PubMed] [Google Scholar]
- 10.Stone CD. The economic burden of inflammatory bowel disease: clear problem, unclear solution. Dig Dis Sci. 2012;57:3042–3044. doi: 10.1007/s10620-012-2417-8. [DOI] [PubMed] [Google Scholar]
- 11.Main AN, Morgan RJ, Russell RI, Hall MJ, MacKenzie JF, Shenkin A, Fell GS. Mg deficiency in chronic inflammatory bowel disease and requirements during intravenous nutrition. JPEN J Parenter Enteral Nutr. 1981;5:15–19. doi: 10.1177/014860718100500115. [DOI] [PubMed] [Google Scholar]
- 12.Weglicki WB. Hypomagnesemia and inflammation: clinical and basic aspects. Annu Rev Nutr. 2012;32:55–71. doi: 10.1146/annurev-nutr-071811-150656. [DOI] [PubMed] [Google Scholar]
- 13.Moorthy D, Cappellano KL, Rosenberg IH. Nutrition and Crohn’s disease: an update of print and Web-based guidance. Nutr Rev. 2008;66:387–397. doi: 10.1111/j.1753-4887.2008.00048.x. [DOI] [PubMed] [Google Scholar]
- 14.Hwang C, Ross V, Mahadevan U. Micronutrient deficiencies in inflammatory bowel disease: from A to zinc. Inflamm Bowel Dis. 2012;18:1961–1981. doi: 10.1002/ibd.22906. [DOI] [PubMed] [Google Scholar]
- 15.Filippi J, Al-Jaouni R, Wiroth JB, Hébuterne X, Schneider SM. Nutritional deficiencies in patients with Crohn’s disease in remission. Inflamm Bowel Dis. 2006;12:185–191. doi: 10.1097/01.MIB.0000206541.15963.c3. [DOI] [PubMed] [Google Scholar]
- 16.Romagnani P, Annunziato F, Baccari MC, Parronchi P. T cells and cytokines in Crohn’s disease. Curr Opin Immunol. 1997;9:793–799. doi: 10.1016/s0952-7915(97)80180-x. [DOI] [PubMed] [Google Scholar]
- 17.Zorzi F, Monteleone I, Sarra M, Calabrese E, Marafini I, Cretella M, Sedda S, Biancone L, Pallone F, Monteleone G. Distinct profiles of effector cytokines mark the different phases of Crohn’s disease. PLoS One. 2013;8:e54562. doi: 10.1371/journal.pone.0054562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bamias G, Sugawara K, Pagnini C, Cominelli F. The Th1 immune pathway as a therapeutic target in Crohn’s disease. Curr Opin Investig Drugs. 2003;4:1279–1286. [PubMed] [Google Scholar]
- 19.Strober W, Zhang F, Kitani A, Fuss I, Fichtner-Feigl S. Proinflammatory cytokines underlying the inflammation of Crohn’s disease. Curr Opin Gastroenterol. 2010;26:310–317. doi: 10.1097/MOG.0b013e328339d099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Monteleone I, Pallone F, Monteleone G. Th17-related cytokines: new players in the control of chronic intestinal inflammation. BMC Med. 2011;9:122. doi: 10.1186/1741-7015-9-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gougeon PY, Lourenssen S, Han TY, Nair DG, Ropeleski MJ, Blennerhassett MG. The pro-inflammatory cytokines IL-1β and TNFα are neurotrophic for enteric neurons. J Neurosci. 2013;33:3339–3351. doi: 10.1523/JNEUROSCI.3564-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lakhan SE, Kirchgessner A. Neuroinflammation in inflammatory bowel disease. J Neuroinflammation. 2010;7:37. doi: 10.1186/1742-2094-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garrett WS, Gordon JI, Glimcher LH. Homeostasis and inflammation in the intestine. Cell. 2010;140:859–870. doi: 10.1016/j.cell.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu C, Shan T, Feng A, Li Y, Zhu W, Xie Y, Li N, Li J. Triptolide ameliorates Crohn’s colitis is associated with inhibition of TLRs/NF-κB signaling pathway. Fitoterapia. 2011;82:709–715. doi: 10.1016/j.fitote.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 25.Chen Y, Wang C, Liu Y, Tang L, Zheng M, Xu C, Song J, Meng X. miR-122 targets NOD2 to decrease intestinal epithelial cell injury in Crohn’s disease. Biochem Biophys Res Commun. 2013;438:133–139. doi: 10.1016/j.bbrc.2013.07.040. [DOI] [PubMed] [Google Scholar]
- 26.Szebeni B, Veres G, Dezsõfi A, Rusai K, Vannay A, Mraz M, Majorova E, Arató A. Increased expression of Toll-like receptor (TLR) 2 and TLR4 in the colonic mucosa of children with inflammatory bowel disease. Clin Exp Immunol. 2008;151:34–41. doi: 10.1111/j.1365-2249.2007.03531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hart AL, Al-Hassi HO, Rigby RJ, Bell SJ, Emmanuel AV, Knight SC, Kamm MA, Stagg AJ. Characteristics of intestinal dendritic cells in inflammatory bowel diseases. Gastroenterology. 2005;129:50–65. doi: 10.1053/j.gastro.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 28.Rubino SJ, Selvanantham T, Girardin SE, Philpott DJ. Nod-like receptors in the control of intestinal inflammation. Curr Opin Immunol. 2012;24:398–404. doi: 10.1016/j.coi.2012.04.010. [DOI] [PubMed] [Google Scholar]
- 29.Sugimoto J, Romani AM, Valentin-Torres AM, Luciano AA, Ramirez Kitchen CM, Funderburg N, Mesiano S, Bernstein HB. Magnesium decreases inflammatory cytokine production: a novel innate immunomodulatory mechanism. J Immunol. 2012;188:6338–6346. doi: 10.4049/jimmunol.1101765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nolan JD, Johnston IM, Walters JRF. Physiology of malabsorption. Surgery. 2012;30:268–274. [Google Scholar]
- 31.Lomer MC. Dietary and nutritional considerations for inflammatory bowel disease. Proc Nutr Soc. 2011;70:329–335. doi: 10.1017/S0029665111000097. [DOI] [PubMed] [Google Scholar]
- 32.de Baaij JHF, Hoenderop JGJ, Bindels RJM. Regulation of magnesium balance: lessons learned from human genetic disease. Clin Kidney J. 2012:5 (Suppl 1): i15–i24. doi: 10.1093/ndtplus/sfr164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amasheh S, Fromm M, Günzel D. Claudins of intestine and nephron - a correlation of molecular tight junction structure and barrier function. Acta Physiol (Oxf) 2011;201:133–140. doi: 10.1111/j.1748-1716.2010.02148.x. [DOI] [PubMed] [Google Scholar]
- 34.Ismail Y, Ismail AA, Ismail AA. The underestimated problem of using serum magnesium measurements to exclude magnesium deficiency in adults; a health warning is needed for “normal” results. Clin Chem Lab Med. 2010;48:323–327. doi: 10.1515/CCLM.2010.077. [DOI] [PubMed] [Google Scholar]
- 35.Laires MJ, Monteiro CP, Bicho M. Role of cellular magnesium in health and human disease. Front Biosci. 2004;9:262–276. doi: 10.2741/1223. [DOI] [PubMed] [Google Scholar]
- 36.Touyz RM. Transient receptor potential melastatin 6 and 7 channels, magnesium transport, and vascular biology: implications in hypertension. Am J Physiol Heart Circ Physiol. 2008;294:H1103–H1118. doi: 10.1152/ajpheart.00903.2007. [DOI] [PubMed] [Google Scholar]
- 37.Barbagallo M, Dominguez LJ. Magnesium metabolism in type 2 diabetes mellitus, metabolic syndrome and insulin resistance. Arch Biochem Biophys. 2007;458:40–47. doi: 10.1016/j.abb.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 38.Nielsen FH. Magnesium, inflammation, and obesity in chronic disease. Nutr Rev. 2010;68:333–340. doi: 10.1111/j.1753-4887.2010.00293.x. [DOI] [PubMed] [Google Scholar]
- 39.Rochelson B, Dowling O, Schwartz N, Metz CN. Magnesium sulfate suppresses inflammatory responses by human umbilical vein endothelial cells (HuVECs) through the NFkappaB pathway. J Reprod Immunol. 2007;73:101–107. doi: 10.1016/j.jri.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 40.Rice NR, Ernst MK. In vivo control of NF-kappa B activation by I kappa B alpha. EMBO J. 1993;12:4685–4695. doi: 10.1002/j.1460-2075.1993.tb06157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luan J, Shattuck-Brandt R, Haghnegahdar H, Owen JD, Strieter R, Burdick M, Nirodi C, Beauchamp D, Johnson KN, Richmond A. Mechanism and biological significance of constitutive expression of MGSA/GRO chemokines in malignant melanoma tumor progression. J Leukoc Biol. 1997;62:588–597. doi: 10.1002/jlb.62.5.588. [DOI] [PubMed] [Google Scholar]
- 42.WEBB M. The influence of magnesium on cell division. IV. The specificity of magnesium. J Gen Microbiol. 1951;5:480–484. doi: 10.1099/00221287-5-3-480. [DOI] [PubMed] [Google Scholar]
- 43.Pachikian BD, Neyrinck AM, Deldicque L, De Backer FC, Catry E, Dewulf EM, Sohet FM, Bindels LB, Everard A, Francaux M, et al. Changes in intestinal bifidobacteria levels are associated with the inflammatory response in magnesium-deficient mice. J Nutr. 2010;140:509–514. doi: 10.3945/jn.109.117374. [DOI] [PubMed] [Google Scholar]
- 44.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 45.Griffiths EA, Duffy LC, Schanbacher FL, Qiao H, Dryja D, Leavens A, Rossman J, Rich G, Dirienzo D, Ogra PL. In vivo effects of bifidobacteria and lactoferrin on gut endotoxin concentration and mucosal immunity in Balb/c mice. Dig Dis Sci. 2004;49:579–589. doi: 10.1023/b:ddas.0000026302.92898.ae. [DOI] [PubMed] [Google Scholar]
- 46.Cavicchia PP, Steck SE, Hurley TG, Hussey JR, Ma Y, Ockene IS, Hébert JR. A new dietary inflammatory index predicts interval changes in serum high-sensitivity C-reactive protein. J Nutr. 2009;139:2365–2372. doi: 10.3945/jn.109.114025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pearson TA, Mensah GA, Alexander RW, Anderson JL, Cannon RO, Criqui M, Fadl YY, Fortmann SP, Hong Y, Myers GL, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: A statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation. 2003;107:499–511. doi: 10.1161/01.cir.0000052939.59093.45. [DOI] [PubMed] [Google Scholar]
- 48.Galland L. Diet and inflammation. Nutr Clin Pract. 2010;25:634–640. doi: 10.1177/0884533610385703. [DOI] [PubMed] [Google Scholar]
- 49.Kiss LS, Papp M, Lovasz BD, Vegh Z, Golovics PA, Janka E, Varga E, Szathmari M, Lakatos PL. High-sensitivity C-reactive protein for identification of disease phenotype, active disease, and clinical relapses in Crohn’s disease: a marker for patient classification? Inflamm Bowel Dis. 2012;18:1647–1654. doi: 10.1002/ibd.21933. [DOI] [PubMed] [Google Scholar]
- 50.Vermeire S, Van Assche G, Rutgeerts P. Laboratory markers in IBD: useful, magic, or unnecessary toys? Gut. 2006;55:426–431. doi: 10.1136/gut.2005.069476. [DOI] [PMC free article] [PubMed] [Google Scholar]