

Abstract
The estrogen receptor (ER) pathway plays a critical role in breast cancer development and progression. Endocrine therapy targeting estrogen action is the most important systemic therapy for ER positive breast cancer. However its efficacy is limited by intrinsic and acquired resistance. Mechanisms responsible for endocrine resistance include deregulation of the ER pathway itself, including loss of ER expression, post-translational modification of ER, deregulation of ER co-activators; increased receptor tyrosine kinase signaling leading to activation of various intracellular pathways involved in signal transduction, proliferation and cell survival, including growth factor receptor tyrosine kinases human epidermal growth factor receptor-2, epidermal growth factor receptor, PI3K/AKT/mammalian target of rapamycin (mTOR), Mitogen activated kinase (MAPK)/ERK, fibroblast growth factor receptor, insulin-like growth factor-1 receptor; alterations in cell cycle and apoptotic machinery; Epigenetic modification including dysregulation of DNA methylation, histone modification, and nucleosome remodeling; and altered expression of specific microRNAs. Functional genomics has helped us identify a catalog of genetic and epigenetic alterations that may be exploited as potential therapeutic targets and biomarkers of response. New treatment combinations targeting ER and such oncogenic signaling pathways which block the crosstalk between these pathways have been proven effective in preclinical models. Results of recent clinical studies suggest that subsets of patients benefit from the combination of inhibitor targeting certain oncogenic signaling pathway with endocrine therapy. Especially, inhibition of the mTOR signaling pathway, a key component implicated in mediating multiple signaling cascades, offers a promising approach to restore sensitivity to endocrine therapy in breast cancer. We systematically reviewed important publications cited in PubMed, recent abstracts from ASCO annual meetings and San Antonio Breast Cancer Symposium, and relevant trials registered at ClinicalTrials.gov. We present the molecular mechanisms contributing to endocrine resistance, in particular focusing on the biological rationale for the clinical development of novel targeted agents in endocrine resistant breast cancer. We summarize clinical trials utilizing novel strategies to overcome therapeutic resistance, highlighting the need to better identify the appropriate patients whose diseases are most likely to benefit from these specific strategies.
Keywords: Endocrine therapy, Endocrine resistance, Breast cancer, Therapeutic advances, Targeted therapy
Core tip: Endocrine therapy is the important systemic therapy for hormone receptor positive breast cancer. However, treatment resistance is common. Multiple mechanisms responsible for endocrine resistance have been identified over the past decade. New treatment combinations targeting estrogen receptor and growth factor receptor signaling which block the crosstalk between these pathways are effective in preclinical models and clinical studies. In this review, we summarize the complex genomic and epigenetic regulatory pathways involved in endocrine resistance, in particular focusing on the clinical trials utilizing novel strategies to overcome therapeutic resistance.
INTRODUCTION
Estrogen receptor (ER) is expressed in about 75% of human breast cancers which is the one of the leading cause of death for women globally. The estrogen-bound ER functions through ligand-activated transcriptional regulation (genomic actions) and by acting as a component of signaling cascades outside of the nucleus (non-genomic actions)[1-4]. Clinical observations and laboratory studies suggest ER signaling pathway is the major driver in promoting proliferation, survival and invasion of ER-positive breast cancer cells[3]. Endocrine therapy is the mainstay of treatment for patients with ER-positive breast cancer, especially those with metastatic disease. Endocrine therapies include treatments which target ER by blocking receptor binding with an antagonist or by depriving the tumor of estrogen. The three broad groups of currently approved anti-estrogen therapies are selective estrogen receptor modulators (SERMs) such as tamoxifen, raloxifene and toremifene, which block activity of ER; selective estrogen receptor down regulators (SERDs) such as fulvestrant, which induce destabilization and degradation of ER; and aromatase inhibitors (AIs), including steroidal/irreversible (anastrozole and letrozole) and nonsteroidal/reversible (exemestane) inhibitors, which decrease estrogen production in peripheral tissues and within the tumors through inhibition of the enzyme aromatase[5-11]. Endocrine therapy as the first targeted therapy in cancer treatment has successfully improved outcome of millions of breast cancer patients in the past 30 years[5,12].
There is evidence that some breast tumors are more resistant to endocrine therapy than others, despite expressing ER. This is supported by stratification of ER positive tumors into luminal A and luminal B subtypes based on molecular profiling studies over the last decade. The luminal B subtype is more aggressive and less endocrine sensitive, while the luminal A subtype is more indolent and endocrine responsive[13-15]. Recently The Cancer Genome Atlas (TCGA) data reinforces that luminal B cancers represent a unique subtype of breast cancer, with a distinctive biology from that of luminal A cancers. Multigene tests performed on the primary breast tumor are increasingly utilized in clinical practice to assist in adjuvant therapy decision making and to distinguish which patients might benefit most from a combination of endocrine therapy plus chemotherapy, rather than endocrine therapy alone. For example, the 21-gene (OncotypeDx) and 70-gene (MammaPrint) assays can classify ER positive tumors according to their aggressiveness, risk of recurrence, and likelihood of benefitting from adjuvant endocrine or chemotherapy. PAM50 is a 50 gene expression assay to separate breast tumor samples into known intrinsic molecular subtypes (basal-like, HER-2 enriched, luminal A and luminal B) and correlate with risk of relapse. The progesterone receptor (PR) is expressed in half of patients with ER+ breast tumors[16]. Clinical studies have shown that ER+/PR+ tumors are more responsive to endocrine therapy than ER+/PR- tumors[17]. Furthermore, down-regulation of PR correlates with high growth factor activity, indicating that loss of PR in ER positive breast tumors could serve as a predictor of endocrine therapy outcome[16,17]. However, no biomarkers that predict resistance to endocrine therapy with certainty are available currently. Therefore most patients with ER positive breast cancers are treated with endocrine therapy, in adjuvant and/or metastatic setting. Tamoxifen is the treatment of choice in premenopausal patients. And aromatase inhibitors (e.g., letrozole and anastrozole) have become the treatment of choice as first-line therapy in postmenopausal patients. On disease progression, second-line treatment options include other classes of AIs (steroidal or nonsteroidal) and the ER antagonists, fulvestrant and tamoxifen[18]. But the effectiveness of endocrine therapy is limited by high rates of de novo or intrinsic resistance (existing before any treatment is given) and acquired resistance during treatment (resistance that develops during a given therapy after an initial period of response). One third of patients will have recurrent disease within 15 years after being treated with tamoxifen for 5 years[11]. About 50% of patients with metastatic disease do not respond to initial endocrine treatment[8]. Inevitably the vast majority of patients with ER-positive advanced breast cancer will become refractory to endocrine therapy.
A plethora of mechanisms have been proposed to explain resistance to endocrine therapy, including deregulation of various components of the ER pathway itself[11,14,19], activation of escape pathways that provide tumors with alternative cell proliferative and survival stimuli[20-24], alterations in cell cycle and apoptotic machinery[3,25], modulation in epigenetics and microRNA profile[1,4,6,26]. In this review, we summarize the key mechanisms that have been implicated in the development of endocrine resistance in breast cancer. We give an overview of the completed and ongoing clinical trials with novel agents targeting these alternative mechanisms, with the goal to overcome endocrine resistance in breast cancer.
LITERATURE SEARCH
PubMed was searched for articles in English published between January, 2000 to February, 2014 using the terms “breast cancer”, “endocrine resistance”, as well as the individual terms of the molecular components under molecular mechanism listed in this Review. Reference lists from key articles were searched for additional material. Abstracts from the ASCO annual meetings and the San Antonio Breast Cancer Symposium were considered (2010-2012). ClinicalTrials.gov was searched for relevant trials. Articles were identified on the basis of the authors’ knowledge of the advances in endocrine resistant breast cancer research.
MOLECULAR MECHANISM OF ENDOCRINE RESISTANCE
De novo resistance in breast cancer is characterized by loss of ER (the ERα isoform) expression and ER gene mutations such as deletion and point mutation. Patients carrying inactive alleles of cytochrome P4502D6 (CYP2D6) deficiency cannot convert tamoxifen to its active metabolite, endoxifen, therefore are resistant to tamoxifen[27]. By contrast, multiple mechanisms have been detected to account for the acquired resistance to endocrine therapies. Although it is beyond the focus of this review to summarize all of the known mechanism of endocrine resistance in breast cancer, we can focus on the molecular changes in some of the key pathways involved and their clinical implications (Figure 1).
Figure 1.
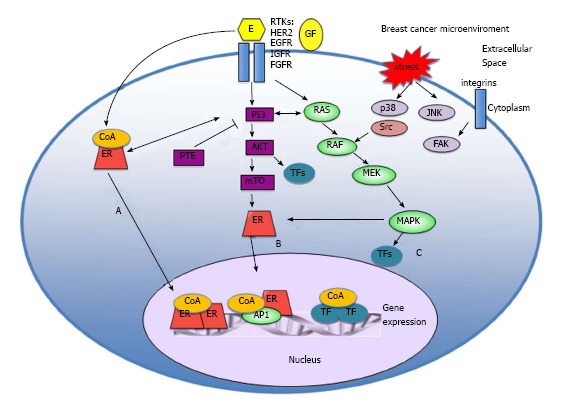
Estrogen receptor action at molecular level. A: Ligand dependent activation: in classic estrogen signaling, ligand-bound ER activates gene expression-either through direct binding of dimeric ER to specific DNA response elements in complexes including co-activators, or function as a coregulator through protein - protein interactions with other transcription factors to facilitate binding to serum response elements and activation of transcription; B: Ligand independent activation: the ER can also be activated by ligand independent fashion, as a consequence of signaling events downstream of membrane receptor tyrosine kinases (RTKs); C: Non-genomic mechanisms: signaling can be mediated through non-genomic mechanisms by ER that is localized at the cell membrane or in the cytoplasm. ER: Estrogen receptor; mTOR: Mammalian target of rapamycin; FGFR: Fibroblast growth factor receptor; IGF-1R: Insulin-like growth factor-1 receptor; EGFR: Epidermal growth factor receptor.
DEREGULATION OF CLASSIC ESTROGEN SIGNALING
The classic function of ER is its nuclear function, also known as genomic activity, to regulate the expression of genes important for normal and cancer cell proliferation and survival[3]. The nuclear estrogen receptors (ERα and ERβ) have similar structure, consisting of a central DNA-binding domain flanked by two autonomous transcriptional activation domains. In classic estrogen signaling, ligand-bound ER activates gene expression-either through direct binding of dimeric ER to specific DNA response elements in complexes including co-activators, or function as a coregulator through protein-protein interactions with other transcription factors, such as activation protein 1 (Ap1), specificity protein 1 (Sp1) and nuclear factor (NF-κB) to facilitate binding to serum response elements and activation of transcription[28-30].
Mechanisms of endocrine resistance include the loss of ERα expression which occurs in 15%-20% of resistant breast cancers, ERα mutations which present in < 1% of ER-positive tumors, the expression of ER splicing variants, specifically the truncated variant ERα36, and estrogen related receptors (ERR)[11,14,31-33]. Deregulation of ER co-regulators has been implicated in endocrine resistance as well. For example, increased Ap1 and NF-κB transcriptional activity are associated with endocrine resistance. Overexpression of nuclear receptor co-activator 3 (nCOA3, also known as AIB1 or SRC3), detected in two-thirds of all breast cancers, has been implicated in clinical and experimental tamoxifen resistance[3,21,34].
Post-translational modifications (phosphorylation, methylation and ubiquitination) of ER and its co-regulators are regulated to influence ER activity, interactions with other proteins including cytoplasmic signaling molecules[21,35,36]. Aberrant regulations at this post-translational level contribute to endocrine resistance as well[3].
ACTIVATION OF GROWTH FACTOR RECEPTOR PATHWAYS
The ER can also be activated by ligand independent fashion, as a consequence of signaling events downstream of membrane receptor tyrosine kinases (RTKs). RTKs are the intracellular portions of a class of growth factor receptors including HER2 (ERBB2), epidermal growth factor receptor (EGFR) and insulin-like growth factor receptor (IGFR). The bidirectional crosstalk between the RTK signaling and ER pathways has been implicated in the development of resistance to endocrine therapy in preclinical studies. Many clinical trials have begun to test several attractive strategies, such as manipulation of growth factor signaling networks and the use of tyrosine kinase and multikinase inhibitors that may delay or even overcome the resistance of breast cancers to endocrine therapy.
HER2 pathway
HER2 (Human epidermal growth factor receptor 2/ERBB2) is a member of the HER receptor tyrosine kinase family, which plays an important role in promoting cell proliferation and malignant growth in breast cancer. Over-expression of HER2 occurs in approximately 30% of metastatic breast cancers (MBC) and is associated with aggressive disease course and poor outcome with reduced disease-free and overall survival rates. Both preclinical and clinical evidence suggested that HER2 over-expression confers resistance to anti-estrogen agents in ER positive tumors[10]. Activation of the Her2 pathway, even without HER overexpression, confers tamoxifen resistance in ER positive cancer cells[37]. Preclinical studies demonstrated that tamoxifen resistant cells have the ability to switch between HER2 and the ER pathway for cell growth and survival. Upregulation of HER2 signaling occurs in some tumors with disease progression during endocrine therapy. Recent studies show that HER2 gene expression is repressed by the PAX2-ER-tamoxifen complex in sensitive breast cancer cell lines; while in tamoxifen resistant cell lines, the ER coactivator AIB-1/SRC-3 competes with PAX2 for binding, leading to increased HER2 transcription[38]. In addition, HER2 activation decreases ER level and increase ER phosphorylation, even in the absence of estrogen[38-40]. HER2 signaling alters ER mediated transcription through disrupting the interaction between ER and its coregulators (corepressors and coactivators). HER2 also activates downstream signaling pathways, such as the phosphoinositide 3-kinase (PI3K)/AKT pathway and mitogen activated kinase (MAPK) pathway, as discussed later[3,15,19].
The interdependence of ER and HER2 pathways is highlighted by examples in which treatment with AIs or downregulation of ER with fulvestrant has inhibited the growth of HER2-positive tumors that had progressed with trastuzumab or lapatinib. In addition, HER2 inhibition with trastuzumab or lapatinib restores or upregulates ER levels or transcriptional activity in breast cancer cells[24,41]. These data provide rationale for combined inhibition of ER and HER2 pathway, and clinical studies have demonstrated the benefit of targeting both the ER and HER2 in ER positive/HER2 positive breast cancer. In the phase III TAnDEM (Trastuzumab in Dual HER2 positive ER positive Metastatic Breast Cancer) trial, 207 postmenopausal women with HER2 positive ER positive MBC were randomized to anstrozole alone or anastrozole plus trastuzumab. The combination arm was clearly associated with a longer progression free survival (PFS) (4.8 mo vs 2.4 mo, P = 0.0016) and a higher clinical benefit rate (CBR) (42.7% vs 20.3%)[42]. Similarly, in the randomized, double-blind phase III study of letrozole with or without lapatinib in MBC, PFS and clinical benefit were superior in the combination arm compared with the AI-alone arm in 219 patients with ER positive/HER2 positive MBC[43]. Both trials suggest that both HER2 and ER should be simultaneously targeted for maximal therapeutic efficacy.
EGFR pathway
Among the four HER family members (HER1-4), HER1 is better known as epidermal growth factor receptor (EGFR). Binding of EGF-related growth factors leads to receptor homo and/or heterodimerization (with HER2) and activation of downstream signaling cascades including PI3K/AKT and MARK pathways. In breast cancer, overexpression of EGFR and subsequently increased activity of MAPK and PI3K/AKT signaling pathways confer estrogen independency, resistance to endocrine therapy and poorer prognosis[44-46]. For example, activation of ErbB3, EGFR and Erk is shown to be essential for growth of human breast cancer cell lines with acquired resistance to fulvestrant[47]. In preclinical study, Gefitinib, a small molecule inhibitor of EGFR, effectively inhibited EGFR-HER2 heterodimerization, phosphorylation and downstream signaling in the tamoxifen resistant MCF-7 cell line[48,49].
Lapatinib is a dual tyrosine kinase inhibitor blocking EGFR and HER2. In cell models of HER2 positive breast cancer with acquired endocrine resistance, lapatinib restores hormone sensitivity[50]. Johnston et al[51] reports in the randomized, double-blind phase III study, 1286 postmenopausal women with ER positive MBC, were randomized to receive letrozole with or without lapatinib. The benefit of combination therapy was observed in the ER positive, HER2 positive, but not in the ER positive, HER2 negative group. Letrozole plus lapatinib significantly increased PFS vs letrozole-placebo (8.2 mo vs 3.0 mo, HR = 0.71; 95%CI: 0.53-0.96, P = 0.019) in HER2 positive population[46,51]. There was also a trend toward a prolonged PFS for the combination observed in patients who experienced relapse less than 6 mo since prior adjuvant tamoxifen discontinuation. These data suggest that there is benefit with the addition of an EGFR/HER2- targeted therapy to an AI in patients who experience relapse early during prior tamoxifen therapy which is consistent with preclinical models where EGFR activity is enhanced in association with endocrine resistance[51].
Several selected EGFR inhibitors are being investigated as monotherapy or in combination with endocrine therapy in an attempt to overcome or prevent endocrine resistance. However, clinical trials that target EGFR in ER positive breast cancer have yielded mixed results. In the randomized placebo controlled phase II trial of tamoxifen with or without geftinib, 290 patients were stratified into an endocrine naïve group who had not received endocrine therapy within one year prior to enrollment, and another group who had developed recurrence during or after AI therapy. PFS was not significantly prolonged in the endocrine naïve group (8.8 mo vs 10.9 mo; P = 0.31) or the group who had AI[52]. Another small randomized placebo controlled phase II trial enrolled a total of 93 ER + metastatic breast cancer patients with or without prior endocrine therapy. In this study, combination of anastrozole with geftinib showed a statistically significant increases in PFS compared to anastrozole plus placebo (14.7 mo vs 8.4 mo, HR = 0.55; 95%CI: 0.32-0.94). Similarly, subset analysis of PFS for patients who had received prior endocrine treatment compared with those who were endocrine therapy naïve showed a more pronounced benefit for patients that had not previously received endocrine therapy[47]. These trials have suggested targeting EGFR could delay resistance to endocrine therapy in endocrine naïve patients.
Strategy of combined targeting the ER and EGFR was assessed in the neoadjuvant setting as well. Polychronis and colleague conducted the double-blind, placebo -controlled Phase II trial[53]. 56 patients with ER and EGFR expressing breast cancer were randomized to receive gefitinib and placebo, or geftinib plus anastrozole, for 4-6 wk prior to surgery. The combination arm showed a significant reduction in Ki67, which is the primary end point, than the monotherapy arm (5.6% difference, P = 0.0054). In contrast, Smith et al[54] reported a separate randomized phase II trial of neoadjuvant anstrozole alone or with gefitinib, in which 206 postmenousal women with early stage ER positive breast cancer were randomized to receive 16 wk of anastrozole monotherapy, 16 wk of anstrozole with 14 wk of geftinib (preceded by two weeks of placebo) or 16 wk of geftinib before surgery. There was no difference in proliferation index as measured by Ki67 for either geftinib regimen when compared to anastrozole alone. Moreover, there was no difference in overall objective response (48% vs 61%, P = 0.08). The authors concluded that addition of gefitinib/EGFR inhibitor to neoadjuvant anastrozole did not improve clinical or biologic effect[54]. The selection of EGFR overexpressing breast cancer cases in Polychronis et al’s study might account for the difference in these trial results. One could postulate that the ideal setting for testing combination of endocrine therapy and EGFR inhibitors is in the patients with acquired resistance since it is associated with adaptive upregulation of growth factor receptor signaling. Further biomarker studies in patients who had prior endocrine therapy are clearly warranted to identify a phenotype that may predict relapse and subsequent benefit from combined endocrine therapy and EGFR inhibitors.
Mitogen activated kinase pathway
The mitogen activated kinase pathway (MAPK) pathway is stimulated by the RAF serine/threonine kinase, and signals to additional downstream cytoplasmic serine-threonine kinases that ultimately activate MAP kinases such as, ERKs, c-jun N-teminal kinases, and p38MAPKs with resultant downstream phosphorylation of transcription factors. As discussed earlier, the MAPK pathway is important in mediating HER2-and EGFR-induced endocrine resistance. In addition, studies show that ERK and p38 phosphorylate AIB1 and ER coactivators[3,8]. Clinical trials targeting the MAPK pathway directly using MAPK inhibitors in combination with endocrine therapy are ongoing. Results on the randomized phase II trial, fulvestrant with or without AZD6244 (selumetinib, a MAPK Inhibitor) in advanced stage breast cancer progressing after aromatase inhibitor are awaited (NCT01160718).
The PI3K-AKT- mammalian target of rapamycin pathway
The PI3K-AKT (a serine/threonine kinase) pathway plays a central role in cell survival, proliferation and angiogenesis and is frequently deregulated in cancer[45]. Phosphatidylinositol 3-kinase (PI3K) consists of a regulatory subunit (p85) and a catalytic subunit (p110). PI3K is activated by growth factor RTKs and G-protein-coupled receptors (GPCRs). PI3K phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2) to produce phosphatidylinositol 3,4,5- trisphosphate (PIP3). In turn, PIP3 recruits several adaptor proteins such as phosphatidylinositol-dependent kinase 1 (PDK1) and AKT (a serine/threonine kinase), which when activated, drive cell proliferation and survival. Through dephosphorylation of PIP3 and PIP2 respectively, PTEN and INPP4B provide negative regulation of this pathway. AKT activates the mammalian target of rapamycin (mTOR) -containing complex 1 (mTORC1), which regulates protein synthesis[25]. Activating mutations or genetic amplification of PI3K catalytic subunit, amplification of downstream targets such as Akt, amplification of upstream receptors such as erbB2/HER2 and loss of negative regulators such as PTEN have all been described in breast cancer[55-57]. The Cancer Genome Atlas (TCGA) analysis confirms the high mutation frequency of PIK3CA in luminal/ER-positive breast cancer. PIK3CA somatic mutation is present in approximately 32% of luminal B subgroup, 49% of luminal A, 42% of HER2-enriched, and only 7% of basal-like breast cancer. Within the same pathway, PTEN mutation/loss and INPP4B loss were observed in more luminal B (24%, 16% each) than luminal A subtype (13% and 9% respectively)[14,58]. The PI3K-AKT pathway is widely viewed as an important therapeutic target and PI3K pathway inhibitors are being studied in clinical trials.
Preclinical studies have associated PI3K pathway activation with de novo and acquired resistance to endocrine therapy. Increased phosphorylation of mTOR substrates and AKT is observed in estradiol deprived breast cancer cell lines. Oncogene overexpression that activate PI3K/AKT signaling (e.g., HER2, type 1 insulin-like growth factor receptor (IGF1R), activated mutant AKT1) and RNAi-mediated knockdown of PTEN lead to resistance to tamoxifen, fulvestrant, and estrogen deprivation in ER-positive breast cancer cells. Studies using long-term estrogen-deprived (LTED) ER-positive breast cancer cell lines have shown that endocrine resistance develops concomitantly with amplification of PI3K/AKT/mTOR signaling[59]. Similar changes haven been observed with chronic exposure of MCF-7 cells and xenografts to fulvestrant[23].
Moreover, inhibition of PI3K has reversed antiestrogen resistance in experimental models. For example, treatment with the PI3K/mTOR inhibitor BEZ235 or the mTOR inhibitor everolimus prevents the growth of LTED cell lines in the absence of estrogen[60]. Everolimus in combination with tamoxifen had an additive anti-tumor effect in breast cancer cells in vitro[61]. In another study, the combination of temsirolimus with an ER antagonist synergistically inhibited the growth of breast cancer cells in vitro and growth in a xenograft model of breast cancer (mTOR)[62]. In a separate study, high levels of AKT activity conferred resistance to letrozole and fulvestrant through alteration of the cell cycle and apoptotic response in an in vitro breast cancer cell model[63]. Treatment with everolimus plus either letrozole or fulvestrant restored responsiveness in the resistant cells and results in synergistic inhibition of the proliferation and induction of apoptosis[60].
These preclinical studies indicated the promise of drugs targeting PI3K network (PI3K, AKT, mTOR) in ER positive breast cancer resistant to endocrine therapy. Table 1 summarizes the randomized trials in which inhibitors of PI3k pathway have been combined with endocrine therapy. Neoadjuvant treatment with letrozole and the mTOR inhibitor everolimus more effectively reduced tumor cell proliferation and improved clinical response compared with letrozole alone in patients with early-stage ER-positive breast cancer[64]. Two studies (BOLERO-2 and TAMRAD trials) have demonstrated superior benefit of mTOR inhibition in combination with endocrine therapy in advanced resistant ER positive breast cancers. In the phase III randomized BOLERO-2 trial, 724 patients with ER positive metastatic breast cancer (MBC) who had recurrence or progression while receiving previous therapy with a nonsteroidal aromatase inhibitor (either letrozole or anastrozole) were randomly assigned to everolimus and exemestane vs exemestane and placebo. The median PFS was significantly longer in the combination arm (10.6 mo vs 4.1 mo, HR 0.36; 95%CI: 0.27-0.47; P < 0.001, according to central assessment)[65]. The combination of exemestane and everolimus has been approved for ER positive advanced breast cancer in United States and Europe based on the magnitude of these positive results. TAMRAD is a randomized phase II trial of tamoxifen with or without everolimus in patients with aromatase inhibitor (AI)-resistant metastatic breast cancer. Patients in the combination arm showed an improved clinical benefit rate (61% vs 42%), time to progression (8.6 mo vs 4.5 mo), and overall survival compared with patients receiving tamoxifen alone. Notably patients with acquired endocrine resistance (relapse > 6 mo after AI treatment) derived the greatest benefit from the combination compared with those with primary resistance (relapse during adjuvant AI or within 6 mo of AI treatment in the metastatic setting) with an improvement in the median PFS of 12.4 mo vs 1.5 mo, respectively[66].
Table 1.
Clinical trials of targeted agents in endocrine resistant breast cancer
| Agent | Class | Type of study | Study design | Patient population | Status/Results | Ref. |
| Targeting receptor tyrosine kinases signaling pathway | ||||||
| PI3K/AKT/mTOR | ||||||
| Everolimus | mTOR inhibitor | Phase III randomized | Exemestane +/- everolimus | ER+/HER2- LABC/MBC pts failed previous therapy with a nonsteroidal AI | PFS: 10.6 vs 4.1 mo, HR 0.36; P < 0.001, favoring combination arm | [76] |
| Everolimus | mTOR inhibitor | Phase II randomized | Tamoxifen +/- everolimus | ER+/HER2- MBC pts after previous therapy with AI | CBR: 61% vs 42%; TTP: 8.5 vs 4.5 mo, P = 0.008, favoring combination arm | [77] |
| Temsirolimus | mTOR inhibitor | Phase III randomized | Letrozole +/- temsirolimus | First line therapy for patients with ER positive MBC | No difference in CBR, terminated early | [78] |
| Everolimus | mTOR inhibitor | Phase II randomized | Letrozole +/- everolimus | Neoadjuvant therapy in ER + breast cancer | RR (by U/S): 58% vs 47%; P = 0.035, favoring combination arm | [75] |
| Sirolimus | mTOR inhibitor | Phase I/II | Tamoxifen +/- sirolimus | Pts with ER+ MBC | N = 400, TAM + SIR: 193; TAM alone: 207), ORR: TAM + SIR 40%; TAM alone 4%; Time to progression: TAM + SIR: 11 mo TAM alone: 3 mo | Bhattacharyya et al Eur.J.Cancer 47, Abstract 16LBA (2011) |
| Bkm120 | Pan-PI3K inhibitor | Phase III randomized | Fulvestrant + BMK120 | ER+/HER2- LABC/MBC Postmenopausal pts, AI Treated, Progressed on or After mtor Inhibitor | NCT01633060 | |
| Bkm120 | Pan-PI3K inhibitor | Phase Ib | Fulvestrant + BMK120 | Postmenopausal pts with ER+ MBC | Ongoing, to determine the maximum tolerated dose of BKM120 | NCT01339442 |
| Bez235 | Dual PI3K-mTOR inhibitor | Phase Ib | Letrozole + BEZ235 | Postmenopausal pts with ER+ MBC | NCT01248494 | |
| BMK120 or BEZ235 | Pan-PI3K inhibitor | Phase Ib | Letrozole +BMK120 or BEZ235 | Postmenopausal pts with ER+ MBC | NCT01248494 | |
| XL147 or XL765 | Pan-PI3K inhibitors/dual PI3K/mTOR inhibitor | Phase I/II | Letrozole +XL147 or XL765 | ER+/HER2- MBC pts refractory to a previous AI therapy | NCT01082068 | |
| GDC-0941 or GDC-0980 | dual PI3K/mTOR inhibitor | Phase II randomized | Fulvestrant +GDC-0941 or GDC-0980 | Part I: ER+/HER2- | NCT01437566 | |
| postmenopausal LABC/MBC | ||||||
| refractory to AI; part II: part I | ||||||
| criteria pluspik3 camutation | ||||||
| Gdc-0032 | PI3K inhibitor | Phase I/II | GDC-0032 + fulvestrant | ER+/HER2- LABC/MBC Postmenopausal pts | NCT01296555 | |
| Byl719 | PI3K-α inhibitor | Phase I | BYL719 + letrozole or exemestane | ER+/HER2- LABC/MBC pts | NCT01870505 | |
| Mk2206 | AKT inhibitor | Phase I | Endocrine therapy + MK2206 | Postmenopausal pts with ER+ MBC | NCT01344031 | |
| Mk2206 | AKT inhibitor | Phase II | MK2206 monotherapy | LABC/LRBC/MBC withpik3ca mutation or AKT mutation or PTEN loss | NCT01277757 | |
| Azd5363 | AKT inhibitor | Phase I/II | Paclitaxel +/- AZD5363 | Parta: all MBC, partb: ER+ MBC, stratified by PIK3CA mutation | NCT01625286 | |
| Igf-1r | ||||||
| Amg 479 | IGF1R mAB | Phase II randomized | Addition of AMG 479 to either exemestane or fulvestrant | MBC or LABC pts who had progressed on prior endocrine therapy | No statistically significant difference in PFS (PFS: 3.9 vs 5.7 mo, favoring placebo arm, P = 0.44), OS or CBT between two arms | [87] |
| Bms-754807 | dual IGF-1R/insulin receptor kinase inhibitor | Phase II randomized | BMS-754807 +/- letrozole | MBC or LA BC pts who had progressed on prior nonsteroidal AI | NCT01225172 | |
| Dalotuzumab (MK-0646) | IGF1R mAB | Phase I/II | MK-0646 and fulvestrant and dasatinib | ER+/HER2- MBC pts without prior therapy in metastatic setting | NCT00903006 | |
| Cixutumumab | IGF1R mAB | Phase I/II | Cixutumumab and temsirolimus | MBC or LA BC pts progressed on on one to two chemotherapy | NCT00699491 | |
| Ridaforolimus (mk-8669) with dalotuzumab (mk-0646) Fgf | mTOR inhibitor and IGF-1R mAB | Ridaforolimus and dalotuzumab vs standard care | Er + bc | NCT01234857 | ||
| Dovitinib (TKI258) | TKI inhibits FGFR1–3, VEGFR and PDGFR | Phase II Phase I/II | Dovitinib monotherapy, stratified by FGF amplification | 4 groups of MBC pts: (group 1: FGFR1+, HR+), (group 2: FGFR1+, HR-) (group 3: FGFR1-, HR+), (group 4: FGFR1-, HR-) | Dovitinib has activity in breast cancers with amplified FGF pathway | [94] |
| Dovitinib (TKI258) | TKI inhibits FGFR1–3, VEGFR and PDGFR | Dovitinib(TKI258) + AI | ER+/HER2- postmenopausal MBC resistant to AI with fgfr1 amplification status confirmed | NCT01484041 | ||
| Dovitinib (TKI258) | TKI inhibits FGFR1–3, VEGFR and PDGFR | Phase II randomized | Fulvestrant +/- Dovitinib,stratified by FGF | Postmenopausal pts with HER2-/HR+ LA BC or MBC progressing within 12 mos of completion of adjuvant endocrine therapy or after ≤ 1 prior endocrine therapy in the advanced setting | NCT01528345 | |
| Azd4547 | Phase II | amplification | HER2-MBC with fgfr1 amplification | NCT01795768 | ||
| Azd4547 | Phase II | Fulvestrant +/- AZD4547 | ER+ postmenopausal LABC or MBC with fgfr1 polysomy or gene amplification resistant to endocrine treatment (Adjuvant or First-line Metastatic) | NCT01202591 | ||
| Targeting cell cycle regulators | ||||||
| Pd 0332991 | CDK4/6 inhibitor | Phase I/II randomized | Letrozole +/- PD 0332991 | First line therapy for postmenopausal pts with ER+/HER2- MBC | [99] | |
| Pd-0332991 (palbociclib) | CDK4/6 inhibitor | Phase III randomized | Letrozole +/- PD 0332991 | First line therapy for postmenopausal pts with ER+/HER2- MBC | NCT01740427 | |
| Lee011 | Phase Ib/II | LEE011 + exemestane +/-everolimus | Postmenopausal pts with ER+/HER2- LABC/MBC | NCT01857193 | ||
| Epigenetic therapy | ||||||
| Vorinostat | HDAC inhibitor | Phase II | Vorinostat + tamoxifen | ER+ MBC progressed on previous endocrine therapy | N = 43; 34 evaluable, 7 (21%) PR; 4 (29%) SD; ORR 19%, CBR 40% | [105] |
| Entinostat | HDAC inhibitor | phase II randomized | Exmestane+/- entinostat | MBC or LA BC pts who had progressed on prior nonsteroidal AI | N = 130; PFS: 4.3 vs 2.3 mo ( HR 0.73, 95%CI: 0.50 to 1.07; P = 0.06); OS: 28.1 vs 19.8 mo (HR 0.59, CI, 0.36 to 0.97; P = .036), favoring combination | [106] |
| Panobinostat | HDAC inhibitor | Phase I/II | Panobinostat + letrozole | MBC, triple negative phase II portion | NCT01105312 | |
| Vorinostat | HDAC inhibitor | phase II | Vorinostat + AI | ER + MBC pts who previously derived benefit from AI | NCT01153672 | |
| Vorinostat | phase II | Vorinostat/placebo + nab-paclitaxel + carboplatin (n = 62) | Primary operable breast cancer, triple-negative or high grade ER-positive, HER2-negative | Ongoing | NCT00616967 | |
MBC: Metastatic BC; LABC: Locally advanced BC; mAB: monoclonal antibody; ORR: Objective response rate; CBR: Clinical benefit rate response or stable disease >24 wk; PR: Partial response; SD: Stable disease; TKI: Tyrosine Kinase Inhibitor; PI3K/AKT/mTOR: PI3K-AKT- mammalian target of rapamycin (mTOR) pathway; IGF1R: Insulin-like growth factor-1 receptor pathway; FGF: Fibroblast growth factor. signaling.
In contrast, Wolff et al[67] examined letrozole with or without temsirolimus as first line therapy for patients with ER positive MBC who had no prior endocrine therapy for advanced disease in a randomized phase III trial. The study was terminated early due to lack of efficacy in the combination arm. Differences in results between the temsirolimus trial and the everolimus trials are likely attributable to different dosing schedules and pharmacokinetics, as well as different patient populations. It is possible that by selecting the more resistant cases, the TAMARD and BOLERO-2 trials were enhanced with breast cancers that are likely to be driven by PI3K-mTOR signaling. Studies to identify predictive biomarker that could be used to select patients who would likely benefit from the combined mTOR and ER targeting approach are needed. In addition to mTOR inhibitors, drugs targeting other components of the PI3K pathway are in clinical development. Furthermore, isozyme-specific PI3K inhibitors have been developed in the hope of increasing therapeutic benefit while decreasing toxicity. Pan-PI3K inhibitors BKM120 and XL-147, dual PI3K/mTOR inhibitors BEZ235and XL-765, and AKT inhibitor MK2206 have entered phase I, or phase I/II trials in combination of endocrine therapy.
Hedgehog signaling
The hedgehog (Hh) signaling pathway is highly conserved and plays a critical role in embryonic development. The Hh pathway has been increasingly recognized as playing a crucial role in carcinogenesis in the last decade. Three mammalian Hh ligands have been identified in humans, as denoted by the prefixes Sonic, Indian, and Desert (SHH, IHH, and DHH). They activate the Hh signaling pathway by binding to the cell surface receptor Patched (PTCH), which otherwise represses the activity of the transmembrane receptor like protein Smoothened (SMO). Release of SMO from PTCH-mediated repression subsequently leads to the modulation of GLI (glioma-associated oncogene homolog) transcription factors. There are three mammalian GLI proteins, GLI1, GLI2 and GLI3. GLI1 is a transcriptional activator; GLI2 can either activate or repress gene expression; GLI3 acts as a transcriptional repressor. Aberrant activation of the Hh pathway has been reported in several malignancies including breast cancer[68,69].
Traditionally, four major mechanisms have been proposed account for aberrant activation of the Hh pathway: (1) Hh ligand-independent mechanism - Loss of function mutations in PTCH or gain of function mutations in SMO lead to constitutive activation of this pathway; (2) Autocrine signaling- tumor cells produce Hh ligand to activate the Hh signaling; (3) Paracrine signaling - Hh ligand produced by tumor cell stimulates stromal and endothelial cells that produce growth factors supporting tumor growth and survival; and (4) Reverse paracrine signaling-Hh ligand produced by stromal cells support tumor growth and survival. Upon the pathway activation, the GLI transcription factors activate or inhibit transcription by binding to their responsive genes and interacting with the transcriptional complex. A ligand-dependent autocrine model of activating the Hh signaling has been described in breast cancer[69,70].
We recently show noncanonical Hh signaling as an alternative growth promoting mechanism that is activated in tamoxifen-resistant breast tumors. Importantly PI3K/AKT pathway plays a critical role in regulating Hh signaling by protecting key components of this pathway from proteasomal degradation. We showed that activation of Hh signaling correlated inversely with disease-free and overall survival in a cohort of 315 patients with breast cancer with poor disease outcome. Furthermore, we observed that among ER positive, node-positive patients, Hh activation in the primary tumors was an independent prognostic factor for worse disease-free survival. Add treatment of tamoxifen-resistant xenografts with anti-Hh compound GDC-0449 blocked tumor growth in mice. These promising preclinical results describe a signaling event linking PI3K/AKT pathway with Hh signaling that promotes endocrine resistance[71]. Targeting Hh pathway alone or in combination with PI3K/AKT pathway could therefore be a novel therapeutic option in treating endocrine resistant breast cancer. We are currently planning a phase I/II clinical trial using GDC-0449 (vismodegib), an oral compound approved for the management advanced basal cell carcinomas in patients with ER positive MBC that are resistant to endocrine therapy. Interestingly, Hh signalling has been shown to condition the bone microenvironment for osteolytic metastasis of breast cancer[45], therefore Hedgehog inhibitors are candidate drugs for the treatment of patients with bone metastases which is the most common site of metastasis in ER positive breast cancer.
Insulin-like growth factor-1 receptor pathway
Studies have shown that ligand activation of Insulin-like growth factor-1 receptor (IGF-1R) and its downstream pathways stimulate tumor growth by inhibition of apoptosis and promotion of transformation, metastasis and angiogenesis[72]. IGF-1R is expressed in 90% to 95% of breast cancer and is often co-expressed with ER[73]. The crosstalk between IGF-1R and ER pathway is critical for the development of IGF-1R -medicated endocrine resistance in breast cancer. For example, estrogen activates IGF1R pathway through genomic and nongenomic mechanism. IGF-1R plays a direct role in ER phosphorylation. In addition, activation of IGF-1R signaling is associated with loss of PR expression, which itself is associated with high proliferative ER positive breast cancer[74]. IGF1R overexpression also renders resistance to tamoxifen and fulvestrant through activation of MAPK and PI3K pathway.
Multiple agents interrupting the IGF-1 signaling pathway are developed and tested in clinical trials. AMG 479, a humanized monoclonal antibody antagonist of IGF1R, is tested with exemestane or fulvestrant in postmenopausal women with ER positive locally advanced or metastatic breast cancer who had disease progression on prior endocrine therapy in a randomized phase II trial. No statistically significant difference in PFS (PFS: 3.9 mo vs 5.7 mo, favoring placebo arm, P = 0.44), OS or CBT between two arms in this study[75]. Ongoing trails with IGF-1R inhibitors are listed in Table 1. Correlative studies of these trials will be critical to determine whether there is a benefit adding IGF-1R inhibition to anti-estrogen therapy in patient cases with aggressive features, such as increased proliferation.
Fibroblast growth factor signaling
Fibroblast growth factor receptor (FGFR) signaling system includes at least 18 FGF ligands and four transmembrane tyrosine kinase FGF receptors, and it is involved in cancer cell proliferation, migration, angiogenesis, and survival[76]. Multiple studies indicate that deregulated FGFRs can function as driving oncogenes stimulating tumorigenesis in a variety of human malignancies in addition to its role as an escape mechanism of anti-VEGF (vascular endothelial growth factor) therapies[76,77]. A variety of FGFR pathway alterations have been identified in cancer and include activating mutations; chromosomal translocations resulting in expression of FGFR-fusion proteins with constitutive FGFR kinase activity; aberrant splicing of FGFR and isoform switching which substantially alter ligand specificity; gene amplifications or receptor overexpression through post-transcriptional regulation. Subsequently, aberrant activation of downstream pathways results in mitogenic and antiapoptotic responses in cells[78,79].
FGFR family members are frequently overexpressed in breast cancer[28]. FGFR1 is the most commonly amplified genes following erb2/HER2 in breast cancer, present in about in 8%-15% of all breast cancer[14,76]. Large series have shown that FGFR1 amplification is associated with high proliferation as assessed by Ki-67 immunostaining, drives resistance to endocrine therapy and is an independent predictive factor of poor prognosis[22].
Preclinical models of breast cancer cells with amplification of FGFR1 or FGFR2 have demonstrated sensitivity to inhibition of FGFR[80]. Several antibodies and small molecule inhibitors of FGFR are currently in early-phase clinical trials. Dovitinib (TKI258) is a first generation oral tyrosine kinase inhibitor (TKI) which inhibits FGFR1-3, VEGFR and platelet-derived growth factor receptor (PDGFR). Dovitinib inhibits proliferation in FGFR1- and FGFR2- amplified, but not FGFR-normal, breast cancer cell lines. Dovitinib monotherapy was evaluated in the phase II trial selecting patients on the basis of hormone receptor (HR) status and FGFR1 amplification status. The mean reduction in target lesions was 21.1% in patients with FGF pathway-amplified breast cancer based on qPCR assay, compared with a 12.0% increase in target lesions in patients who did not present with FGF pathway-amplified breast cancer. Therefore, preliminary results suggest Dovitinib has antitumor activity in advanced breast cancer with FGF pathway alterations and warrants further investigation[81].
CELL CYCLE SIGNALING AND APOPTOSIS
Experimental model data and clinical correlations indicate anti-estrogen treatment leads to a G1 phase-specific cell cycle arrest and reduction in growth rate. Several molecular consequences that result in apoptosis have been documented. Aberrant regulation of positive and negative regulators of the cell cycle has been shown to interrupt and inhibit the antiproliferative effects of endocrine therapy, leading to treatment resistance[3]. For example, overexpression of the positive regulators MYC, cyclins E1 and D1 cause endocrine resistance either by activating cyclin-dependent kinases critical for G1 phase or by relieving the inhibitory effects of the negative cell cycle regulators p21 and p27[3,74]. Importantly, expression and activity of these negative cell cycle regulators are downregulated by multiple growth factor receptors and their downstream signaling pathways by modulating specific transcription factors, microRNAs, or by interfering protein phosphorylation. Moreover, increased expression of anti-apoptotic molecules such as BCl-2 and BCl-Xl and decreased expression of pro-apoptotic molecules such as BAK, BiK and caspase 9 lead to endocrine resistance as well[82]. Of note, activation of growth factor receptor signaling via the PI3K/AKT pathway is critical modulators of many apoptotic/survival molecules[83]. Cyclin D1 is a well-studied ER target gene that is required for estrogen-induced cell proliferation. Cyclin D1 binds to and activates cell cycle-dependent protein kinases four and six (CDK4/6) essential for mediating RB-induced cell cycle progression at the G1/S checkpoint[53,74]. Cyclin D1 amplification and overexpression was a common oncogenic event in breast cancer and preferentially occurred within luminal tumors, and more specifically within luminal B subtype. In the Cancer Genome Atlas (TCGA) network studies, Cyclin D1 is amplified in 58% of luminal B breast cancers with CDK4 gain in 25% of this subtype. In comparison, only 29% of luminal A tumors has Cyclin D1 amplification with 14% has CDK4 gain [14]. Furthermore, Wang et al[84] report that the alternatively spliced message, cyclin D1b, is aberrantly regulated in response to therapeutic challenge and promotes resistance to estrogen antagonists. Recently, Thangavel et al[85] noted that a unique gene signature indicative of RB protein loss of function could identify luminal B breast cancers most likely to be resistant to endocrine therapies. Therefore targeting cyclin D1 and its downstream mediators of ER action CDK4/6 may provide a viable strategy to treat endocrine resistant breast cancers.
A phase I/II clinical trial testing the efficacy of letrozole with or without PD-0332991 (an oral CDK4/6 inhibitor) was conducted as first-line treatment of ER-positive advanced breast cancer (NCT00721409). This trial excluded patients who have previously been treated for advanced breast cancer. Thus the patient population is not determined to be endocrine resistant. The preliminary results were very impressive and showed significant prolongation of median PFS with the combination when compared to letrozole alone (26.2 mo vs 7.5 mo; HR = 0.32,95%CI: 0.19-0.56, P < 0.001)[86]. The result of the randomized, multicenter, double-blind phase III study of palbociclib (PD-0332991), plus letrozole vs placebo plus letrozole for postmenopausal women with ER positive, HER2 negative MBC who have not received any prior systemic treatment for advanced disease is awaited (NCT01740427)[87]. Trials using other CDK inhibitors (Novartis) are also underway.
EPIGENETICS AND ENDOCRINE RESISTANCE
Epigenetics is defined as reversible changes in gene expression without change in the DNA sequence. DNA methylation is mediated by the action of DNA methyltransferases (DNMTs). DNMTs directly interact with histone deacetylases (HDACs) and the methyl-CpG-binding domain (MBD) family of proteins at the promoter regions to form a repressive transcription complex. DNA methylation, histone modification, and nucleosome remodeling are the major epigenetic changes that are dysregulated in breast cancer. Several genes involved in proliferation, anti-apoptosis, invasion, and metastasis have been shown to undergo epigenetic changes in breast cancer[88,89].
There is increasing evidence that epigenetic modification plays a potential role in the development of endocrine resistance in breast cancer. The epigenetic regulation of ER is mediated though the recruitment of multi-molecular complexes containing HDAC1, DNMT1 and other co-repressors to the promoter region. Methylation of the gene encoding ER-α is one of the mechanisms of loss of ER expression in ER negative breast cancer cell. The epigenetic silencing of ER target genes is crucial to the development of ER independent growth and endocrine treatment resistance. A number of preclinical studies have shown that epigenetic therapy can impact expression of ER. For example, inhibition of DNMTs in ER negative breast cancer cells leads to induction of ER expression[90,91]. HDAC inhibitors can restore ER expression, either alone via chromatin remodeling or in combination with DNMT inhibitors[89]. The TCGA study highlights the finding that breast cancer molecular subtypes harbor specific patterns of epigenetic hardwiring and further demonstrates luminal B is a distinct subtype from luminal A not only based on the mRNA-based assay but also at the methylation and protein levels[14]. Five DNA methylation groups were identified from 802 patient samples. Interestingly, the hypermethylated group 3 was significantly related to Luminal B subtype. Comparison between DNA methylation status and mRNA expression profile of group 3 with other groups led to identification of over 4000 differentially methylated genes and almost 2000 differentially expressed genes[14]. Collectively, these data provide basis for the biological rationale for combining endocrine therapy with epigenetic-targeted therapies.
A phase II study of vorinostat, a HDAC inhibitor, in combination with tamoxifen was conducted in MBC patients who had progressed on previous lines of hormone therapy[92]. The overall response rate was 19% and CBR was 40% (defined as Complete Response, Partial Response or Stable Disease of > 6 mo in duration) in 43 patients treated. The results from the randomized double blind phase II study of exmestane with or without entinostat, a benzamide HDAC inhibitor, are promising for reversal of AI endocrine therapy resistance. 130 postmenopausal women with locally recurrent or metastatic ER-positive breast cancer progressing on treatment with a nonsteroidal AI were enrolled In this study, PFS was 4.3 mo vs 2.3 mo ( HR 0.73, 95%CI: 0.50-1.07, P = 0.055) and OS was 28.1 mo vs 19.8 mo (HR = 0.59, 95%CI: 0.36-0.97) for the group receiving combination therapy vs. exmestane alone[93]. Trials combining letrozole and panobinostat, vorinostat and AI therapy in metastatic breast cancer, vorinostat and tamoxifen in early stage breast cancer, are ongoing. Based on the higher frequency of methylation observed in Luminal B tumors, it is possible that luminal B breast cancers may represent a better target for epigenetic therapy than other subtypes.
MICRO RNA
Micro RNA (miRNAs) is a class of small noncoding, single-stranded, highly conserved RNAs (19-25 nucleotides) involved in essentially all aspects of physiological and pathological cellular processes, such as development, proliferation, differentiation and apoptosis. MiRNA can either cleave mature mRNA molecules or inhibit their translation through base-pairing within the 3’-UTR of protein coding genes. Research over the past decade has demonstrated that about one third of human genes appear to be targeted by miRNAs and each miRNA is thought to regulate multiple genes. Interestingly, specific miRNA signatures have been associated with different molecular subtypes of breast cancer. In the Cancer Genome Atlas network analysis, 7 breast cancer subtypes were identified on the basis of MiRNAs expression and correlated with molecular subgroups[14]. We have explored the potential role of specific miRNAs in endocrine resistance, especially resistance to tamoxifen, in breast cancer. Studies from our and other groups showed miR-221, miR-222 and miR-181b are up-regulated, whereas miR-21, miR-342 and miRNA-489 are downregulated in the tamoxifen -resistant cells. Multiple mechanisms of these miRNAs in conferring resistance to tamoxifen have been published. Mir-221 and -222 target the cell cycle inhibitor, p27/Kip1 through posttranslational modification and sequestration of p27 protein, or through miRNA - mediated suppression. Mir-221 and -222 overexpression is known to suppress ERɑ expression at protein level which leads to tamoxifen resistance in ER positive breast cancer[19] . We recently reported that TIMP3, a tissue metalloproteinase inhibitor, is down-regulated by miR-221, -222 and -181b. We showed miRNA-mediated regulation of TIMP3 level and inhibition of metalloproteases contribute to tamoxifen resistance in cell culture models, mouse xenograft models, as well as in primary breast tumors. Direct injection of antago miRNA-221/222 to tamoxifen resistant xenografts in mice caused decrease in miRNA-221/222 level and restoration sensitivity to tamoxifen[94]. Other groups subsequently reported upregulation of miR-221and -222 is implicated in resistance to fulvestrant as well[32] .
Investigation during the last decade demonstrate emerging regulatory role of miRNAs in endocrine resistant breast cancer. Future studies evaluating miRNAs as prognostic and predictive markers, as well as novel therapeutic targets to overcome resistance are warranted.
CONCLUSION
Recent progress in the field of endocrine therapy has produced a significant number of active compounds. Patients with ER-positive advanced breast cancer are treated with different endocrine agents serially at tumor progression, often resulting in long periods of disease control with no significant toxicity. Inevitably, however, vast majority of patients will become refractory to endocrine therapy. Therefore resistance to endocrine therapy continues to be a subject of great importance. In this review, we have summarized the complex genomic and epigenetic regulatory pathways involved in endocrine resistance. A combination of ER-targeted and HER2- targeted therapies is our current standard-of-care therapy in ER positive, HER2 positive breast cancer. Early results from clinical trials suggest that subsets of patients may benefit from a combination of inhibitor targeting certain growth factor pathway with endocrine therapy. The combination of exemestane and mTOR inhibitor everolimus has been approved for ER positive advanced breast cancer in USA and Europe based on the magnitude of positive results in two randomized phase III trials. The use of epigenetic therapy or miRNA/antimiRNA-based therapy with existing endocrine therapy in breast cancer is a topic of active interest.
Many challenges still remain as we try to identify the subsets of patients most likely to benefit from these novel targeted agents. Efforts should be directed at defining biological markers that could predict the efficacy of a specific agent. The use of genome-wide approaches in detecting gene alterations that drive resistance to endocrine therapy will hopefully promote personalized cancer medicine in management of endocrine resistance breast cancer. Clearly, future clinical trials with prospective patient selection based on predictive biomarkers are needed.
Footnotes
P- Reviewer: Braga S, Specchia ML, Vetvicka V S- Editor: Wen LL L- Editor: A E- Editor: Lu YJ
References
- 1.Colditz GA. Relationship between estrogen levels, use of hormone replacement therapy, and breast cancer. J Natl Cancer Inst. 1998;90:814–823. doi: 10.1093/jnci/90.11.814. [DOI] [PubMed] [Google Scholar]
- 2.McDonnell DP, Norris JD. Connections and regulation of the human estrogen receptor. Science. 2002;296:1642–1644. doi: 10.1126/science.1071884. [DOI] [PubMed] [Google Scholar]
- 3.Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9:631–643. doi: 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- 4.Clark GM, Osborne CK, McGuire WL. Correlations between estrogen receptor, progesterone receptor, and patient characteristics in human breast cancer. J Clin Oncol. 1984;2:1102–1109. doi: 10.1200/JCO.1984.2.10.1102. [DOI] [PubMed] [Google Scholar]
- 5.Baum M, Buzdar A, Cuzick J, Forbes J, Houghton J, Howell A, Sahmoud T. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early-stage breast cancer: results of the ATAC (Arimidex, Tamoxifen Alone or in Combination) trial efficacy and safety update analyses. Cancer. 2003;98:1802–1810. doi: 10.1002/cncr.11745. [DOI] [PubMed] [Google Scholar]
- 6.Coates AS, Keshaviah A, Thürlimann B, Mouridsen H, Mauriac L, Forbes JF, Paridaens R, Castiglione-Gertsch M, Gelber RD, Colleoni M, et al. Five years of letrozole compared with tamoxifen as initial adjuvant therapy for postmenopausal women with endocrine-responsive early breast cancer: update of study BIG 1-98. J Clin Oncol. 2007;25:486–492. doi: 10.1200/JCO.2006.08.8617. [DOI] [PubMed] [Google Scholar]
- 7.Regan MM, Neven P, Giobbie-Hurder A, Goldhirsch A, Ejlertsen B, Mauriac L, Forbes JF, Smith I, Láng I, Wardley A, et al. Assessment of letrozole and tamoxifen alone and in sequence for postmenopausal women with steroid hormone receptor-positive breast cancer: the BIG 1-98 randomised clinical trial at 8·1 years median follow-up. Lancet Oncol. 2011;12:1101–1108. doi: 10.1016/S1470-2045(11)70270-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011;62:233–247. doi: 10.1146/annurev-med-070909-182917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arpino G, Green SJ, Allred DC, Lew D, Martino S, Osborne CK, Elledge RM. HER-2 amplification, HER-1 expression, and tamoxifen response in estrogen receptor-positive metastatic breast cancer: a southwest oncology group study. Clin Cancer Res. 2004;10:5670–5676. doi: 10.1158/1078-0432.CCR-04-0110. [DOI] [PubMed] [Google Scholar]
- 10.Arpino G, Wiechmann L, Osborne CK, Schiff R. Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: molecular mechanism and clinical implications for endocrine therapy resistance. Endocr Rev. 2008;29:217–233. doi: 10.1210/er.2006-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Early Breast Cancer Trialists' Collaborative Group (EBCTCG) Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365:1687–1717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- 12.Bliss JM, Kilburn LS, Coleman RE, Forbes JF, Coates AS, Jones SE, Jassem J, Delozier T, Andersen J, Paridaens R, et al. Disease-related outcomes with long-term follow-up: an updated analysis of the intergroup exemestane study. J Clin Oncol. 2012;30:709–717. doi: 10.1200/JCO.2010.33.7899. [DOI] [PubMed] [Google Scholar]
- 13.Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 14.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Britton DJ, Hutcheson IR, Knowlden JM, Barrow D, Giles M, McClelland RA, Gee JM, Nicholson RI. Bidirectional cross talk between ERalpha and EGFR signalling pathways regulates tamoxifen-resistant growth. Breast Cancer Res Treat. 2006;96:131–146. doi: 10.1007/s10549-005-9070-2. [DOI] [PubMed] [Google Scholar]
- 16.Creighton CJ. The molecular profile of luminal B breast cancer. Biologics. 2012;6:289–297. doi: 10.2147/BTT.S29923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cui X, Schiff R, Arpino G, Osborne CK, Lee AV. Biology of progesterone receptor loss in breast cancer and its implications for endocrine therapy. J Clin Oncol. 2005;23:7721–7735. doi: 10.1200/JCO.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 18.Burstein HJ, Prestrud AA, Seidenfeld J, Anderson H, Buchholz TA, Davidson NE, Gelmon KE, Giordano SH, Hudis CA, Malin J, et al. American Society of Clinical Oncology clinical practice guideline: update on adjuvant endocrine therapy for women with hormone receptor-positive breast cancer. J Clin Oncol. 2010;28:3784–3796. doi: 10.1200/JCO.2009.26.3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gutierrez MC, Detre S, Johnston S, Mohsin SK, Shou J, Allred DC, Schiff R, Osborne CK, Dowsett M. Molecular changes in tamoxifen-resistant breast cancer: relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. J Clin Oncol. 2005;23:2469–2476. doi: 10.1200/JCO.2005.01.172. [DOI] [PubMed] [Google Scholar]
- 20.Koch L. Pharmacotherapy: Small-molecule disruptors of glucokinase inhibition. Nat Rev Endocrinol. 2014;10:66. doi: 10.1038/nrendo.2013.236. [DOI] [PubMed] [Google Scholar]
- 21.Ali S, Coombes RC. Endocrine-responsive breast cancer and strategies for combating resistance. Nat Rev Cancer. 2002;2:101–112. doi: 10.1038/nrc721. [DOI] [PubMed] [Google Scholar]
- 22.Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, Natrajan R, Marchio C, Iorns E, Mackay A, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010;70:2085–2094. doi: 10.1158/0008-5472.CAN-09-3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller TW, Hennessy BT, González-Angulo AM, Fox EM, Mills GB, Chen H, Higham C, García-Echeverría C, Shyr Y, Arteaga CL. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest. 2010;120:2406–2413. doi: 10.1172/JCI41680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller TW, Balko JM, Arteaga CL. Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. J Clin Oncol. 2011;29:4452–4461. doi: 10.1200/JCO.2010.34.4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 26.Majumder S, Jacob ST. Emerging role of microRNAs in drug-resistant breast cancer. Gene Expr. 2011;15:141–151. doi: 10.3727/105221611x13176664479287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoskins JM, Carey LA, McLeod HL. CYP2D6 and tamoxifen: DNA matters in breast cancer. Nat Rev Cancer. 2009;9:576–586. doi: 10.1038/nrc2683. [DOI] [PubMed] [Google Scholar]
- 28.Frasor J, Stossi F, Danes JM, Komm B, Lyttle CR, Katzenellenbogen BS. Selective estrogen receptor modulators: discrimination of agonistic versus antagonistic activities by gene expression profiling in breast cancer cells. Cancer Res. 2004;64:1522–1533. doi: 10.1158/0008-5472.can-03-3326. [DOI] [PubMed] [Google Scholar]
- 29.Klinge CM. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001;29:2905–2919. doi: 10.1093/nar/29.14.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou Y, Yau C, Gray JW, Chew K, Dairkee SH, Moore DH, Eppenberger U, Eppenberger-Castori S, Benz CC. Enhanced NF kappa B and AP-1 transcriptional activity associated with antiestrogen resistant breast cancer. BMC Cancer. 2007;7:59. doi: 10.1186/1471-2407-7-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Riggins RB, Schrecengost RS, Guerrero MS, Bouton AH. Pathways to tamoxifen resistance. Cancer Lett. 2007;256:1–24. doi: 10.1016/j.canlet.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herynk MH, Fuqua SA. Estrogen receptor mutations in human disease. Endocr Rev. 2004;25:869–898. doi: 10.1210/er.2003-0010. [DOI] [PubMed] [Google Scholar]
- 33.Creighton CJ, Casa A, Lazard Z, Huang S, Tsimelzon A, Hilsenbeck SG, Osborne CK, Lee AV. Insulin-like growth factor-I activates gene transcription programs strongly associated with poor breast cancer prognosis. J Clin Oncol. 2008;26:4078–4085. doi: 10.1200/JCO.2007.13.4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Osborne CK, Schiff R. Growth factor receptor cross-talk with estrogen receptor as a mechanism for tamoxifen resistance in breast cancer. Breast. 2003;12:362–367. doi: 10.1016/s0960-9776(03)00137-1. [DOI] [PubMed] [Google Scholar]
- 35.Le Romancer M, Treilleux I, Leconte N, Robin-Lespinasse Y, Sentis S, Bouchekioua-Bouzaghou K, Goddard S, Gobert-Gosse S, Corbo L. Regulation of estrogen rapid signaling through arginine methylation by PRMT1. Mol Cell. 2008;31:212–221. doi: 10.1016/j.molcel.2008.05.025. [DOI] [PubMed] [Google Scholar]
- 36.Ring A, Dowsett M. Mechanisms of tamoxifen resistance. Endocr Relat Cancer. 2004;11:643–658. doi: 10.1677/erc.1.00776. [DOI] [PubMed] [Google Scholar]
- 37.Loi S, Sotiriou C, Haibe-Kains B, Lallemand F, Conus NM, Piccart MJ, Speed TP, McArthur GA. Gene expression profiling identifies activated growth factor signaling in poor prognosis (Luminal-B) estrogen receptor positive breast cancer. BMC Med Genomics. 2009;2:37. doi: 10.1186/1755-8794-2-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hurtado A, Holmes KA, Geistlinger TR, Hutcheson IR, Nicholson RI, Brown M, Jiang J, Howat WJ, Ali S, Carroll JS. Regulation of ERBB2 by oestrogen receptor-PAX2 determines response to tamoxifen. Nature. 2008;456:663–666. doi: 10.1038/nature07483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hutcheson IR, Knowlden JM, Madden TA, Barrow D, Gee JM, Wakeling AE, Nicholson RI. Oestrogen receptor-mediated modulation of the EGFR/MAPK pathway in tamoxifen-resistant MCF-7 cells. Breast Cancer Res Treat. 2003;81:81–93. doi: 10.1023/A:1025484908380. [DOI] [PubMed] [Google Scholar]
- 40.Knowlden JM, Hutcheson IR, Jones HE, Madden T, Gee JM, Harper ME, Barrow D, Wakeling AE, Nicholson RI. Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology. 2003;144:1032–1044. doi: 10.1210/en.2002-220620. [DOI] [PubMed] [Google Scholar]
- 41.Xia W, Bacus S, Hegde P, Husain I, Strum J, Liu L, Paulazzo G, Lyass L, Trusk P, Hill J, et al. A model of acquired autoresistance to a potent ErbB2 tyrosine kinase inhibitor and a therapeutic strategy to prevent its onset in breast cancer. Proc Natl Acad Sci USA. 2006;103:7795–7800. doi: 10.1073/pnas.0602468103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaufman B, Mackey JR, Clemens MR, Bapsy PP, Vaid A, Wardley A, Tjulandin S, Jahn M, Lehle M, Feyereislova A, et al. Trastuzumab plus anastrozole versus anastrozole alone for the treatment of postmenopausal women with human epidermal growth factor receptor 2-positive, hormone receptor-positive metastatic breast cancer: results from the randomized phase III TAnDEM study. J Clin Oncol. 2009;27:5529–5537. doi: 10.1200/JCO.2008.20.6847. [DOI] [PubMed] [Google Scholar]
- 43.Schwartzberg LS, Franco SX, Florance A, O’Rourke L, Maltzman J, Johnston S. Lapatinib plus letrozole as first-line therapy for HER-2+ hormone receptor-positive metastatic breast cancer. Oncologist. 2010;15:122–129. doi: 10.1634/theoncologist.2009-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miller DL, el-Ashry D, Cheville AL, Liu Y, McLeskey SW, Kern FG. Emergence of MCF-7 cells overexpressing a transfected epidermal growth factor receptor (EGFR) under estrogen-depleted conditions: evidence for a role of EGFR in breast cancer growth and progression. Cell Growth Differ. 1994;5:1263–1274. [PubMed] [Google Scholar]
- 45.Fedele P, Calvani N, Marino A, Orlando L, Schiavone P, Quaranta A, Cinieri S. Targeted agents to reverse resistance to endocrine therapy in metastatic breast cancer: where are we now and where are we going? Crit Rev Oncol Hematol. 2012;84:243–251. doi: 10.1016/j.critrevonc.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 46.Mueller H, Flury N, Eppenberger-Castori S, Kueng W, David F, Eppenberger U. Potential prognostic value of mitogen-activated protein kinase activity for disease-free survival of primary breast cancer patients. Int J Cancer. 2000;89:384–388. doi: 10.1002/1097-0215(20000720)89:4<384::aid-ijc11>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 47.Cristofanilli M, Valero V, Mangalik A, Royce M, Rabinowitz I, Arena FP, Kroener JF, Curcio E, Watkins C, Bacus S, et al. Phase II, randomized trial to compare anastrozole combined with gefitinib or placebo in postmenopausal women with hormone receptor-positive metastatic breast cancer. Clin Cancer Res. 2010;16:1904–1914. doi: 10.1158/1078-0432.CCR-09-2282. [DOI] [PubMed] [Google Scholar]
- 48.Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, Weiss H, Rimawi M, Schiff R. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008;68:826–833. doi: 10.1158/0008-5472.CAN-07-2707. [DOI] [PubMed] [Google Scholar]
- 49.Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, Schiff R. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst. 2004;96:926–935. doi: 10.1093/jnci/djh166. [DOI] [PubMed] [Google Scholar]
- 50.Leary AF, Drury S, Detre S, Pancholi S, Lykkesfeldt AE, Martin LA, Dowsett M, Johnston SR. Lapatinib restores hormone sensitivity with differential effects on estrogen receptor signaling in cell models of human epidermal growth factor receptor 2-negative breast cancer with acquired endocrine resistance. Clin Cancer Res. 2010;16:1486–1497. doi: 10.1158/1078-0432.CCR-09-1764. [DOI] [PubMed] [Google Scholar]
- 51.Johnston S, Pippen J, Pivot X, Lichinitser M, Sadeghi S, Dieras V, Gomez HL, Romieu G, Manikhas A, Kennedy MJ, et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J Clin Oncol. 2009;27:5538–5546. doi: 10.1200/JCO.2009.23.3734. [DOI] [PubMed] [Google Scholar]
- 52.Osborne CK, Neven P, Dirix LY, Mackey JR, Robert J, Underhill C, Schiff R, Gutierrez C, Migliaccio I, Anagnostou VK, et al. Gefitinib or placebo in combination with tamoxifen in patients with hormone receptor-positive metastatic breast cancer: a randomized phase II study. Clin Cancer Res. 2011;17:1147–1159. doi: 10.1158/1078-0432.CCR-10-1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Polychronis A, Sinnett HD, Hadjiminas D, Singhal H, Mansi JL, Shivapatham D, Shousha S, Jiang J, Peston D, Barrett N, et al. Preoperative gefitinib versus gefitinib and anastrozole in postmenopausal patients with oestrogen-receptor positive and epidermal-growth-factor-receptor-positive primary breast cancer: a double-blind placebo-controlled phase II randomised trial. Lancet Oncol. 2005;6:383–391. doi: 10.1016/S1470-2045(05)70176-5. [DOI] [PubMed] [Google Scholar]
- 54.Smith IE, Walsh G, Skene A, Llombart A, Mayordomo JI, Detre S, Salter J, Clark E, Magill P, Dowsett M. A phase II placebo-controlled trial of neoadjuvant anastrozole alone or with gefitinib in early breast cancer. J Clin Oncol. 2007;25:3816–3822. doi: 10.1200/JCO.2006.09.6578. [DOI] [PubMed] [Google Scholar]
- 55.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–644. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tran B, Bedard PL. Luminal-B breast cancer and novel therapeutic targets. Breast Cancer Res. 2011;13:221. doi: 10.1186/bcr2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Trotman LC, Pandolfi PP. PTEN and p53: who will get the upper hand? Cancer Cell. 2003;3:97–99. doi: 10.1016/s1535-6108(03)00022-9. [DOI] [PubMed] [Google Scholar]
- 58.Dean JL, Thangavel C, McClendon AK, Reed CA, Knudsen ES. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene. 2010;29:4018–4032. doi: 10.1038/onc.2010.154. [DOI] [PubMed] [Google Scholar]
- 59.DeGraffenried LA, Fulcher L, Friedrichs WE, Grünwald V, Ray RB, Hidalgo M. Reduced PTEN expression in breast cancer cells confers susceptibility to inhibitors of the PI3 kinase/Akt pathway. Ann Oncol. 2004;15:1510–1516. doi: 10.1093/annonc/mdh388. [DOI] [PubMed] [Google Scholar]
- 60.Ghayad SE, Bieche I, Vendrell JA, Keime C, Lidereau R, Dumontet C, Cohen PA. mTOR inhibition reverses acquired endocrine therapy resistance of breast cancer cells at the cell proliferation and gene-expression levels. Cancer Sci. 2008;99:1992–2003. doi: 10.1111/j.1349-7006.2008.00955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Treeck O, Wackwitz B, Haus U, Ortmann O. Effects of a combined treatment with mTOR inhibitor RAD001 and tamoxifen in vitro on growth and apoptosis of human cancer cells. Gynecol Oncol. 2006;102:292–299. doi: 10.1016/j.ygyno.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 62.Sadler TM, Gavriil M, Annable T, Frost P, Greenberger LM, Zhang Y. Combination therapy for treating breast cancer using antiestrogen, ERA-923, and the mammalian target of rapamycin inhibitor, temsirolimus. Endocr Relat Cancer. 2006;13:863–873. doi: 10.1677/erc.1.01170. [DOI] [PubMed] [Google Scholar]
- 63.Beeram M, Tan QT, Tekmal RR, Russell D, Middleton A, DeGraffenried LA. Akt-induced endocrine therapy resistance is reversed by inhibition of mTOR signaling. Ann Oncol. 2007;18:1323–1328. doi: 10.1093/annonc/mdm170. [DOI] [PubMed] [Google Scholar]
- 64.Baselga J, Semiglazov V, van Dam P, Manikhas A, Bellet M, Mayordomo J, Campone M, Kubista E, Greil R, Bianchi G, et al. Phase II randomized study of neoadjuvant everolimus plus letrozole compared with placebo plus letrozole in patients with estrogen receptor-positive breast cancer. J Clin Oncol. 2009;27:2630–2637. doi: 10.1200/JCO.2008.18.8391. [DOI] [PubMed] [Google Scholar]
- 65.Baselga J, Campone M, Piccart M, Burris HA, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366:520–529. doi: 10.1056/NEJMoa1109653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bachelot T, Bourgier C, Cropet C, Ray-Coquard I, Ferrero JM, Freyer G, Abadie-Lacourtoisie S, Eymard JC, Debled M, Spaëth D, et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: a GINECO study. J Clin Oncol. 2012;30:2718–2724. doi: 10.1200/JCO.2011.39.0708. [DOI] [PubMed] [Google Scholar]
- 67.Wolff AC, Lazar AA, Bondarenko I, Garin AM, Brincat S, Chow L, Sun Y, Neskovic-Konstantinovic Z, Guimaraes RC, Fumoleau P, et al. Randomized phase III placebo-controlled trial of letrozole plus oral temsirolimus as first-line endocrine therapy in postmenopausal women with locally advanced or metastatic breast cancer. J Clin Oncol. 2013;31:195–202. doi: 10.1200/JCO.2011.38.3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sahebjam S, Siu LL, Razak AA. The utility of hedgehog signaling pathway inhibition for cancer. Oncologist. 2012;17:1090–1099. doi: 10.1634/theoncologist.2011-0450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Takebe N, Hunsberger S, Yang SX. Expression of Gli1 in the hedgehog signaling pathway and breast cancer recurrence. Chin J Cancer Res. 2012;24:257–258. doi: 10.3978/j.issn.1000-9604.2012.09.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ellis MJ, Tao Y, Young O, White S, Proia AD, Murray J, Renshaw L, Faratian D, Thomas J, Dowsett M, et al. Estrogen-independent proliferation is present in estrogen-receptor HER2-positive primary breast cancer after neoadjuvant letrozole. J Clin Oncol. 2006;24:3019–3025. doi: 10.1200/JCO.2005.04.3034. [DOI] [PubMed] [Google Scholar]
- 71.Ramaswamy B, Lu Y, Teng KY, Nuovo G, Li X, Shapiro CL, Majumder S. Hedgehog signaling is a novel therapeutic target in tamoxifen-resistant breast cancer aberrantly activated by PI3K/AKT pathway. Cancer Res. 2012;72:5048–5059. doi: 10.1158/0008-5472.CAN-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8:915–928. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- 73.Shimizu C, Hasegawa T, Tani Y, Takahashi F, Takeuchi M, Watanabe T, Ando M, Katsumata N, Fujiwara Y. Expression of insulin-like growth factor 1 receptor in primary breast cancer: immunohistochemical analysis. Hum Pathol. 2004;35:1537–1542. doi: 10.1016/j.humpath.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 74.Lukas J, Bartkova J, Bartek J. Convergence of mitogenic signalling cascades from diverse classes of receptors at the cyclin D-cyclin-dependent kinase-pRb-controlled G1 checkpoint. Mol Cell Biol. 1996;16:6917–6925. doi: 10.1128/mcb.16.12.6917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kaufman JF, H Bourgeois, H Kennecke. A Randomized, Double-Blind, Placebo-Controlled, Phase 2 Study of AMG 479 With Exemestane (E) or Fulvestrant (F) in Postmenopausal Women With Hormone-Receptor Positive (HR ) Metastatic (M) or Locally Advanced (LA) Breast Cancer (BC) Cancer Res. 2010;70:S1–4. [Google Scholar]
- 76.Dienstmann R, Rodon J, Prat A, Perez-Garcia J, Adamo B, Felip E, Cortes J, Iafrate AJ, Nuciforo P, Tabernero J. Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors. Ann Oncol. 2014;25:552–563. doi: 10.1093/annonc/mdt419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10:116–129. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 78.Heinzle C, Sutterlüty H, Grusch M, Grasl-Kraupp B, Berger W, Marian B. Targeting fibroblast-growth-factor-receptor-dependent signaling for cancer therapy. Expert Opin Ther Targets. 2011;15:829–846. doi: 10.1517/14728222.2011.566217. [DOI] [PubMed] [Google Scholar]
- 79.Wesche J, Haglund K, Haugsten EM. Fibroblast growth factors and their receptors in cancer. Biochem J. 2011;437:199–213. doi: 10.1042/BJ20101603. [DOI] [PubMed] [Google Scholar]
- 80.Hynes NE, Dey JH. Potential for targeting the fibroblast growth factor receptors in breast cancer. Cancer Res. 2010;70:5199–5202. doi: 10.1158/0008-5472.CAN-10-0918. [DOI] [PubMed] [Google Scholar]
- 81.André F, Bachelot T, Campone M, Dalenc F, Perez-Garcia JM, Hurvitz SA, Turner N, Rugo H, Smith JW, Deudon S, et al. Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin Cancer Res. 2013;19:3693–3702. doi: 10.1158/1078-0432.CCR-13-0190. [DOI] [PubMed] [Google Scholar]
- 82.Mandlekar S, Kong AN. Mechanisms of tamoxifen-induced apoptosis. Apoptosis. 2001;6:469–477. doi: 10.1023/a:1012437607881. [DOI] [PubMed] [Google Scholar]
- 83.Riggins RB, Bouton AH, Liu MC, Clarke R. Antiestrogens, aromatase inhibitors, and apoptosis in breast cancer. Vitam Horm. 2005;71:201–237. doi: 10.1016/S0083-6729(05)71007-4. [DOI] [PubMed] [Google Scholar]
- 84.Wang Y, Dean JL, Millar EK, Tran TH, McNeil CM, Burd CJ, Henshall SM, Utama FE, Witkiewicz A, Rui H, et al. Cyclin D1b is aberrantly regulated in response to therapeutic challenge and promotes resistance to estrogen antagonists. Cancer Res. 2008;68:5628–5638. doi: 10.1158/0008-5472.CAN-07-3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Thangavel C, Dean JL, Ertel A, Knudsen KE, Aldaz CM, Witkiewicz AK, Clarke R, Knudsen ES. Therapeutically activating RB: reestablishing cell cycle control in endocrine therapy-resistant breast cancer. Endocr Relat Cancer. 2011;18:333–345. doi: 10.1530/ERC-10-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Finn RS, Crown JP, Lang I. Results of a randomized phase 2 study of PD 0332991, a cyclin-dependent kinase (CDK) 4/6 inhibitor, in combination with letrozole vs letrozole alone for first-line treatment of ER /HER2– advanced breast cancer (BC) Cancer Res. 2012:24 Suppl: Abstract nr S1–6. [Google Scholar]
- 87.Finn RS, Dieras V, Gelmon KA. A randomized, multicenter, double-blind phase III study of palbociclib (PD-0332991), an oral CDK 4/6 inhibitor, plus letrozole vs placebo plus letrozole for the treatment of postmenopausal women with ER( ), HER2(–) breast cancer who have not received any prior systemic anticancer treatment for advanced disease. J Clin Oncol. 2013:14. [Google Scholar]
- 88.Frogne T, Benjaminsen RV, Sonne-Hansen K, Sorensen BS, Nexo E, Laenkholm AV, Rasmussen LM, Riese DJ, de Cremoux P, Stenvang J, et al. Activation of ErbB3, EGFR and Erk is essential for growth of human breast cancer cell lines with acquired resistance to fulvestrant. Breast Cancer Res Treat. 2009;114:263–275. doi: 10.1007/s10549-008-0011-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lustberg MB, Ramaswamy B. Epigenetic Therapy in Breast Cancer. Curr Breast Cancer Rep. 2011;3:34–43. doi: 10.1007/s12609-010-0034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ferguson AT, Lapidus RG, Baylin SB, Davidson NE. Demethylation of the estrogen receptor gene in estrogen receptor-negative breast cancer cells can reactivate estrogen receptor gene expression. Cancer Res. 1995;55:2279–2283. [PubMed] [Google Scholar]
- 91.Yang X, Phillips DL, Ferguson AT, Nelson WG, Herman JG, Davidson NE. Synergistic activation of functional estrogen receptor (ER)-alpha by DNA methyltransferase and histone deacetylase inhibition in human ER-alpha-negative breast cancer cells. Cancer Res. 2001;61:7025–7029. [PubMed] [Google Scholar]
- 92.Munster PN, Thurn KT, Thomas S, Raha P, Lacevic M, Miller A, Melisko M, Ismail-Khan R, Rugo H, Moasser M, et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br J Cancer. 2011;104:1828–1835. doi: 10.1038/bjc.2011.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yardley DA, Ismail-Khan RR, Melichar B, Lichinitser M, Munster PN, Klein PM, Cruickshank S, Miller KD, Lee MJ, Trepel JB. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J Clin Oncol. 2013;31:2128–2135. doi: 10.1200/JCO.2012.43.7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lu Y, Roy S, Nuovo G, Ramaswamy B, Miller T, Shapiro C, Jacob ST, Majumder S. Anti-microRNA-222 (anti-miR-222) and -181B suppress growth of tamoxifen-resistant xenografts in mouse by targeting TIMP3 protein and modulating mitogenic signal. J Biol Chem. 2011;286:42292–42302. doi: 10.1074/jbc.M111.270926. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]