

Abstract
There have been significant improvements in the detection and treatment of breast cancer in recent decades. However, there is still a need to develop more effective therapeutic techniques that are patient specific with reduced toxicity leading to further increases in patients’ overall survival; the ongoing progress in understanding recurrence, resistant and spread also needs to be maintained. Better understanding of breast cancer pathology, molecular biology and progression as well as identification of some of the underlying factors involved in breast cancer tumourgenesis and metastasis has led to the identification of novel therapeutic targets. Over a number of years interest has risen in breast tumour kinase (Brk) also known as protein tyrosine kinase 6; the research field has grown and Brk has been described as a desirable therapeutic target in relation to tyrosine kinase inhibition as well as disruption of its kinase independent activity. This review will outline the current “state of play” with respect to targeted therapy for breast cancer, as well as discussing Brk’s role in the processes underlying tumour development and metastasis and its potential as a therapeutic target in breast cancer.
Keywords: Breast tumour kinase, Protein tyrosine kinase 6, Breast neoplasms, Targeted molecular therapy, Intracellular signaling peptides and proteins, Protein kinase inhibitors
Core tip: Breast tumour kinase/protein tyrosine kinase 6 is overexpressed in up to 86% of invasive breast cancers. It plays a key role in regulating a number of cell processes that are involved in the metastatic process. As a kinase involved in both epidermal growth factor and insulin like growth factors signaling, inhibiting its activity could prove to be an effective way to enhance the effects of current targeted treatments. In addition, disrupting the protein-protein interactions central for the kinase-independent aspects of its function may generate an alternative mechanism for selective targeting of breast cancer cells.
INTRODUCTION
Exactly how breast cancer therapeutics has changed over the years is an intriguing aspect of the progress and evolution that has been made in research for cancer cures and treatments. Looking at the incidence and mortality rates over the years in the United Kingdom and worldwide indicates the significant impact that continued improvements in detection and treatment of breast cancer have made. Statistically breast cancer incidence rates have been increasing since the 1970s[1]. Approximately 75000 cases of breast cancer were diagnosed in 1975 compared to approximately 126000 in 2010[1]. The increased frequency may be due to improved diagnostic techniques that contribute towards the rise in incidence; thus detecting cancers that may have remained unnoticed until much later. Breast cancer is the most common cancer in the United Kingdom with more than 48000 people being diagnosed each year[1] and is the most recognized cause for the mortality rates of women aged between 33 and 55. This indicates room for improvement in relation to enhanced and effective therapeutic techniques that are patient specific, relatively non-toxic and contribute towards patients overall survival. Not only that, but there is ongoing progress needed towards understanding recurrence, resistant and spread[2] .
A closer look at breast cancer pathology, molecular biology and progression may lead to a better understanding of the underlying factors involved in breast cancer tumourgenesis and metastasis. A particular interest has risen in breast tumour kinase (Brk) also known as protein tyrosine kinase 6 (PTK6). Over a number of years the Brk research field has grown and Brk has been hinted at, as being a desirable therapeutic target in relation to tyrosine kinase inhibition as well as disruption of its kinase independent activity. Here, we outline Brk’s role in the pathways essential for breast tumour development and discuss its potential as a therapeutic target in breast cancer.
BRK
Brk, also known as PTK6, was originally identified in a study involving human melanocytes and subsequently isolated from breast cancer cell lines in a study identifying novel kinases with therapeutic potential[3]. The ptk6 gene encodes the non-receptor tyrosine kinase, which consists of SH2, SH3 a linker region and catalytic domains. Total activity of Brk is significantly higher in malignancy than in normal mammary tissue, and over-expression of the protein has been noted in more than 80% of invasive ductal breast tumors[4]. Brk expression so far has been detected in the majority of breast cancer cell lines with differing intensities[5]. Gene sequencing indicated similarities with the SRC-family of protein tyrosine kinases, however there are distinct differences such as the lack of N-terminal extension and consensus sequences for fatty acylation and membrane association[6]. Furthermore, its genomic structure is quite distinct from the SRC-family PTKs, which demonstrates an evolutionary divergence[7]. Brk has also been shown to have a significant degree of similarity with the Drosophila src related gene known as Dsrc41, with six out of seven of Brk’s exon boundaries conserved with the Dsrc41 gene which has 9 exons. This could indicate Brk is likely to share a common ancestor with Dsrc41. The ptk6 gene comprises 8 exons and encodes a 451 amino acid protein and FISH studies indicated localisation to chromosome 20q13.3[6]. The protein product has a predicted molecular weight of 50 kDa, which generally resolves to around 48 kDa on an SDS-PAGE gel.
SH3 domains are small protein nodules made up of β-sheets. SH3 domains allow the assembly of specific protein complexes via proline-rich peptide binding, reviewed in[8]. Brk’s SH2 and SH3 domains are used for substrate recognition[9,10]. The Brk SH3 domain has been known to undergo conformational changes due to pH fluctuations indicating that its structure could determine substrate and protein interaction, thus influencing its varied role in diverse cellular environments. The SH3 domain may have a role in enzyme regulation[11]. The SH2 domain contains α/β folds and a phosphotyrosine binding surface with two α-helices opposite a central βα-sheet made up of four anti-parallel strands[12]. This domain plays a role in protein-protein interactions and is important for regulation of catalytic activity[10]. Due to the lack of myristoylation and a nuclear localization sequence, Brk’s regulation is difficult to determine which allows for more flexibility with its subcellular localization, reviewed in[13].
REASONS FOR NEW CANCER THERAPIES
Chemotherapy and radiotherapy have been recognised to target normal rapidly dividing cells such as bone marrow, gastrointestinal tract or hair follicles; the range and intensity of adverse effects reduces the specificity and increases toxicity of these therapies. Both these types of treatments therefore have a limited therapeutic index and can often be palliative in use as reviewed in[14]. Hormonal therapies have also been in use; for example Tamoxifen, which has been used for early stage and metastatic breast cancer since its licence in 1972[15]. Although proven to be effective against breast cancer, especially those that are oestrogen receptor positive, there are still many issues that need to be overcome for maximum effect of the drug to be achieved, whether by itself or as combination therapy. These include the diverse adverse toxicities such as thrombosis, strokes and development of secondary cancers. Other issues include resistance against Tamoxifen and subsequent recurrence in some patients. Furthermore, this drug is only effective against oestrogen or progesterone receptor positive breast cancers thus making it unsuitable for other types of breast cancer such as HER2 positive/ER/PR negative and triple negative breast cancers (reviewed in[16]). However there are treatments available for HER2 positive cancers such as Herceptin, a monoclonal antibody that binds to HER2 thus negatively effecting receptor function, as well as a number of kinase inhibitors. Unfortunately the more advanced stages of breast cancer do not always respond to Herceptin therapy and those that do, often progress in 12 mo from the start of the treatment[17]. In addition resistance may occur due to the involvement of a number of signaling pathway molecules such as activation of the PI3K/AKT pathway, loss of PTEN and activation of PIK3CA, reviewed in[18]. Brk is expressed in a wide range of cancer types and its expression appears to be independent of ER/PR/HER2 positivity, thus making it an ideal candidate for therapeutic intervention[5,19].
TYROSINE KINASE INHIBITION
Targeted therapy is largely directed specifically towards tumour cells thus reducing many side effects and providing a wider therapeutic window. They can also be used in combination with traditional chemotherapy or radiotherapy to enhance anticancer effects. Further patient benefit includes convenience with oral consumption rather than intravenous administration as is the case for many chemotherapy drugs.
Tyrosine kinases have been implicated in a range of cancers; they are involved in cellular signalling and play an important role in growth factor signalling. In their active forms tyrosine kinases can promote tumour cell proliferation, growth and induce anti-apoptotic effects, as well as promote angiogenesis and tumour cell metastasis. Since most of these cellular events contribute to tumour progression and decreased patient prognosis, tyrosine kinases are considered ideal candidates for targeted therapy.
There are two main types of kinases; these can be categorized as receptor protein kinases that are generally membrane spanning, or non-receptor protein kinases that relay intracellular signals from the receptors and, of which, Brk is one. Briefly, when ligands bind to their cognate receptors, they stimulate receptor dimerization followed by autophosphorylation and activation of tyrosine kinase activity. As a result, multiple signalling pathways are activated and intracellular mediators in these pathways transduce signals from membrane receptors through the cytosol and into the nucleus which ultimately alters DNA synthesis and cell division as well as a wide range of biological processes as reviewed in[14]. Protein tyrosine kinase activity within breast tumour tissues has been reported to be significantly higher in comparison with benign or normal breast tissues[20]. For example, the tyrosine kinase activity of the product of c-src proto-oncogene has been found to be elevated in human breast tumors[21], and several tyrosine kinase receptors have also been implicated in breast cancer development and progression.
These include members of the erbB Type 1 transmemebrane receptor family such as epidermal growth factor receptor (EGFR) and HER2/neu transmembrane tyrosine kinase receptor, as well as receptors for insulin like growth factors (IGF) such as IGF-1R[22,23]. Others include fibroblast growth factor receptor[24] and met receptor tyrosine kinase[25]. Of the substrates and proteins interacting with Brk[13], many are linked to signalling from these receptors, thus indicating a role for Brk in these signalling pathways.
EXAMPLES OF TYROSINE KINASE INHIBITORS IN BREAST CANCER-LAPATINIB, ERLOTINIB AND GETFINTIB
Gefitinib is an EGFR inhibitor, the first of its kind, that is also known to have an effect in HER2-overexpressing cell lines probably due to the reduction in phosphorylation of HER2/EGFR heterodimer[26]. Gefitinib is especially effective in ER-positive and Tamoxifen resistant tumours indicating a target group of patients for this type of treatment[27]. Erlotinib works in much the same way as Gefitinib, as a reversible EGFR inhibitor. It is clinically effective in locally advanced and metastatic non-small cell lung cancer[28] but its benefit in breast is only recently becoming clearer, and there is some indication for Erlotinib in triple negative breastcancers[29]. Erlotinib inhibits triple negative breast cancer as shown by Ueno and Zhang[30] when they generated a SUM149 xenograft model by implanting luciferase expressing SUM149 cells into mammary pads of athymic nude mice. The results indicated significant inhibition of tumour growth at doses of 50 and 100 mg/kg.
There is a strong correlation between HER2 and EGFR in terms of dimerization, expression as well as activity[31] since HER2 is EGFR’s most common heterodimerization partner and HER2 potentiates EGFR signalling by enhancing EGF binding affinity[32]. A dual inhibitor for HER2 and EGFR may therefore be of greater clinical benefit than individual therapies. When using an inhibitor with a single mode of action, therapeutic resistance can be increased whereas dual inhibition may reduce the chance of this happening[32]. Other benefits of dual inhibition may include targeting a wider range of cancers since single inhibitors are, in some cases, specific for one particular type of cancer as well as a more effective inhibition in cancer cell growth overall[33].
One such inhibitor already in use is Lapatinib, which reversibly inhibits the tyrosine kinase activity of both HER2 and EGFR, reviewed in[32]. It is an orally available inhibitor that binds to EGFR in its inactive form in comparison to other EGFR inhibitors such as Erlotinib and Geftinib, which bind EGFR in its active form. This allows for a greater duration of effect at the target site[34]. So far Lapatinib has proven to be well tolerated with rare occurrences of high-grade toxicities. In relation to Brk, Lapatinib has been recognised to have reduced efficacy with increased levels of Brk in HER2-transfected mammary epithelial cells[35]. This may suggest a role for Brk in acquired resistance against HER2 targeted therapy when both proteins are over-expressed. Targeting Brk alongside HER2 inhibition could overcome resistance as well as increase efficacy of HER2-targetted treatment.
Both Gefitinib and Erlotinib initially indicated no real clinical benefit for metastatic breast cancer patients and had little efficacy in phase II studies[27]. This indicates lack of correlation between EGFR expression level with response to treatment even though many breast cancer cells express EGFR[36]. Previously there was strong evidence for combination therapy with tyrosine kinase inhibitors and hormone therapy[32], thus allowing for a greater clinical benefit. However more recently, combination therapy using Geftinib and anti-hormonal therapy (fulvestrant and anastrozole) demonstrated only a modest increase in clinical benefit compared to Geftinib or hormonal therapy alone[37].
Erlotinib resistance is linked to CDK2 activity; cell proliferation is induced in Erlotinib treated cells when CDK2 is expressed and, conversely, there is increased sensitivity to Erlotinib when CDK2 activity is suppressed. Erlotinib has also shown to upregulate p27 and nuclear translocation in association with cell growth inhibition in non-small cell lung cancer[38]. Cyclin dependent kinases regulate cell cycle progression from quiescence to mitosis by activating the transcription factor E2F leading to activation of genes needed for progression from G1 to S phase[36]. Furthermore, in relation to breast cancer it has been shown that inhibition of the erbB2 pathway by EGFR inhibitors in erbB2-overexpressing breast cancer cell lines caused G1 phase arrest with accumulation of p27 and reduced Cyclin D1[39]. This is further verified by Nahta et al[40] who showed that downregulation of p27 in breast cancer cells was accompanied by an increased S-phase fraction and increased resistance to Herceptin therapy. The induction of p27 is therefore necessary in Erlotinib inhibition of cell growth.
Since cell cycle deregulation is implicated in development of neoplasia and may contribute towards development of breast cancer, as well as response to therapy the discovery that Brk is involved in cell cycle regulation was of importance[41]. Brk deregulates cell cycle progression; an inverse correlation was shown between Brk expression levels and p27 expression levels. With increasing Brk expression there was reduced p27, and in cells lacking p27 and Fox03a, Brk induced a decreased level of cell proliferation indicating it may need the p27/Fox03a pathway to promote cell growth. This indicates the importance of Brk’s role in cell cycle progression and suggests a role for Brk in mediating cell responses to EGFR/HER2 inhibitors through regulation of p27.
BRK AND HER2 INTERACTIONS
In cancer cells, alterations in HER receptors or in their downstream signalling components are known to occur; HER2 is a negative prognostic factor, the presence of which indicates aggressive phenotype and reduced overall survival rate as reviewed in[42], although it should be noted that survival rates for HER2 positive cancers are improving due to the introduction of targeted therapies[43].
HER2 is overexpressed in approximately 20%-30% of breast cancers[44] and it has been suggested that Brk may be involved in regulation of signal transduction from HER tyrosine kinases. Several studies have shown a link between HER2/neu expression and Brk/PTK6[35,45].
This is of interest because, as discussed above, HER2 targeted therapy although clinically proven to be effective does pose some restrictions such as lack of effect in some HER2/neu positive cancers as well as resistance. Therefore, Brk targeted therapy maybe of clinical benefit especially when used in combination with existing HER2/neu targeted therapy. Due to the strong correlation between PTK6 and HER2, there is evidence suggesting that it may also be linked with prognosis thus indicating a role for Brk as a prognostic factor in breast cancer. PTK6 expression studied in 426 breast cancer cases further supports its potential as a independent prognostic factor regardless of morphological and molecular markers such as lymph node involvement, tumour size and HER2 status[46]. Co-expression of Brk and HER2 has been linked to co-amplification of both the ErbB2 and ptk6 genes[35], although this is not a consistent finding in other patient studies. It is worth noting that although HER2 is over expressed in 20%-30% of breast cancers[44], Brk is over expressed in upto 86% of breast cancers[47], which indicates that in the majority of breast cancers Brk is over expressed independently of HER2 status.
At a protein level, Aubele and colleagues showed that PTK6 forms protein complexes with HER2 in paraffin tissues from invasive breast carcinomas[48]. Recently, the effect of simultaneous knockdown of PTK6 and HER2 has been analysed in the herceptin-resistant cell line JIMT-1[49]. The results indicated significant reduction in phosphorylation of important signalling intermediates, namely MAPK, ERK and p38MAPK and PTEN, which are involved in tumourgenesis. Furthermore there was reduced migration and invasion of JIMT-1 cells when expression of both proteins was suppressed. HER2 may also be involved in elevating Brk levels via upregulating calpastatin and inhibiting calpain-1 activity in breast cancer cells[50].
These combined data further support the study of dual inhibition of Brk and HER2, firstly for a greater clinical effect and secondly to overcome anti-HER2 therapeutic resistance.
BRK AND EGFR INTERACTIONS
ErbB signalling in breast tumour progression has been extensively documented. EGFR tyrosine kinases have been involved in regulation of normal and abnormal cellular proliferation and survival (reviewed in[51]). Both EGFR and HER2 are intrinsically linked; HER2 potentiates EGFR signalling by enhancing binding ability of EGF and reducing its degradation and studies have indicated that HER2 signalling is reduced via EGFR-specific inhibitors[52].
The EGF receptor family are linked to the tumourgenic transformation of breast epithelial cells. When Brk Transfected mammary epithelial cell lines, MCF10A and Hb4a, were treated with human growth factors (EGF), mitogenic activity of Brk was observed. This reveals Brk’s role in sensitizing human mammary epithelial cells to growth factors such as EGF[53]. Furthermore, Brk association with the EGF receptor has also been detected, even in the absence of EGF, leading to proliferation of epithelial cells. EGFR expression also plays a role in keratinocyte differentiation[54], a process in which Brk is involved[55]. Inhibition of EGFR signalling during differentiation induces growth arrest, however Brk’s promotion of EGFR signalling suggests that one of its roles may involve promoting cell survival during early differentiation.
EGF stimulation results in rapid phosphorylation of Brk indicating the involvement of Brk in the EGF signalling complex[56]. EGF binding to its receptor, EGFR, not only induces its phosphorylation but also the phosphorylation of other EGFR family members (HER2, 3 and 4). The expression of Brk therefore not only increases phosphorylation of EGFR and HER2 but also of HER3 (erbB3) in breast cell lines[56]. It may do this via direct interaction with an erbB3-containing complex in response to EGF stimulation. Brk’s ability to enhance EGFR signalling could be mediated by inhibition of EGFR downregulation[57], which is achieved by phosphorylation of either ARAP-1[58] or c-Cbl[59], thereby prolonging EGFR signalling.
Expression of Brk enhances EGF-induced ErbB3 phosphorylation and recruitment of P13K to ErbB3 thus inducing P13K activity[56]. Since the P13K pathway is implicated in breast cancer and is linked with resistance to HER2 targeted therapies, its interaction with Brk may give a wider overview of the mechanisms involved in developing resistance. P13K has been a desirable therapeutic target of its own and current therapies, as reviewed in[60], focus on anti-mTOR agents, tyrosine kinase inhibitors and P13K inhibitors. However to create a greater clinical effect, combinations of these treatments including a potential Brk tyrosine kinase inhibitor maybe more useful and reduce the activation of compensatory feedback loops which could decrease efficacy of single agents. Since Akt is a known substrate of Brk, and Brk negatively regulates its phosphorylation in epithelial cells, the potential consequences of this therapy may need to be fully assessed[61]. Nonetheless, due to most of the normal cells that express Brk being outside the proliferative areas of the tissues Brk targeted therapy may still be highly specific[62].
Overall Brk has shown to prolong EGFR signalling, sensitise tumour cells to EGF stimulation, inhibit degradation of EGFR and reduce sensitivity of EGFR inhibitors in Brk overexpressing cells. This suggests Brk inhibiton could reduce tumour proliferation and growth via reduced EGFR signalling and increase EGFR inhibitor sensitivity.
BRK AND IGF INTERACTIONS
Insulin receptor substrate 4 (IRS-4) is part of the insulin receptor substrate family, which also contains IRS-1, IRS-2 and IRS-3. Their function includes a variety of biological effects such as cell proliferation, growth, survival and differentiation downstream of insulin and insulin-like growth factor 1 (IGF-1) receptors[63]. IRS-4, upon IGF-1 stimulation, is phosphorylated which leads it to binding to P13K activating the MAPK pathway. The link between insulin-like growth factor (IGF) and breast cancer has been well documented; the IGF-1 receptor is significantly overexpressed in tumour cell lines compared to normal breast cancer epithelial tissue and benign tumours[64] and there is also a clear correlation between poor breast cancer prognosis and overexpression of IGF-1R[65]. In the MCF-7 breast carcinoma cell line, IGF stimulation results in Akt activation leading to cell proliferation through phosphorylation of Raf kinase. Along with this, IGF stimulation leads to resistance to anoikis[66], a process whereby epithelial cells undergo apoptosis due to loss of interaction with neighbouring cells and the basement membrane[67]. Overexpression of IGF-1R is also implicated in resistance against breast cancer therapy, especially against Herceptin[17].
Immunoprecipation and mass spectrometry have shown IRS-4 substrate interaction with Brk in co-transfected HEK 293 cells[68]. In addition to this interaction with IRS-4, Brk also co-precipitates with IRS-1 and IGF-IR[66]. The interaction of Brk with IGF-IR as well as EGFR and HER2 gives an indication of the range of Brk activity in regulating signalling. EGFR, HER2 and IGF-1R are overexpressed in different subsets of breast cancers whereas Brk is overexpressed in majority of breast cancers and has a role in each of these signalling pathways, thus making it an attractive therapeutic target for disrupting signalling cross-talk. Since Brk interacts with IGF-1R, disrupting this association may also reduce the chance of developing resistance against current breast cancer therapies such as Herceptin, through HER2-IGF-R1 heterodimer formation resulting in phosphorylation and activation of HER2 as reviewed in[69].
BRK AND ITS INTERACTION ALTERNATIVELY SPLICED ISOFORM, ALT-PTK6
During the characterisation and chromosome mapping of Brk, an alternatively spliced short isoform was identified in T47D cells, which was originally called λm5 and then later renamed to ALT-PTK6[6,70]. ALT-PTK6 is 134 amino acids in size (15 kDa) and is expressed from an alternatively spliced transcript that has a 122 base pair deletion at the 3’ end of the SH3 coding region; thus it lacks the SH2 and kinase domains and has an alternative C-terminal proline rich sequence (Figure 1). The protein level of ALT-PTK6 was lower than full length Brk in T47D breast cancer cells[6]. There is still speculation of the role of ALT-PTK6 and the extent of its effect in the overall process of cancer pathology, however, there has been indication that the short isoform may act as a competitive inhibitor for the SH3 binding partners[70] since both contain the SH3 domain[6].
Figure 1.
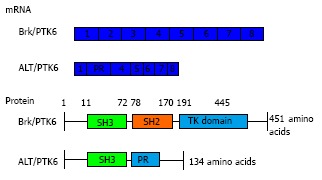
Breast tumour kinase domain structure. Full length Brk/PTK6 and short isoform ALT-PTK6 structure indicating the SH3 domain (green), SH2 domain (orange) and a tyrosine kinase domain or proline rich sequence (blue). Underneath is indicated the cDNA structure of both forms, with each box indicating each exon (total of 8 exons). ALT-PTK6 has deletion at exon 2 and lacks SH2 domain due to shift in open reading frame, which gives a proline rich sequence at exon 3 instead. PTK6: Protein tyrosine kinase 6; Brk: Breast tumour kinase.
The Tyner laboratory were the first to describe a function of ALT-PTK6, albeit in prostate cancer cell lines[70]. β-catenin has been identified as a direct substrate of Brk[71] and ALT-PTK6 may influence Brk’s ability to regulate β-catenin/TCF transcription[70]. The Wnt/β-catenin pathway promotes cancer cell growth and its deregulation is involved in pathogenesis of various cancers including breast cancer, reviewed by[72]. ALT-PTK6 has also been shown to inhibit the phosphorylation of Brk to some extent, since the presence of ALT-PTK6 reduced PTK6 interaction with tyrosine-phosphorylated proteins in what appears to be a dose-dependent manner[70].
Further to this, in co-transfection experiments, increased ALT-PK6 expression resulted in an increase of constitutively active form of Brk in the nucleus with a decreased proportion at the membrane[70]. There was also reduced proliferation in ALT-PTK6 expressing prostate tumour cells compared to those without this short isoform. These data therefore suggest that ALT-PTK6 may contribute towards limiting Brk localisation to the nucleus and acting as a competitive inhibitor thus reducing the ability of Brk to interact with its substrates that promote tumour cell growth.
The localisation of Brk therefore may play an important role in development of cancer since Brk’s arrangement in the cellular environment may affect it’s role due to the variety of substrates available in nucleus and cytoplasm[13]. Also Brk’s role in various tissue types has shown to be dramatically different, for example, in normal tissues it is involved in regulation of differentiation process whereas in tumour cells it promotes cell proliferation and survival. Therefore, Brk’s function may depend on the tissue it is expressed in, its intracellular location and the substrate it interacts with[70].
BRK’S ROLE IN CELL MIGRATION, TUMOUR FORMATION AND METASTASIS
To a large extent Brk is involved in breast cancer cell proliferation[73], however it is also involved in cell migration. A reduced ability for T47D and JIMT-1 breast cancer cell migration was observed in response to Brk suppression[49]. Paxillin is a multidomain protein that is recruited to edges of the cell once migration is initiated, as reviewed in[74], and Brk has been recognised as a novel paxillin tyrosine kinase that binds to, as well as phosphorylates, paxillin[75]. Brk also acts a mediator of EGF-induced paxillin phosphorylation, thus promoting activation of Rac1 and stimulating cell migration and invasion. KAP3A is a subunit of the kinesin-2 heterotrimeric complex and binds microtubule-based subunits KIF3A/3B to various cargo proteins, which enable membrane morphogenesis[76]. KAP3A has also been identified as a substrate of Brk and is phosphorylated at its C-terminus, a process required for Brk-induced cell migration[77]. Along with this, p190, another substrate of Brk, once phosphorylated is associated with p120 leading to Rho inactivation and Ras activation[78]. Migratory effects of Brk are greatly impaired in cells lacking p190. These interactions indicate an important function of Brk in regulating tumour cell migration via various systems, thus a Brk-targeted therapy could potentially cause disruption in these processes.
STAT3 and STAT5 are also recognised as substrates of Brk and are involved in cellular processes leading to cell proliferation, migration and survival[79-81]. STAT3 is regarded as an oncogene, with tyrosine phosphorylation of STAT3 connected to breast cancer development as discussed in[79]. Activation of STAT3 by Brk may contribute towards cell transformation and uncontrolled growth in early stages of breast cancer. Brk also mediates STAT3 regulation in established tumours[80], and constitutive activation of Brk accelerated cell migration and tumour growth in vivo[82].
Angiogenesis has been recognised as an essential process in survival of cancerous cells in vivo; it is involved in tumour growth, progression and metastasis[83]. One of the main pro-angiogenic factors involved in promoting tumour angiogenesis is vascular endothelial factor (VEGF)[84]. Osteopontin is a secreted non-collagenous chemokine-like protein that regulates VEGF expression via a Brk/nuclear factor-kappaB (NF-κB)/ ATF-4 signalling cascade[85]. Higher levels of expression of Brk, NF-κB and ATF-4 correlated with higher tumour grades. As osteopontin is secreted from the bone and expressed in brain[86,87], which are frequent sites for breast cancer metastasis[88], this study provides a mechanism whereby Brk could be involved in the formation of metastases.
BRK AND CELL DEATH
Brk’s proliferative ability when coupled with its ability to protect cancerous cells from cell death promotes tumour cell survival. Brk has shown association with the P13-K/Akt pathway which is not only involved in cell proliferation but in apoptosis; thus activation of this pathway may reduce the ability of cells to undergo apoptosis[56]. Furthermore, Brk can protect breast cancer cells from autophagy; reduced expression of Brk coupled with suspension of culture increased the number of dead cells compared to controls[89]. Brk also increases phoshoporylation of p38 MAPK which is associated with pro-survival cell phenotypes in breast cancer[90]. Brk’s ability to protect cancer cells from cell death is further enchanced due to its ability to protect cells from DNA damage-induced apoptosis in colon cancer cells. Knockdown of Brk in HCT116 cells led to increased apoptosis following γ-irradiation[91]. In addition p53 was recognised as a possible positive regulator of Brk/PTK6 activity in response to DNA damage. Reduced expression of p21 and STAT3 were also noticed in Brk-suppressed cells, leading to increased apoptosis and decreased survival[91]. This gives indication for PTK6 inhibitors reducing tumour cell growth and survival.
As discussed above, Brk also protected cells from anoikis via IGF-I signalling[66]. Further to this, recently PTK6 was described to protect cells from anoikis via direct phosphorylation of focal adhesion kinase (FAK) and activating Akt[92]. Knockdown of PTK6 in PC3 prostate cancer cell line disrupted FAK and Akt activation which in consequence promoted anoikis thus indicating important promotional role of Brk in anchorage independent survival.
Within a different cellular context such as non transformed rat fibroblasts, Brk has shown a role contradictory to that seen in breast cancer since it sensitises cells to apoptosis[93]. Furthermore, it also promotes apoptosis in crypt epithelial cells in response to DNA damage[94]. This may show a role for Brk as a damage sensor that promotes apoptosis in response to cellular stress. These differences in Brk’s role may again depend on its cellular localisation, substrates and protein interaction as well as the tissue in which it is expressed[74].
FUTURE PERSPECTIVES
Given that Brk’s roles extend to regulation of several cancer hallmarks (Figure 2), Brk has promise as an ideal therapeutic target in breast cancer. Brk’s therapeutic potential could also extend to non-small cell lung cancer, prostate and colon cancer therapy[70,91,95].
Figure 2.
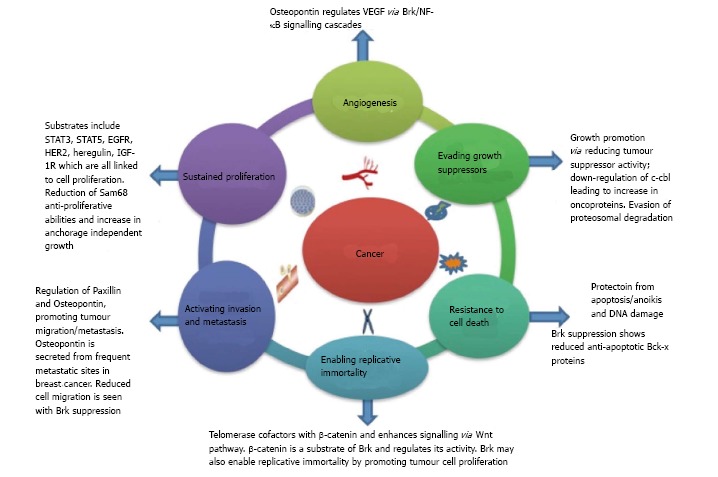
Breast tumour kinase and cancer hallmarks. Brk and cancer hallmarks. The original six cancer hallmarks described by Hannahan and Weinberg (2001)[96] are illustrated along with a summary of Brk’s role in their regulation. Brk: Breast tumour kinase.
It was recognised as one of the tyrosine kinases expressed in breast cancer due to the immense interest in tyrosine kinase inhibitors[23]. So far tyrosine kinase inhibitors have proven to be well tolerated with manageable side effects, selective and relatively effective in a range of cancers[97]. PTK6-null mice have shown to grow relatively healthily into adulthood indicating potential tolerability to Brk inhibitors in breast cancer patients[61]. These properties still make tyrosine kinase inhibition an attractive prospect despite Brk’s kinase independent functions. Nonetheless, many of Brk’s activities involve its kinase catalytic domain including EGFR signalling, cell migration and cell death[13], thus targeting Brk’s kinase activity may still prove to be effective. Furthermore, in comparison to HER2, EGFR, IGFR and p53 its expression is much higher in many types of breast cancer indicating targeting Brk may benefit a wider range of patients[62].
Further study into the regulation of Brk may provide greater understanding in its therapeutic value. So far studies focusing on discovery of selective Brk inhibitors have shown effective inhibition with imidazo[1,2-a]pyrazin-8-amines[98]. Recently, Brk’s importance in anti-cancer therapy was uncovered in a study involving heat shock protein 90 (Hsp90) which has shown to regulate Brk since it is involved in folding and stability of many proteins[99]. A potent Hsp90 inhibitor, geldanamycin decreased PTK6 expression in T47D and BT474 cell lines indicating the requirement of Brk’s interaction with Hsp90 for stability. Further investigations in a clinical setting regarding these therapies may show their efficacy in treating subtypes of breast cancers including triple negative basal like breast cancer.
Brk has many substrates and protein interactions; some have been reported to require Brk’s kinase domain whereas others may require protein-protein interactions and some through indirect association via a third party[13]. Therefore, Brk kinase-independent activity does need consideration when looking at tyrosine kinase inhibitors. Interfering with Brk’s protein-protein interactions by disrupting its conformational activation may also be therapeutically effective, including disruptions in the SH2 and SH3 domains[62].
Additionally, ALT-PTK6 the short isoform of Brk may contribute towards its cellular localisation as increased ALT-PTK6 expression resulted in decreased Brk at the membrane and an increase in nuclear Brk[70]. Normally Brk is active in differentiating, non-dividing epithelial cells in the small intestine where it is a negative regulator of growth, however, due to an external stimuli such as DNA damage by irradiation, it contributes towards apoptosis[94]. This shows Brk’s differing role within the same tissue but in different conditions. Thus Brks functions may also depend on external stimuli and the short isoform ALT-PTK6. This needs further investigation and clarification within breast cancers.
Brk’s role as a potential oncogene in relation to breast cancer is further supported by a recent study, which indicated Brk catalytically phosphorylated c-Cbl, a E3 ubiquitin ligase that downregualtes oncoproteins, resulting in c-Cbl degradation via auto-ubiquitination[59]. Of the multiple signalling pathways that Brk is involved in, this is yet another pathway in which Brk plays a key role thereby enhancing tumour survival. Therefore Brk’s involvement is extensive which suggests targeting Brk may allow targeting of a wider range of pathways that lead to tumour growth, survival and progression.
In the immediate future, most progress could probably be made in combination treatments. Brk’s role in mediating cell responses to current EGFR/HER2 therapies is becoming known[35,49]. Further work in this area could pave the way for improvement to these therapies. Investigations into Brk’s role in tumour progression should allow for pre-clinical studies of breast cancer metastasis and determine whether any therapeutic intervention has potential clinical benefit.
CONCLUSION
Brk has been implicated in tumorigenesis, survival and progression as well as diagnosis and prognosis of breast cancer despite the fact there is no indication of expression of Brk in normal mammary tissue. Thus making it a viable target of breast cancer therapy and an interesting protein to investigate. Brk expression changes have not been associated with soley with an increase in ptk6 gene copy number, which allows for study of alternative mechanisms by which Brk expression and breast cancer develops. Ongoing research may still reveal more associations of Brk with, as yet, uncharacterised substrates. A wider view of Brk’s interacting proteins and substrates therefore will prove to be useful in developing anti-Brk therapies for breast cancers.
Footnotes
P- Reviewer: Wu KM S- Editor: Song XX L- Editor: A E- Editor: Lu YJ
References
- 1.Office for National Statistics. Breast cancer incidence, mortality and survival, england, 1971-2011 infographic. England: Part of Cancer Statistics Registrations;; 2011. Available from: http: //www.ons.gov.uk/ons/rel/vsob1/cancer-statistics-registrations--england--series-mb1-/no--42--2011/info-breast-cancer.html. [Google Scholar]
- 2.Eccles SA, Aboagye EO, Ali S, Anderson AS, Armes J, Berditchevski F, Blaydes JP, Brennan K, Brown NJ, Bryant HE, et al. Critical research gaps and translational priorities for the successful prevention and treatment of breast cancer. Breast Cancer Res. 2013;15:R92. doi: 10.1186/bcr3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mitchell PJ, Barker KT, Martindale JE, Kamalati T, Lowe PN, Page MJ, Gusterson BA, Crompton MR. Cloning and characterisation of cDNAs encoding a novel non-receptor tyrosine kinase, brk, expressed in human breast tumours. Oncogene. 1994;9:2383–2390. [PubMed] [Google Scholar]
- 4.Ostrander JH, Daniel AR, Lofgren K, Kleer CG, Lange CA. Breast tumor kinase (protein tyrosine kinase 6) regulates heregulin-induced activation of ERK5 and p38 MAP kinases in breast cancer cells. Cancer Res. 2007;67:4199–4209. doi: 10.1158/0008-5472.CAN-06-3409. [DOI] [PubMed] [Google Scholar]
- 5.Barker KT, Jackson LE, Crompton MR. BRK tyrosine kinase expression in a high proportion of human breast carcinomas. Oncogene. 1997;15:799–805. doi: 10.1038/sj.onc.1201241. [DOI] [PubMed] [Google Scholar]
- 6.Mitchell PJ, Barker KT, Shipley J, Crompton MR. Characterisation and chromosome mapping of the human non receptor tyrosine kinase gene, brk. Oncogene. 1997;15:1497–1502. doi: 10.1038/sj.onc.1201292. [DOI] [PubMed] [Google Scholar]
- 7.Mitchell PJ, Sara EA, Crompton MR. A novel adaptor-like protein which is a substrate for the non-receptor tyrosine kinase, BRK. Oncogene. 2000;19:4273–4282. doi: 10.1038/sj.onc.1203775. [DOI] [PubMed] [Google Scholar]
- 8.Mayer BJ. SH3 domains: complexity in moderation. J Cell Sci. 2001;114:1253–1263. doi: 10.1242/jcs.114.7.1253. [DOI] [PubMed] [Google Scholar]
- 9.Qiu H, Miller WT. Role of the Brk SH3 domain in substrate recognition. Oncogene. 2004;23:2216–2223. doi: 10.1038/sj.onc.1207339. [DOI] [PubMed] [Google Scholar]
- 10.Hong E, Shin J, Bang E, Kim MH, Lee ST, Lee W. Complete sequence-specific 1H, 13C and 15N resonance assignments of the human PTK6 SH2 domain. J Biomol NMR. 2001;19:291–292. doi: 10.1023/a:1011221125013. [DOI] [PubMed] [Google Scholar]
- 11.Qiu H, Miller WT. Regulation of the nonreceptor tyrosine kinase Brk by autophosphorylation and by autoinhibition. J Biol Chem. 2002;277:34634–34641. doi: 10.1074/jbc.M203877200. [DOI] [PubMed] [Google Scholar]
- 12.Hong E, Shin J, Kim HI, Lee ST, Lee W. Solution structure and backbone dynamics of the non-receptor protein-tyrosine kinase-6 Src homology 2 domain. J Biol Chem. 2004;279:29700–29708. doi: 10.1074/jbc.M313185200. [DOI] [PubMed] [Google Scholar]
- 13.Harvey A, Burmi R. Future therapeutic strategies: Implications for brk targeting. In: Esra Gunduz, Mehmet Gunduz, editors. Breast cancer - current and alternative therapeutic modalities; 2011. [Google Scholar]
- 14.Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther. 2005;315:971–979. doi: 10.1124/jpet.105.084145. [DOI] [PubMed] [Google Scholar]
- 15.Smith E. Tamoxifen - the start of something big. 2012(1): 10. Available from: http: //scienceblog.cancerresearchuk.org/2012/10/15/high-impact-science-tamoxifen-the-start-of-something-big/
- 16.den Hollander P, Savage MI, Brown PH. Targeted Therapy for Breast Cancer Prevention. Front Oncol. 2013;3:250. doi: 10.3389/fonc.2013.00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fink MY, Chipuk JE. Survival of HER2-Positive Breast Cancer Cells: Receptor Signaling to Apoptotic Control Centers. Genes Cancer. 2013;4:187–195. doi: 10.1177/1947601913488598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harvey A, editor . Cancer cell signalling. 1st ed. UK: Wiley;; 2013. Available from: http://eu.wiley.com/WileyCDA/WileyTitle/productCd-1119967570.html. [Google Scholar]
- 19.Brauer PM, Tyner AL. Building a better understanding of the intracellular tyrosine kinase PTK6 - BRK by BRK. Biochim Biophys Acta. 2010;1806:66–73. doi: 10.1016/j.bbcan.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Romain S, Chinot O, Klijn JG, van Putten WL, Guirou O, Look M, Martin PM, Foekens JA. Prognostic value of cytosolic tyrosine kinase activity in 249 node-positive breast cancer patients. Br J Cancer. 1994;70:304–308. doi: 10.1038/bjc.1994.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ottenhoff-Kalff AE, Rijksen G, van Beurden EA, Hennipman A, Michels AA, Staal GE. Characterization of protein tyrosine kinases from human breast cancer: involvement of the c-src oncogene product. Cancer Res. 1992;52:4773–4778. [PubMed] [Google Scholar]
- 22.Nielsen TO, Andrews HN, Cheang M, Kucab JE, Hsu FD, Ragaz J, Gilks CB, Makretsov N, Bajdik CD, Brookes C, et al. Expression of the insulin-like growth factor I receptor and urokinase plasminogen activator in breast cancer is associated with poor survival: potential for intervention with 17-allylamino geldanamycin. Cancer Res. 2004;64:286–291. doi: 10.1158/0008-5472.can-03-1242. [DOI] [PubMed] [Google Scholar]
- 23.Meric F, Lee WP, Sahin A, Zhang H, Kung HJ, Hung MC. Expression profile of tyrosine kinases in breast cancer. Clin Cancer Res. 2002;8:361–367. [PubMed] [Google Scholar]
- 24.Fearon AE, Gould CR, Grose RP. FGFR signalling in women’s cancers. Int J Biochem Cell Biol. 2013;45:2832–2842. doi: 10.1016/j.biocel.2013.09.017. [DOI] [PubMed] [Google Scholar]
- 25.Ponzo MG, Park M. The Met receptor tyrosine kinase and basal breast cancer. Cell Cycle. 2010;9:1043–1050. doi: 10.4161/cc.9.6.11033. [DOI] [PubMed] [Google Scholar]
- 26.Anderson NG, Ahmad T, Chan K, Dobson R, Bundred NJ. ZD1839 (Iressa), a novel epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor, potently inhibits the growth of EGFR-positive cancer cell lines with or without erbB2 overexpression. Int J Cancer. 2001;94:774–782. doi: 10.1002/ijc.1557. [DOI] [PubMed] [Google Scholar]
- 27.Agrawal A, Gutteridge E, Gee JM, Nicholson RI, Robertson JF. Overview of tyrosine kinase inhibitors in clinical breast cancer. Endocr Relat Cancer. 2005;12 Suppl 1:S135–S144. doi: 10.1677/erc.1.01059. [DOI] [PubMed] [Google Scholar]
- 28.Herbst RS, Bunn PA. Targeting the epidermal growth factor receptor in non-small cell lung cancer. Clin Cancer Res. 2003;9:5813–5824. [PubMed] [Google Scholar]
- 29.Sharma P, QJ Khan, BF Kimler, JR Klemp, CJ Connor, MK McGinness, JW Mammen, OW Tawfik, F Fan, Fabian CJ. Results of a phase II study of neoadjuvant Platinum/Taxane based chemotherapy and erlotinib for triple negative breast cancer. Cancer Research. 2010:70. [Google Scholar]
- 30.Ueno NT, Zhang D. Targeting EGFR in Triple Negative Breast Cancer. J Cancer. 2011;2:324–328. doi: 10.7150/jca.2.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, Opresko L, Shankaran H, Chrisler WB, Wiley HS, Resat H. HER/ErbB receptor interactions and signaling patterns in human mammary epithelial cells. BMC Cell Biol. 2009;10:78. doi: 10.1186/1471-2121-10-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bilancia D, Rosati G, Dinota A, Germano D, Romano R, Manzione L. Lapatinib in breast cancer. Ann Oncol. 2007;18 Suppl 6:vi26–vi30. doi: 10.1093/annonc/mdm220. [DOI] [PubMed] [Google Scholar]
- 33.Rusnak DW, Affleck K, Cockerill SG, Stubberfield C, Harris R, Page M, Smith KJ, Guntrip SB, Carter MC, Shaw RJ, et al. The characterization of novel, dual ErbB-2/EGFR, tyrosine kinase inhibitors: potential therapy for cancer. Cancer Res. 2001;61:7196–7203. [PubMed] [Google Scholar]
- 34.Wood ER, Truesdale AT, McDonald OB, Yuan D, Hassell A, Dickerson SH, Ellis B, Pennisi C, Horne E, Lackey K, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64:6652–6659. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- 35.Xiang B, Chatti K, Qiu H, Lakshmi B, Krasnitz A, Hicks J, Yu M, Miller WT, Muthuswamy SK. Brk is coamplified with ErbB2 to promote proliferation in breast cancer. Proc Natl Acad Sci USA. 2008;105:12463–12468. doi: 10.1073/pnas.0805009105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamasaki F, Zhang D, Bartholomeusz C, Sudo T, Hortobagyi GN, Kurisu K, Ueno NT. Sensitivity of breast cancer cells to erlotinib depends on cyclin-dependent kinase 2 activity. Mol Cancer Ther. 2007;6:2168–2177. doi: 10.1158/1535-7163.MCT-06-0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carlson RW, O’Neill A, Vidaurre T, Gomez HL, Badve SS, Sledge GW. A randomized trial of combination anastrozole plus gefitinib and of combination fulvestrant plus gefitinib in the treatment of postmenopausal women with hormone receptor positive metastatic breast cancer. Breast Cancer Res Treat. 2012;133:1049–1056. doi: 10.1007/s10549-012-1997-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ling YH, Li T, Yuan Z, Haigentz M, Weber TK, Perez-Soler R. Erlotinib, an effective epidermal growth factor receptor tyrosine kinase inhibitor, induces p27KIP1 up-regulation and nuclear translocation in association with cell growth inhibition and G1/S phase arrest in human non-small-cell lung cancer cell lines. Mol Pharmacol. 2007;72:248–258. doi: 10.1124/mol.107.034827. [DOI] [PubMed] [Google Scholar]
- 39.Lenferink AE, Busse D, Flanagan WM, Yakes FM, Arteaga CL. ErbB2/neu kinase modulates cellular p27(Kip1) and cyclin D1 through multiple signaling pathways. Cancer Res. 2001;61:6583–6591. [PubMed] [Google Scholar]
- 40.Nahta R, Takahashi T, Ueno NT, Hung MC, Esteva FJ. P27(kip1) down-regulation is associated with trastuzumab resistance in breast cancer cells. Cancer Res. 2004;64:3981–3986. doi: 10.1158/0008-5472.CAN-03-3900. [DOI] [PubMed] [Google Scholar]
- 41.Chan E, Nimnual AS. Deregulation of the cell cycle by breast tumor kinase (Brk) Int J Cancer. 2010;127:2723–2731. doi: 10.1002/ijc.25263. [DOI] [PubMed] [Google Scholar]
- 42.Montemurro F, Scaltriti M. Biomarkers of drugs targeting HER-family signalling in cancer. J Pathol. 2014;232:219–229. doi: 10.1002/path.4269. [DOI] [PubMed] [Google Scholar]
- 43.Rugo HS, Chien AJ, Franco SX, Stopeck AT, Glencer A, Lahiri S, Arbushites MC, Scott J, Park JW, Hudis C, et al. A phase II study of lapatinib and bevacizumab as treatment for HER2-overexpressing metastatic breast cancer. Breast Cancer Res Treat. 2012;134:13–20. doi: 10.1007/s10549-011-1918-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hayes DF, Thor AD. c-erbB-2 in breast cancer: development of a clinically useful marker. Semin Oncol. 2002;29:231–245. doi: 10.1053/sonc.2002.32899. [DOI] [PubMed] [Google Scholar]
- 45.Born M, Quintanilla-Fend L, Braselmann H, Reich U, Richter M, Hutzler P, Aubele M. Simultaneous over-expression of the Her2/neu and PTK6 tyrosine kinases in archival invasive ductal breast carcinomas. J Pathol. 2005;205:592–596. doi: 10.1002/path.1720. [DOI] [PubMed] [Google Scholar]
- 46.Aubele M, Auer G, Walch AK, Munro A, Atkinson MJ, Braselmann H, Fornander T, Bartlett JM. PTK (protein tyrosine kinase)-6 and HER2 and 4, but not HER1 and 3 predict long-term survival in breast carcinomas. Br J Cancer. 2007;96:801–807. doi: 10.1038/sj.bjc.6603613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aubele M, Walch AK, Ludyga N, Braselmann H, Atkinson MJ, Luber B, Auer G, Tapio S, Cooke T, Bartlett JM. Prognostic value of protein tyrosine kinase 6 (PTK6) for long-term survival of breast cancer patients. Br J Cancer. 2008;99:1089–1095. doi: 10.1038/sj.bjc.6604660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aubele M, Spears M, Ludyga N, Braselmann H, Feuchtinger A, Taylor KJ, Lindner K, Auer G, Stering K, Höfler H, et al. In situ quantification of HER2-protein tyrosine kinase 6 (PTK6) protein-protein complexes in paraffin sections from breast cancer tissues. Br J Cancer. 2010;103:663–667. doi: 10.1038/sj.bjc.6605836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ludyga N, Anastasov N, Rosemann M, Seiler J, Lohmann N, Braselmann H, Mengele K, Schmitt M, Höfler H, Aubele M. Effects of simultaneous knockdown of HER2 and PTK6 on malignancy and tumor progression in human breast cancer cells. Mol Cancer Res. 2013;11:381–392. doi: 10.1158/1541-7786.MCR-12-0378. [DOI] [PubMed] [Google Scholar]
- 50.Ai M, Qiu S, Lu Y, Fan Z. HER2 regulates Brk/PTK6 stability via upregulating calpastatin, an inhibitor of calpain. Cell Signal. 2013;25:1754–1761. doi: 10.1016/j.cellsig.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim H, Muller WJ. The role of the epidermal growth factor receptor family in mammary tumorigenesis and metastasis. Exp Cell Res. 1999;253:78–87. doi: 10.1006/excr.1999.4706. [DOI] [PubMed] [Google Scholar]
- 52.Moasser MM, Basso A, Averbuch SD, Rosen N. The tyrosine kinase inhibitor ZD1839 (“Iressa”) inhibits HER2-driven signaling and suppresses the growth of HER2-overexpressing tumor cells. Cancer Res. 2001;61:7184–7188. [PubMed] [Google Scholar]
- 53.Kamalati T, Jolin HE, Mitchell PJ, Barker KT, Jackson LE, Dean CJ, Page MJ, Gusterson BA, Crompton MR. Brk, a breast tumor-derived non-receptor protein-tyrosine kinase, sensitizes mammary epithelial cells to epidermal growth factor. J Biol Chem. 1996;271:30956–30963. doi: 10.1074/jbc.271.48.30956. [DOI] [PubMed] [Google Scholar]
- 54.Tran QT, Kennedy LH, Leon Carrion S, Bodreddigari S, Goodwin SB, Sutter CH, Sutter TR. EGFR regulation of epidermal barrier function. Physiol Genomics. 2012;44:455–469. doi: 10.1152/physiolgenomics.00176.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tupper J, Crompton MR, Harvey AJ. Breast tumor kinase (Brk/PTK6) plays a role in the differentiation of primary keratinocytes. Arch Dermatol Res. 2011;303:293–297. doi: 10.1007/s00403-010-1118-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kamalati T, Jolin HE, Fry MJ, Crompton MR. Expression of the BRK tyrosine kinase in mammary epithelial cells enhances the coupling of EGF signalling to PI 3-kinase and Akt, via erbB3 phosphorylation. Oncogene. 2000;19:5471–5476. doi: 10.1038/sj.onc.1203931. [DOI] [PubMed] [Google Scholar]
- 57.Li X, Lu Y, Liang K, Hsu JM, Albarracin C, Mills GB, Hung MC, Fan Z. Brk/PTK6 sustains activated EGFR signaling through inhibiting EGFR degradation and transactivating EGFR. Oncogene. 2012;31:4372–4383. doi: 10.1038/onc.2011.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kang SA, Lee ES, Yoon HY, Randazzo PA, Lee ST. PTK6 inhibits down-regulation of EGF receptor through phosphorylation of ARAP1. J Biol Chem. 2010;285:26013–26021. doi: 10.1074/jbc.M109.088971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kang SA, Lee ST. PTK6 promotes degradation of c-Cbl through PTK6-mediated phosphorylation. Biochem Biophys Res Commun. 2013;431:734–739. doi: 10.1016/j.bbrc.2013.01.046. [DOI] [PubMed] [Google Scholar]
- 60.Baselga J. Targeting the phosphoinositide-3 (PI3) kinase pathway in breast cancer. Oncologist. 2011;16 Suppl 1:12–19. doi: 10.1634/theoncologist.2011-S1-12. [DOI] [PubMed] [Google Scholar]
- 61.Haegebarth A, Bie W, Yang R, Crawford SE, Vasioukhin V, Fuchs E, Tyner AL. Protein tyrosine kinase 6 negatively regulates growth and promotes enterocyte differentiation in the small intestine. Mol Cell Biol. 2006;26:4949–4957. doi: 10.1128/MCB.01901-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harvey AJ, Crompton MR. The Brk protein tyrosine kinase as a therapeutic target in cancer: opportunities and challenges. Anticancer Drugs. 2004;15:107–111. doi: 10.1097/00001813-200402000-00002. [DOI] [PubMed] [Google Scholar]
- 63.Zeng X, Yee D. Insulin-like growth factors and breast cancer therapy. Adv Exp Med Biol. 2007;608:101–112. doi: 10.1007/978-0-387-74039-3_7. [DOI] [PubMed] [Google Scholar]
- 64.Sachdev D. Regulation of breast cancer metastasis by IGF signaling. J Mammary Gland Biol Neoplasia. 2008;13:431–441. doi: 10.1007/s10911-008-9105-5. [DOI] [PubMed] [Google Scholar]
- 65.Klinakis A, Szabolcs M, Chen G, Xuan S, Hibshoosh H, Efstratiadis A. Igf1r as a therapeutic target in a mouse model of basal-like breast cancer. Proc Natl Acad Sci USA. 2009;106:2359–2364. doi: 10.1073/pnas.0810221106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Irie HY, Shrestha Y, Selfors LM, Frye F, Iida N, Wang Z, Zou L, Yao J, Lu Y, Epstein CB, et al. PTK6 regulates IGF-1-induced anchorage-independent survival. PLoS One. 2010;5:e11729. doi: 10.1371/journal.pone.0011729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124:619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Qiu H, Zappacosta F, Su W, Annan RS, Miller WT. Interaction between Brk kinase and insulin receptor substrate-4. Oncogene. 2005;24:5656–5664. doi: 10.1038/sj.onc.1208721. [DOI] [PubMed] [Google Scholar]
- 69.Bender LM, Nahta R. Her2 cross talk and therapeutic resistance in breast cancer. Front Biosci. 2008;13:3906–3912. doi: 10.2741/2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brauer PM, Zheng Y, Evans MD, Dominguez-Brauer C, Peehl DM, Tyner AL. The alternative splice variant of protein tyrosine kinase 6 negatively regulates growth and enhances PTK6-mediated inhibition of β-catenin. PLoS One. 2011;6:e14789. doi: 10.1371/journal.pone.0014789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Palka-Hamblin HL, Gierut JJ, Bie W, Brauer PM, Zheng Y, Asara JM, Tyner AL. Identification of beta-catenin as a target of the intracellular tyrosine kinase PTK6. J Cell Sci. 2010;123:236–245. doi: 10.1242/jcs.053264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 73.Harvey AJ, Crompton MR. Use of RNA interference to validate Brk as a novel therapeutic target in breast cancer: Brk promotes breast carcinoma cell proliferation. Oncogene. 2003;22:5006–5010. doi: 10.1038/sj.onc.1206577. [DOI] [PubMed] [Google Scholar]
- 74.Deakin NO, Turner CE. Paxillin comes of age. J Cell Sci. 2008;121:2435–2444. doi: 10.1242/jcs.018044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen HY, Shen CH, Tsai YT, Lin FC, Huang YP, Chen RH. Brk activates rac1 and promotes cell migration and invasion by phosphorylating paxillin. Mol Cell Biol. 2004;24:10558–10572. doi: 10.1128/MCB.24.24.10558-10572.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yamazaki H, Nakata T, Okada Y, Hirokawa N. Cloning and characterization of KAP3: a novel kinesin superfamily-associated protein of KIF3A/3B. Proc Natl Acad Sci USA. 1996;93:8443–8448. doi: 10.1073/pnas.93.16.8443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lukong KE, Richard S. Breast tumor kinase BRK requires kinesin-2 subunit KAP3A in modulation of cell migration. Cell Signal. 2008;20:432–442. doi: 10.1016/j.cellsig.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 78.Shen CH, Chen HY, Lin MS, Li FY, Chang CC, Kuo ML, Settleman J, Chen RH. Breast tumor kinase phosphorylates p190RhoGAP to regulate rho and ras and promote breast carcinoma growth, migration, and invasion. Cancer Res. 2008;68:7779–7787. doi: 10.1158/0008-5472.CAN-08-0997. [DOI] [PubMed] [Google Scholar]
- 79.Gierut J, Zheng Y, Bie W, Carroll RE, Ball-Kell S, Haegebarth A, Tyner AL. Disruption of the mouse protein tyrosine kinase 6 gene prevents STAT3 activation and confers resistance to azoxymethane. Gastroenterology. 2011;141:1371–1380, 1371-1380. doi: 10.1053/j.gastro.2011.06.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu L, Gao Y, Qiu H, Miller WT, Poli V, Reich NC. Identification of STAT3 as a specific substrate of breast tumor kinase. Oncogene. 2006;25:4904–4912. doi: 10.1038/sj.onc.1209501. [DOI] [PubMed] [Google Scholar]
- 81.Weaver AM, Silva CM. Signal transducer and activator of transcription 5b: a new target of breast tumor kinase/protein tyrosine kinase 6. Breast Cancer Res. 2007;9:R79. doi: 10.1186/bcr1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Miah S, Martin A, Lukong KE. Constitutive activation of breast tumor kinase accelerates cell migration and tumor growth in vivo. Oncogenesis. 2012;1:e11. doi: 10.1038/oncsis.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Otrock ZK, Mahfouz RA, Makarem JA, Shamseddine AI. Understanding the biology of angiogenesis: review of the most important molecular mechanisms. Blood Cells Mol Dis. 2007;39:212–220. doi: 10.1016/j.bcmd.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 84.Niu G, Chen X. Vascular endothelial growth factor as an anti-angiogenic target for cancer therapy. Curr Drug Targets. 2010;11:1000–1017. doi: 10.2174/138945010791591395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chakraborty G, Jain S, Kundu GC. Osteopontin promotes vascular endothelial growth factor-dependent breast tumor growth and angiogenesis via autocrine and paracrine mechanisms. Cancer Res. 2008;68:152–161. doi: 10.1158/0008-5472.CAN-07-2126. [DOI] [PubMed] [Google Scholar]
- 86.Oldberg A, Franzén A, Heinegård D. Cloning and sequence analysis of rat bone sialoprotein (osteopontin) cDNA reveals an Arg-Gly-Asp cell-binding sequence. Proc Natl Acad Sci USA. 1986;83:8819–8823. doi: 10.1073/pnas.83.23.8819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Saitoh Y, Kuratsu J, Takeshima H, Yamamoto S, Ushio Y. Expression of osteopontin in human glioma. Its correlation with the malignancy. Lab Invest. 1995;72:55–63. [PubMed] [Google Scholar]
- 88.Irvin W, Muss HB, Mayer DK. Symptom management in metastatic breast cancer. Oncologist. 2011;16:1203–1214. doi: 10.1634/theoncologist.2011-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Harvey AJ, Pennington CJ, Porter S, Burmi RS, Edwards DR, Court W, Eccles SA, Crompton MR. Brk protects breast cancer cells from autophagic cell death induced by loss of anchorage. Am J Pathol. 2009;175:1226–1234. doi: 10.2353/ajpath.2009.080811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lofgren KA, Ostrander JH, Housa D, Hubbard GK, Locatelli A, Bliss RL, Schwertfeger KL, Lange CA. Mammary gland specific expression of Brk/PTK6 promotes delayed involution and tumor formation associated with activation of p38 MAPK. Breast Cancer Res. 2011;13:R89. doi: 10.1186/bcr2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gierut JJ, Mathur PS, Bie W, Han J, Tyner AL. Targeting protein tyrosine kinase 6 enhances apoptosis of colon cancer cells following DNA damage. Mol Cancer Ther. 2012;11:2311–2320. doi: 10.1158/1535-7163.MCT-12-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zheng Y, Gierut J, Wang Z, Miao J, Asara JM, Tyner AL. Protein tyrosine kinase 6 protects cells from anoikis by directly phosphorylating focal adhesion kinase and activating AKT. Oncogene. 2013;32:4304–4312. doi: 10.1038/onc.2012.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Haegebarth A, Nunez R, Tyner AL. The intracellular tyrosine kinase Brk sensitizes non-transformed cells to inducers of apoptosis. Cell Cycle. 2005;4:1239–1246. doi: 10.4161/cc.4.9.1965. [DOI] [PubMed] [Google Scholar]
- 94.Haegebarth A, Perekatt AO, Bie W, Gierut JJ, Tyner AL. Induction of protein tyrosine kinase 6 in mouse intestinal crypt epithelial cells promotes DNA damage-induced apoptosis. Gastroenterology. 2009;137:945–954. doi: 10.1053/j.gastro.2009.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fan C, Zhao Y, Liu D, Zhang X, Wang E. Detection of Brk expression in non-small cell lung cancer: clinicopathological relevance. Tumour Biol. 2011;32:873–880. doi: 10.1007/s13277-011-0188-z. [DOI] [PubMed] [Google Scholar]
- 96.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 97.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 98.Zeng H, Belanger DB, Curran PJ, Shipps GW, Miao H, Bracken JB, Arshad Siddiqui M, Malkowski M, Wang Y. Discovery of novel imidazo[1,2-a]pyrazin-8-amines as Brk/PTK6 inhibitors. Bioorg Med Chem Lett. 2011;21:5870–5875. doi: 10.1016/j.bmcl.2011.07.101. [DOI] [PubMed] [Google Scholar]
- 99.Kang SA, Cho HS, Yoon JB, Chung IK, Lee ST. Hsp90 rescues PTK6 from proteasomal degradation in breast cancer cells. Biochem J. 2012;447:313–320. doi: 10.1042/BJ20120803. [DOI] [PubMed] [Google Scholar]