

Abstract
Organic chemists and metabolic engineers use largely orthogonal technologies to construct essential small molecules like pharmaceuticals and commodity chemicals. While chemists have leveraged the unique capabilities of biological catalysts for small molecule production, metabolic engineers have not likewise integrated reactions from organic synthesis with the metabolism of living organisms. Here we report a method for alkene hydrogenation that utilizes a palladium catalyst and hydrogen gas generated directly by a living microorganism. This biocompatible transformation, which requires both catalyst and microbe and can be used on a preparative scale, represents a new strategy for chemical synthesis that combines organic chemistry and metabolic engineering.
Keywords: biocompatible chemistry, hydrogenation, metabolism, palladium, catalysis
Two scientific disciplines, organic chemistry and metabolic engineering, endeavor to produce small molecules using very different techniques.[1,2] Synthetic organic chemists largely employ non-biological catalysts and reagents to manipulate molecules in multi-step processes, while metabolic engineers harness the reactivity of enzymatic catalysts in the context of living organisms to produce molecules directly from fermentations.[3,4] While organic chemists have been increasingly utilizing enzymes in synthetic efforts[5] such as the industrial-scale synthesis of the diabetes drug sitagliptin (Januvia®),[6] efforts to synthesize molecules by incorporating reactions from organic synthesis into biological systems have lagged behind. We envision achieving this goal using biocompatible chemistry: non-enzymatic transformations that directly interface with the metabolism of living organisms, modifying small molecule metabolites as they are produced and augmenting native biochemistry (Figure 1A).[7]
Figure 1.

Biocompatible chemistry enables the integration of non-enzymatic reactions with microbial metabolism. a) Biocompatible chemistry represents a distinct approach for synthesis that employs chemical tools in a biological environment. b) Design of a biocompatible alkene hydrogenation that uses hydrogen gas produced directly by microbial metabolism for synthesis.
Perhaps the largest obstacle faced in this endeavor is the discordance between the conditions typically required to support life and those often used in organic synthesis (non-aqueous solvents, extreme temperatures, reactive intermediates). For example, the problem of mutual catalyst inactivation can at times complicate efforts to combine transition metal catalysts with purified enzymes in vitro.[8] Additional challenges include the chemical complexity of the cellular and extracellular environments, the typically low concentrations of metabolites, and the potential difficulties associated with accessing intracellular substrates. Encouragingly, similar problems have been surmounted in developing bioorthogonal reactions, which are used to study biological phenomena in living cells and organisms without altering underlying cellular processes.[9] While such transformations illustrate that non-enzymatic chemistry can proceed in the presence of living systems,[10] their application toward small molecule synthesis has been underexplored.
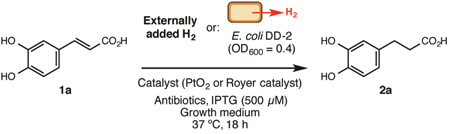
We identified hydrogen gas[11] as a target metabolite for an initial proof-of-concept reaction: a biocompatible alkene hydrogenation that would directly combine hydrogen generated by living bacteria with a transition metal catalyst and could be utilized for preparative scale synthesis (Figure 1B). This choice was motivated by previous reports suggesting transition metal-catalyzed hydrogenation was compatible with living cells. Specifically, unsaturated bacterial membrane lipids can be reduced with transition metal catalysts and added hydrogen gas.[12a] Bacterially produced hydrogen can also directly reduce organic dyes[12b] and ethylene[12c] on an analytical scale using superstoichiometric amounts of catalyst. Though these examples provided important precedent for the desired chemical reactivity, they did not imply or demonstrate synthetic utility (e.g. preparative scale, broad substrate scope). Beyond its potential use for chemical synthesis, we also envisioned using our hydrogenation to elucidate factors influencing the success of biocompatible reactions.
We began by investigating whether hydrogenation could take place in media complex enough to support the growth of E. coli, our intended source of hydrogen (Table 1). We incubated the water-soluble alkene caffeic acid (1a) with platinum(IV)oxide in two types of growth media under an atmosphere of hydrogen gas (Entries 1 and 2) and found that a defined minimal medium (M9 glucose) provided higher conversion than a complex medium (Luria-Bertani (LB) + 0.5% glucose). We then examined the impact of bacteria on these reactions by performing hydrogenations under an atmosphere of hydrogen gas in growth media containing E. coli DD-2 (optical density (OD)600 = 0.4). This engineered strain produces hydrogen from glucose via an inducible pathway consisting of a pyruvate ferredoxin oxidoreductase, a ferredoxin, and a [Fe-Fe] hydrogenase.[13] We observed little change in conversion with organisms in the reaction mixture (Entries 3 and 4). Finally, we incubated catalyst and substrate in the presence of E. coli DD-2 under a nitrogen atmosphere, relying on bacterial production of hydrogen gas (Entries 5 and 6, Figure 1C). We observed 15% conversion for the reaction run in M9 glucose, providing support for our general reaction design.
Table 1.
Proof-of-concept and reaction optimization experiments
 | ||||
|---|---|---|---|---|
| Entry | Growth medium | Cells/H2 added[a] |
Catalyst, mol% | Conversion (%)[b] |
| 1 | LB + glucose | no/yes | PtO2, 40 mol% | 15 |
| 2 | M9 glucose | no/yes | PtO2, 40 mol% | 100 |
| 3 | LB + glucose | yes/yes | PtO2, 40 mol% | 6 |
| 4 | M9 glucose | yes/yes | PtO2, 40 mol% | 91 |
| 5 | LB + glucose | yes/no | PtO2, 40 mol% | 0 |
| 6 | M9 glucose | yes/no | PtO2, 40 mol% | 15 |
| 7 | M9CA glucose + Fe[c] | yes/no | PtO2, 20 mol% | 56 |
| 8 | M9CA glucose + Fe | yes/no | Royer, 8 mol%[d] | 100 |
| 9 | M9CA glucose + Fe | yes/no | Royer, 8 mol% | 100/87[e] |
Reactions were performed at a 5 mM substrate concentration in 5 mL of growth medium containing ampicillin (50 µg/mL), spectinomycin (25 µg/mL), chloramphenicol (12.5 µg/mL), and IPTG (500 µM) under an atmosphere of either hydrogen or nitrogen in 16 mL Hungate tubes with shaking at 190 rpm.
E. coli strain DD-2 was used, OD600 = 0.4.
Determined by 1H NMR.
M9CA glucose + Fe medium contains Fe(NH4)2(SO4)2 (50 µM) and casamino acids (5 g/L).
Royer catalyst is 2.44 wt% palladium on polyethyleneimine/silica gel.
Reaction was performed on a 9 mmol scale (1.6 g of 1a) with 8 mol% Royer catalyst at a substrate concentration of 10 mM for 48 h (87% isolated yield).
Our initial optimization efforts focused on varying growth medium and catalyst (Table 1, Tables S3–4). Based on the increased conversions observed for reactions performed with added hydrogen, we suspected that hydrogen production by E. coli DD-2 was limiting reaction efficiency. We tested various media additives and found that adding either iron or casamino acids (Table S3) to minimal media improved conversion. The combination of both additives provided a further increase (Table 1, Entry 7). These components may boost hydrogen generation by increasing the amount of functional [Fe-Fe] hydrogenase.[14]
We screened a variety of heterogeneous hydrogenation catalysts using our improved reaction media. While most catalysts examined provided no reactivity (Table S4), the Royer palladium catalyst[15] (2.44% palladium on polyethyleneimine (PEI)/silica gel) proved uniquely effective.[16] Using this catalyst, we could double the concentration of substrate and reduce catalyst loading to 8 mol% without impacting conversion (Table 1, Entry 8). Further optimization experiments were carried out with a more challenging substrate (E)-3-(3,4,5-trimethoxyphenyl)acrylic acid (1b) (Figure S2, Tables S5–S11). Ultimately, we identified conditions that were readily scaled to hydrogenate 9 mmol (1.6 g) of 1a (Table 1, Entry 9). The ease with which we could apply this transformation to larger scale reactions is notable, and may indicate that this general approach is suitable for preparative scale synthesis.
We used these optimal reaction conditions to evaluate functional group compatibility, as it was unclear to what extent the presence of living organisms would impact substrate scope. Overall, the hydrogenation displayed broad utility for preparative-scale reactions of water-soluble alkenes (Figure 2). An alkyne substrate (1k) also underwent exhaustive hydrogenation to the corresponding alkane. Most notably, 2-hexenedioic acid (1x) and Z,Z-muconic acid (1y) were converted to adipic acid, an important industrial chemical that is produced on a multimillion ton scale annually and has been a frequent but challenging target for metabolic engineering.[17] These results suggest that adipic acid could be obtained directly from fermentations by combining a biocompatible hydrogenation catalyst with organisms that produce hydrogen and an alkene such as 1y, which has already been generated via engineered microbes.[18]
Figure 2.

Functional group tolerance of the biocompatible hydrogenation. Values shown are isolated yields for preparative scale reactions (5 mM substrate concentration with 8 mol% Royer catalyst in 90 mL of growth medium under an atmosphere of nitrogen in serum bottles shaken at 190 rpm) unless indicated otherwise.x Isolated yield for a preparative scale reaction run with 16 mol% Royer catalyst.y The reaction was run with 2.5 mM substrate.
Finally, we investigated how the biocompatible hydrogenation takes place and its impact on E. coli. A series of control experiments delineated the requirements for a successful reaction (Figure 3A, Table S12). We also quantified the hydrogen and formic acid produced in each reaction mixture, as both metabolites could potentially contribute to hydrogenation.[19] No reaction was observed in the absence of catalyst, confirming that E. coli cannot reduce 1a. The presence of E. coli was essential, indicating that the organisms contribute a key reaction component. The low conversions observed for the –IPTG control and a parental E. coli strain that cannot generate hydrogen support our hypothesis that hydrogen gas is the primary metabolite contributing to the transformation.[20] Finally, we assessed the importance of catalyst-cell contact by sequestering the E. coli in dialysis cassettes. Hydrogenation still occurs, albeit with lower conversion, indicating that physical interaction is not a requirement (Table S12, Entry 11). However, E. coli is known to adsorb onto PEI,[21] and increased catalyst-cell proximity via this mechanism could contribute to the Royer catalyst’s superior utility relative to other heterogeneous catalysts.
Figure 3.

Requirements of the biocompatible hydrogenation and its effect on E.oli. a) Control experiments and metabolite analyses. Reactions were run with 5 mM of substrate 1a and 8 mol% Royer catalyst in 7 mL of growth medium under an atmosphere of nitrogen in 16 mL Hungate tubes shaken at 190 rpm. Conversions were determined by 1H NMR and are the mean of three replicate experiments. Hydrogen and formic acid were quantified after 18 h using GC. b) Survival of E. coli DD-2 after 18 h reactions measured by serial dilution and plate count. Data shown are the mean of three independent experiments.
The application of non-enzymatic catalysts and reagents in metabolic engineering requires that they do not significantly impede host growth and metabolism. To ascertain whether the E. coli survive the hydrogenation, we performed serial dilutions and plate counts directly from spent reaction mixtures, systematically omitting reaction components to assess biocompatibility (Figure 3B). Remarkably, we observed no significant difference in survival between reactions with and without catalyst. Together with the experiment exploring spatial separation, this result may indicate that the hydrogenation occurs outside of cells. Additional experiments to fully elucidate the factors influencing catalyst activity and compatibility will be the focus of future research.
In summary, this work demonstrates that the metabolic output of living microbes and a biocompatible non-enzymatic transformation may be combined to enable preparative scale chemical synthesis. Although this methodology cannot yet match the efficiency of more established approaches,[22,23] this advance represents a crucial first step in merging the fields of organic chemistry and metabolic engineering and complements parallel efforts to engineer non-biological reactivity into enzyme scaffolds.[24] Ultimately, full integration of biocompatible reactions with cellular metabolism could provide access to chemical reactivity that would otherwise be out of reach in a cellular setting and molecules that could not be made using biological chemistry alone.
Experimental Section
General materials and methods, hydrogenation catalyst synthesis and analysis, reaction discovery and optimization, control reactions and metabolite analyses, assessment of catalyst toxicity, and compound characterization data are all described in the Supporting Information.
Supplementary Material
Footnotes
This work was supported by Harvard University, the Corning Foundation, the Searle Scholars Program, and the NIH (DP2 GM105434). We acknowledge Marina Santiago and Katarzyna Hojczyk for assistance with preliminary experiments, Tim Roth for checking the large scale hydrogenation procedure with substrates 1k and 1n, Charles Vidoudez and Peter Girguis for assistance with hydrogen and formic acid quantitation, and Daniel Ducat and Pamela Silver for providing E. coli strain DD-2.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201xxxxxx.
References
- 1.Hoffmann RW. Angew. Chem. Int. Ed. 2013;52:123–130. doi: 10.1002/anie.201203319. Angew. Chem.2013, 125, 133–140. [DOI] [PubMed] [Google Scholar]
- 2.Keasling JD. Science. 2010;330:1355–1358. doi: 10.1126/science.1193990. [DOI] [PubMed] [Google Scholar]
- 3.Jørgensen L, McKerrall SJ, Kuttruff CA, Ungeheuer F, Felding J, Baran PS. Science. 2013;341:878–882. doi: 10.1126/science.1241606. [DOI] [PubMed] [Google Scholar]
- 4.Ro D-K, Paradise EM, Ouellet M, Fisher KJ, Newman KL, Ndungu JM, Ho KA, Eachus RA, Ham TS, Kirby J, Chang MCY, Withers ST, Shiba Y, Sarpong R, Keasling JD. Nature. 2006;440:940–943. doi: 10.1038/nature04640. [DOI] [PubMed] [Google Scholar]
- 5.a) Nestl BM, Nebel BA, Hauer B. Curr. Opin. Chem. Biol. 2011;15:187–193. doi: 10.1016/j.cbpa.2010.11.019. [DOI] [PubMed] [Google Scholar]; b) Bornscheuer UT, Huisman GW, Kazlauskas RJ, Lutz S, Moore JC, Robins K. Nature. 2012;485:185–194. doi: 10.1038/nature11117. [DOI] [PubMed] [Google Scholar]
- 6.Savile CK, Janey JM, Mundorff EC, Moore JC, Tam S, Jarvis WR, Colbeck JC, Krebber A, Fleitz FJ, Brands J, Devine PN, Huisman GW, Hughes GJ. Science. 2010;329:305–309. doi: 10.1126/science.1188934. [DOI] [PubMed] [Google Scholar]
- 7.Lee Y, Umeano A, Balskus EP. Angew. Chem. Int. Ed. 2013;52:11800–11803. doi: 10.1002/anie.201307033. Angew. Chem. 2013, 125, 12016–12019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Martín-Matute B, Bäckvall J-E. Curr. Opin. Chem. Biol. 2007;11:226–232. doi: 10.1016/j.cbpa.2007.01.724. [DOI] [PubMed] [Google Scholar]; b) Denard CA, Hartwig JF, Zhao H. ACS Catalysis. 2013;3:2856–2864. [Google Scholar]; c) Wang ZJ, Clary KN, Bergman RG, Raymond KN, Toste FD. Nat. Chem. 2013;5:100–103. doi: 10.1038/nchem.1531. [DOI] [PubMed] [Google Scholar]; d) Kohler V, Wilson YM, Durrenberger M, Ghislieri D, Churakova E, Quinto T, Knorr L, Haussinger D, Hollmann F, Turner NJ, Ward TR. Nat. Chem. 2013;5:93–99. doi: 10.1038/nchem.1498. [DOI] [PubMed] [Google Scholar]; e) Borchert S, Burda E, Schatz J, Hummel W, Gröger H. J. Mol. Catal. B Enzym. 2012;84:89–93. [Google Scholar]; f) Gröger H, Hummel W. Curr. Opin. Chem. Biol. 2014;19:171–179. doi: 10.1016/j.cbpa.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 9.a) Prescher JA, Bertozzi CR. Nat. Chem. Biol. 2005;1:13–21. doi: 10.1038/nchembio0605-13. [DOI] [PubMed] [Google Scholar]; b) Sletten EM, Bertozzi CR. Acc. Chem. Res. 2011;44:666–667. doi: 10.1021/ar200148z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Sasmal PK, Streu CN, Meggers E. Chem. Comm. 2013;49:1581–1587. doi: 10.1039/c2cc37832a. [DOI] [PubMed] [Google Scholar]; b) Streu C, Meggers E. Angew. Chem. Int. Ed. 2006;45:5645–5648. doi: 10.1002/anie.200601752. Angew. Chem. 2006, 118, 5773–5776. [DOI] [PubMed] [Google Scholar]; c) Yusop RM, Unciti-Broceta A, Johansson EMV, Martin-Sánchez R, Bradley M. Nat. Chem. 2011;3:239–243. doi: 10.1038/nchem.981. [DOI] [PubMed] [Google Scholar]
- 11.a) Lee H-S, Vermaas WFJ, Rittmann BE. Trends Biotechnol. 2010;28:262–271. doi: 10.1016/j.tibtech.2010.01.007. [DOI] [PubMed] [Google Scholar]; b) Kalia VC, Purohit HJ. J. Ind. Microbiol. Biotechnol. 2008;35:403–409. doi: 10.1007/s10295-007-0300-y. [DOI] [PubMed] [Google Scholar]
- 12.a) Vigh L, Los DA, Horváth I, Murata N. Proc. Natl. Acad. Sci. USA. 1993;90:9090–9094. doi: 10.1073/pnas.90.19.9090. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Katsuda T, Ooshima H, Azuma M, Kato J. J. Biosci. Bioengin. 2006;102:220–226. doi: 10.1263/jbb.102.220. [DOI] [PubMed] [Google Scholar]; c) Kaspar HF, Holland AJ, Mountfort DO. Arch. Microbiol. 1987;147:334–339. [Google Scholar]
- 13.Agapakis CM, Ducat DC, Boyle PM, Wintermute EH, Way JC, Silver PA. J. Biol. Eng. 2010;4:3. doi: 10.1186/1754-1611-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JHY, Jo B, Cha H. Microb. Cell. Fact. 2010;9:54. doi: 10.1186/1475-2859-9-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Royer GP, Chow W-S, Hatton KS. J. Mol. Catal. 1985;31:1–13. [Google Scholar]; b) Coleman DR, Royer GP. J. Org. Chem. 1980;45:2268–2269. [Google Scholar]
- 16.After collecting all of the data described in this communication, we discovered differences in activity between batches of commercially available Royer catalyst. Consequently we have developed a procedure for reactivating inactive catalyst lots and our own synthesis of this catalyst. For details see the Supporting Information.
- 17.Polen T, Spelberg M, Bott M. J. Biotechnol. 2013;167:75–84. doi: 10.1016/j.jbiotec.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 18.Niu W, Draths KM, Frost JW. Biotechnol. Prog. 2002;18:201–211. doi: 10.1021/bp010179x. [DOI] [PubMed] [Google Scholar]
- 19.Johnstone RAW, Wilby AH, Entwistle ID. Chem. Rev. 1985;85:129–170. [Google Scholar]
- 20.A reaction run with catalyst and formic acid (100 µM) in media without cells gave 9% conversion (Table S12, Entry 10), suggesting that transfer hydrogenation may make a small contribution to conversion under our standard conditions.
- 21.Treweek GP, Morgan JJ. J. Colloid Interface Sci. 1977;60:258–273. [Google Scholar]
- 22.a) Busacca CA, Fandrick DR, Song JJ, Senanayake CH. Adv. Synth. Catal. 2013;353:1825–1864. [Google Scholar]; b) Hansen KB, Hsiao Y, Xu F, Rivera N, Clausen A, Kubryk M, Krska S, Rosner T, Simmons B, Balsells J, Ikemoto N, Sun Y, Spindler F, Malan C, Grabowski EJJ, Armstrong JD. J. Am. Chem. Soc. 2009;131:8789–8804. doi: 10.1021/ja902462q. [DOI] [PubMed] [Google Scholar]; c) Friedfeld MF, Shelvin M, Hoyt JM, Krska SW, Tudge MT, Chirik PJ. Science. 2013;342:1076–1080. doi: 10.1126/science.1243550. [DOI] [PubMed] [Google Scholar]
- 23.a) Stuermer R, Hauer B, Hall M, Faber K. Curr. Op. Chem. Biol. 2007;11:203–213. doi: 10.1016/j.cbpa.2007.02.025. [DOI] [PubMed] [Google Scholar]; b) Tischer W, Tiemeyer W, Simon H. Biochemie. 1980;62:331–339. doi: 10.1016/s0300-9084(80)80162-3. [DOI] [PubMed] [Google Scholar]; c) Rohdich F, Wiese A, Feicht R, Simon H, Bacher A. J. Biol. Chem. 2001;276:5779–5787. doi: 10.1074/jbc.M008656200. [DOI] [PubMed] [Google Scholar]
- 24.a) Lewis JC. ACS Catalysis. 2013;3:2954–2975. [Google Scholar]; b) Coelho PS, Brustard EM, Kannan A, Arnold FH. Science. 2013;339:307–309. doi: 10.1126/science.1231434. [DOI] [PubMed] [Google Scholar]; c) McIntosh JA, Coelho PS, Farwell CC, Wang ZJ, Lewis JC, Brown TR, Arnold FH. Angew. Chem. Int. Ed. 2013;52:9309–9312. doi: 10.1002/anie.201304401. Angew. Chem.2013, 125, 9479–9482. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.