

Abstract
In a three-stage genome-wide association study among East Asian women including 22,780 cases and 24,181 controls, we identified three novel genetic loci associated with breast cancer risk, including rs4951011 at 1q32.1 (in intron 2 of the ZC3H11A gene, P = 8.82 × 10−9), rs10474352 at 5q14.3 (near the ARRDC3 gene, P = 1.67 × 10−9), and rs2290203 at 15q26.1 (in intron 14 of the PRC1 gene, P = 4.25 × 10−8). These associations were replicated in European-ancestry populations including 16,003 cases and 41,335 controls (P = 0.030, 0.004, and 0.010, respectively). Data from the ENCODE project suggest that variants rs4951011 and rs10474352 may be located in an enhancer region and transcription factor binding sites, respectively. This study provides additional insights into the genetics and biology of breast cancer.
Breast cancer is one of the most common malignancies among women worldwide. Genetic factors play a significant role in breast cancer etiology1, 2. To date, genome-wide association studies (GWAS) have identified approximately 75 genetic loci to be associated with breast cancer risk2–5. With the exception of the studies we have conducted among East Asian women6–9 and one study conducted among women of African ancestry10, all other published GWAS have been conducted among women of European ancestry. Genetic risk variants identified to date from GWAS explain only about 10% of familial risk for breast cancer in East Asian women3. Given the difference in genetic architecture and environmental exposures between women of European and East Asian ancestry, additional GWAS need to be conducted among East Asian women to fully uncover the genetic basis of breast cancer risk.
The current study was conducted as part of the Asia Breast Cancer Consortium (ABCC) to search for additional novel susceptibility loci for breast cancer. Included in this study are data obtained from 22,780 breast cancer cases and 24,181 controls who were recruited in 14 studies conducted in multiple Asian countries (Supplementary Table 1). The discovery stage (Stage 1) included two GWAS, in which 5,285 Chinese women (SBCGS-1) and 4,777 Korean women (SeBCS1) were scanned primarily using the Affymetrix Genome-Wide Human SNP Array 6.0, which consists of 906,602 single nucleotide polymorphisms (SNPs). After applying quality control (QC) filters described previously6, 9, 11, 5,152 Chinese women (2,867 cases and 2,285 controls; 677,157 SNPs) and 4,298 Korean women (2,246 cases and 2,052 controls; 555,117 SNPs) remained in the current analysis. Imputation was conducted for each study following the MACH algorithm12 using HapMap II release 22 CHB and JPT data (2,416,663 SNPs) as the reference. Only SNPs with a high imputation quality score (RSQR ≥ 0.50) were analyzed for associations with breast cancer risk. In the analyses of data from Chinese and Korean women, a total of 1,930,412 SNPs and 1,907,146 SNPs, respectively, were included. A meta-analysis of these GWAS data was conducted using a fixed-effects, inverse-variance meta-analysis with the METAL program13. There was little evidence for inflation in the association test statistics for the studies included in Stage 1 (genomic inflation factors (λ): λ= 1.0426 for SBCGS-1, λ= 1.0431 for SeBCS1, and λ = 1.0499 for both studies combined; Supplementary Figure 1). When scaled to a study of 1,000 cases and 1,000 controls, λ1,000 were 1.02, 1.02, and 1.01, respectively.
To select SNPs for the Stage 2 replication, we used the following criteria: (1) P < 0.05 in the Stage 1 meta results; (2) same direction of association in both Stage 1 studies; (3) no heterogeneity observed between the two Stage 1 studies (P > 0.05 and I2 < 25%); (4) an imputation score of RSQR > 0.5 in both Stage 1 studies; (5) a minor allele frequency (MAF) of > 0.05 in both Stage 1 studies; and (6) not in strong LD (r2 < 0.5) with any of the known breast cancer susceptibility loci or any SNPs we had evaluated previously3, 6–9. For SNPs that met the above criteria but were in LD (r2 > 0.5) with each other, we selected only one SNP for replication. A total of 4,598 SNPs were selected, and assays for 4,071 SNPs were successfully designed using Illumina Infinium assays as a part of large-scale genotyping effort. Of the 4,071 SNPs, 3,850 SNPs were successfully genotyped in an independent set of 3,944 cases and 3,980 controls selected from the Shanghai studies (SBCGS-2). After QC exclusions, 3,678 SNPs were included in the analyses of 3,472 cases and 3,595 controls.
For Stage 3, the top 50 SNPs were selected for further replication in an independent set of 14,195 cases and 16,249 controls from 10 studies participating in the ABCC based on the following criteria: (1) P < 0.005 in the meta-analysis of Stage 1 and 2 data and (2) same direction of association in both Stages 1 and 2. Of the 50 SNPs evaluated in Stage 3, 11 SNPs showed an association with breast cancer risk at P < 0.05 (Supplementary Table 2). Combined analyses of data from all three stages identified three SNPs that were associated with breast cancer risk at the genome-wide significance level (P < 5.0 × 10−8): rs4951011 at 1q32.1, odds ratio (OR) = 1.09, P = 8.82 × 10−9; rs10474352 at 5q14.3, OR = 1.09, P = 1.67 × 10−9; and rs2290203 at 15q26.1, OR = 1.08, P = 4.25 × 10−8 (Table 1). The association between breast cancer risk and each of these three SNPs was consistent across the studies included in the ABCC (Figure 1), and none of the tests for heterogeneity were statistically significant (P > 0.05) (Table 1). No significant heterogeneity was found for the association of these three SNPs with breast cancer risk among Chinese, Japanese, or Korean women (Supplementary Table 3). One additional SNP showed an association with breast cancer risk with a P-value near the conventional GWAS significance level (rs11082321 at 18q11.2, OR = 1.08, P = 6.77× 10−7) (Supplementary Table 2).
Table 1.
Associations of breast cancer risk with newly identified risk variants: Results from the Asia Breast Cancer Consortium
| SNP (Allelesa) | Frequencyb | Locus (Positionc) | Closest gene (Annotation) | Stage | Per-allele association | P for heterogeneityf | |
|---|---|---|---|---|---|---|---|
| OR (95% CI)d | Pe | ||||||
| rs4951011 (G/A) | 0.282 | 1q32.1 (202,032,954) | ZC3H11A (Intron 2) | Stage 1 | 1.09 (1.02–1.17) | 0.007 | |
| Stage 2 | 1.10 (1.02–1.18) | 0.011 | |||||
| Stage 3 | 1.08 (1.05–1.12) | 1.02 × 10−5 | |||||
| Combined | 1.09 (1.06–1.12) | 8.82 × 10−9 | 0.98 | ||||
| rs10474352 (C/T) | 0.482 | 5q14.3 (90,767,981) | ARRDC3 (Intergenic) | Stage 1 | 1.09 (1.03–1.17) | 0.006 | |
| Stage 2 | 1.12 (1.05–1.20) | 7.06 × 10−4 | |||||
| Stage 3 | 1.08 (1.04–1.12) | 1.92 × 10−5 | |||||
| Combined | 1.09 (1.06–1.12) | 1.67 × 10−9 | 0.50 | ||||
| rs2290203 (G/A) | 0.504 | 15q26.1 (89,313,071) | PRC1 (Intron 14) | Stage 1 | 1.08 (1.02–1.14) | 0.012 | |
| Stage 2 | 1.19 (1.10–1.30) | 4.97 × 10−5 | |||||
| Stage 3 | 1.06 (1.03–1.10) | 2.45 × 10−4 | |||||
| Combined | 1.08 (1.05–1.11) | 4.25 × 10−8 | 0.06 | ||||
Abbreviations: OR, odds ratio; CI, confidence interval.
Risk/reference allele; risk allele is shown in bold.
Risk allele frequency in controls from all three stages combined.
Chromosome position (bp) based on NCBI Human Genome Build 36.
Per-allele OR (95% CI) was adjusted for age and principal components in each study; summary OR (95% CI) was obtained using fixed-effect meta-analysis in each stage.
Derived from a weighted z-statistic–based meta-analysis.
P for heterogeneity across studies in all stages was calculated using Cochran’s Q test.
Figure 1. Forest plots for risk variants in the three newly identified breast cancer risk loci by study site and stage.

Per-allele ORs are presented. The size of the box is proportional to the number of cases and controls in each study. (a): rs4951011, (b): rs10474352, (c): rs2290203.
The associations of SNPs rs10474352 and rs2290203 appeared to be stronger for estrogen receptor (ER)-positive breast cancer than for ER-negative breast cancer, and the heterogeneity test was of borderline significance for rs10474352 (P = 0.085) (Supplementary Table 4). The associations of rs4951011 with breast cancer risk were similar for ER-positive and ER-negative breast cancer.
We evaluated the three newly identified risk variants for associations with breast cancer risk in European-ancestry women using data from 16,003 cases and 41,335 controls derived from twelve breast cancer GWAS and included in the DRIVE GAME-ON Consortium. SNPs rs4951011, rs10474352, and rs2290203 were all associated with breast cancer risk in women of European ancestry at P < 0.05 with the same direction of association as observed in East Asian women (Supplementary Table 5). However, the strength of the associations was weaker in European-ancestry women than in East Asian-ancestry women, and the frequencies of the risk alleles were quite different between these two populations.
We evaluated and annotated putative functional variants and candidate genes in each of the three newly identified loci using data from the Encyclopedia of DNA Elements (ENCODE)14, The Cancer Genome Atlas (TCGA) breast cancer project15, expression quantitative trait locus (eQTL) databases16 as well as RegulomeDB17 and HaploReg v218. We summarize the results below for each locus.
SNP rs10474352 is located on 5q14.3, 53,078 bp upstream of the ARRDC3 gene (Figure 2b). The ARRDC3 gene is a member of the arrestin gene family and is suspected of playing a role in breast cancer development. A gene cluster at 5q11-q23 that includes ARRDC3 was found to be deleted in 17% of breast cancer tumor tissue19. Up regulation of the ARRDC3 gene in a breast cancer cell line has been shown to repress cell proliferation, migration, invasion, and in vivo tumorigenesis20. We evaluated ARRDC3 gene expression in 87 breast cancer cases included in TCGA. The expression level of the ARRDC3 gene was significantly lower in tumor tissue than in adjacent normal tissue (P = 1.88 × 10−18) (Supplementary Table 6). This is consistent with a previous study showing that expression levels of the ARRDC3 gene were lower in breast tumor tissue compared with normal tissue and in metastatic tumor tissue compared with primary tumor tissue20. Furthermore, lower ARRDC3 expression in tumor tissue has been associated with poorer disease-free survival in breast cancer patients20. A search of RegulomeDB17 and HaploReg18 indicated that rs10474352 may be located in predicted AP-1and VDR motifs (Supplementary Table 7), suggesting a potential regulatory role. We evaluated whether SNPs in this locus are cis-eQTL for other genes by analyzing TCGA breast cancer data. Our analysis revealed no evidence that this SNP or its correlated SNPs are cis-eQTLs for any genes in this locus. Recently, a SNP located ~596 kb upstream of the ARRDC3 gene, rs421379, was found to be associated with prognosis for early-onset breast cancer in a GWAS21. However, rs421379 is not in LD with rs10474352 (r2=0 in both ASN and CEU data), the SNP in close proximity to ARRDC3 that was identified in our study. Furthermore, in our study, rs421379 had a low MAF (0.03–0.04) and was not associated with breast cancer risk (P = 0.2484 in Stage 1).
Figure 2. Regional plots of association results for the three newly identified risk loci for breast cancer.

For each plot, the −log10 (P values) (y axis) of SNPs are shown according to their chromosomal positions (x axis) in NCBI Build 36. The color of SNPs represents their LD (r2, HapMap Asian) with the index SNP at each locus. With the exception of the index SNPs, which are shown as purple diamonds for Stage 1 and purple circles for the meta-analyses of all studies, data shown for all other SNPs are from Stage 1 only. (a): rs4951011 (1q32.1), (b): rs10474352 (5q14.3), (c): rs2290203 (15q26.1).
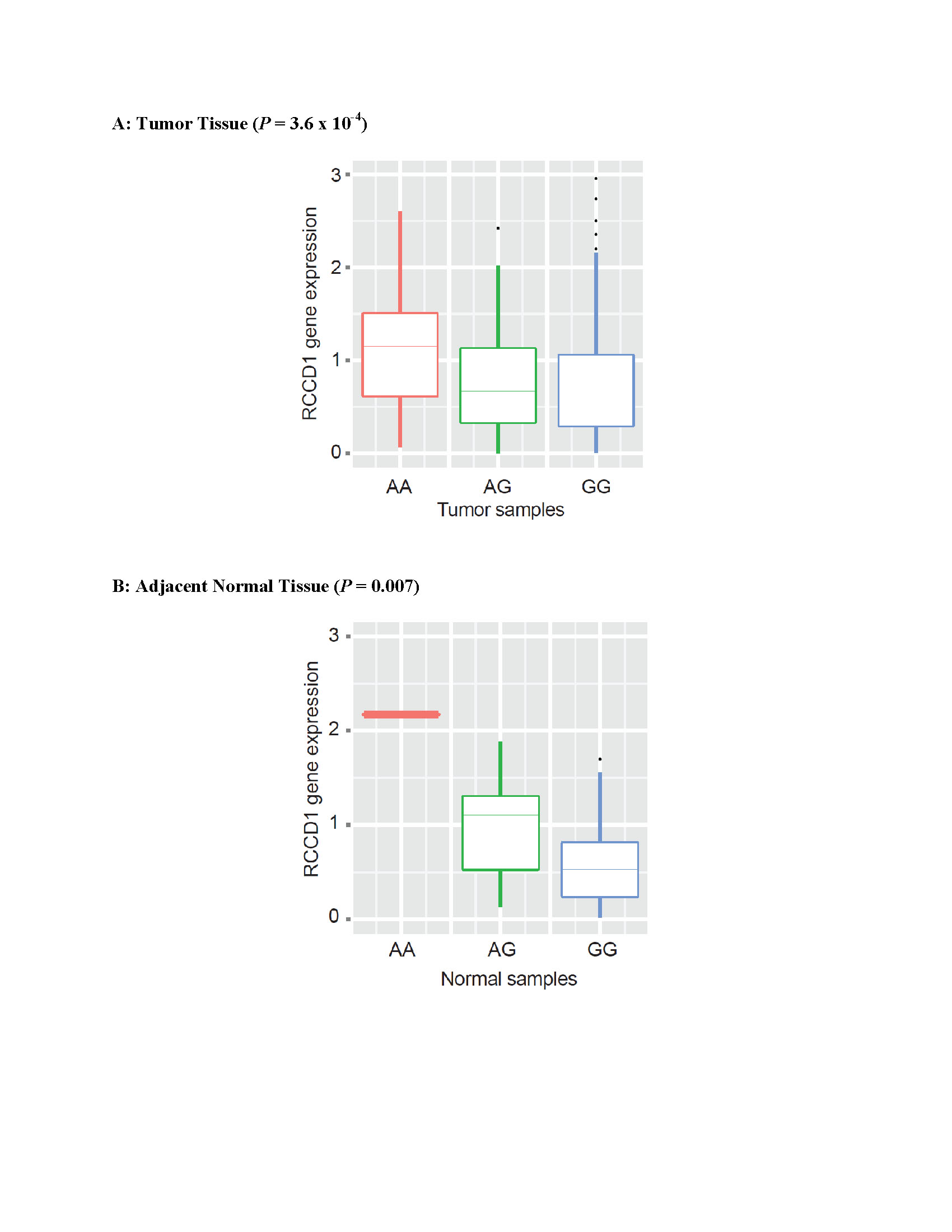
SNP rs2290203 is located in intron 14 of the protein regulator of cytokinesis 1 (PRC1) gene (NM_003981) at 15q26.1 (Figure 2c). This gene encodes the PRC1 protein, which is involved in cytokinesis and is a substrate for several cyclin-dependent kinases22. The PRC1 gene is down-regulated by the TP53 gene, and it is over-expressed in p53-defective cells23. Interestingly, the PRC1 gene is included in a five-gene expression signature that predicted prognosis among breast cancer patients in a recent study24. The expression level of the PRC1 gene was significantly higher in tumor tissue than in adjacent normal tissue (P = 4.62 × 10−30) among breast cancer cases included in TCGA (Supplementary Table 6). Our cis-eQTL analysis using TCGA data showed no association of rs2290203 with PRC1gene expression, but did reveal a correlation with expression of the RCCD1gene, which is 5,712 bp upstream of rs2290203. An eQTL analysis of human monocytes has also indicated that rs2290203 is a cis-eQTL for the RCCD1gene16. In our study, the rs2290203 risk allele (G) was associated with lower RCCD1 expression in both tumor (P = 3.6 × 10−4) and adjacent normal tissue (P = 0.007) (Supplementary Figure 2). However, these associations were no longer statistically significant after adjusting for the most significant cis-eQTL SNPs (rs4544218 for tumor tissue; rs59278520 for normal tissue), which are in strong LD with rs2290203 (Supplementary Figure 3). Variant rs4544218 was not associated with breast cancer risk (P = 0.8925), and rs59278520 was marginally associated with breast cancer risk (P = 0.0518) in the SBCGS-1, Stage 1 samples. The function of the RCCD1 gene is unknown.
SNP rs4951011 is located in intron 2 of the zinc finger CCCH domain-containing protein 11A (ZC3H11A) gene (NM_014827) at 1q32.1 (Figure 2a) and the 5′ untranslated region (UTR) of the zinc finger, BED-type containing 6 (ZBED6) gene (NM_001174108) (not shown in figure). The ZBED6 protein has recently been recognized as a novel transcription factor in placental mammals25. The function of the ZC3H11A gene is not clear. ChromHMM annotation using human mammary epithelial cell (HMEC) data from ENCODE suggests that rs4951011 may be located in a strong enhancer region marked by peaks of several active histone methylation modifications (H3K4me1, H3K4me3, H3K9ac, and H3K27ac). A search of RegulomeDB and HaploReg indicated that rs4951011 may be located in a predicted HNF1 motif and map to a DNase I hypersensitivity site in the MCF-7 cell line (Supplementary Table 7). Expression levels of the ZC3H11A gene were significantly higher in breast tumor tissue than in adjacent normal tissue (P = 0.0049) in TCGA data (Supplementary Table 6). Analyses using TCGA data revealed no evidence that this SNP or other SNPs correlated with it are cis-eQTLs for any genes in this locus. Recently, SNP rs4245739 in the MDM4 gene, ~752 kb downstream of rs4951011 (r2 = 0 in both ASN and CEU data), was associated with ER-negative breast cancer risk5. In our study, rs4245739 had a low MAF (0.03–0.05) and was not associated with breast cancer risk (P = 0.1861 in Stage 1).
In summary, our large GWAS conducted among East Asian women identified three new breast cancer susceptibility loci at 1q32.1, 5q14.3, and 15q26.1 and suggested a possible association with a fourth locus at 18q11.2. The associations of these loci with breast cancer risk may be mediated through cell-growth–control regulation, tumor-cell migration and invasion, or metastasis. Further studies of possible mechanisms through which these loci and genes are involved in breast tumorigenesis are warranted. Results from this study provide additional insights into the genetics and biology of breast cancer.
URLs
1000 Genomes Project, http://www.1000genomes.org/;
The Cancer Genome Atlas (TCGA), http://cancergenome.nih.gov/;
The DRIVE GAME-ON Consortium, http://epi.grants.cancer.gov/gameon/;
EIGENSTRAT, http://genepath.med.harvard.edu/~reich/EIGENSTRAT.htm;
ENCODE: http://www.genome.gov/10005107;
HaploReg, http://www.broadinstitute.org/mammals/haploreg/haploreg.php
HapMap Project, http://hapmap.ncbi.nlm.nih.gov/;
HumanExome Beadchip: http://genome.sph.umich.edu/wiki/Exome_Chip_Design;
LocusZoom, v1.1, http://csg.sph.umich.edu/locuszoom/;
MACH1.0, http://www.sph.umich.edu/csg/abecasis/MaCH/;
Mach2dat, http://genome.sph.umich.edu/wiki/Mach2dat:_Association_with_MACH_output;
METAL, http://www.sph.umich.edu/csg/abecasis/metal;
PLINK version 1.07, http://pngu.mgh.harvard.edu/~purcell/plink/;
R version 3.0.0, http://www.r-project.org/;
RegulomeDB, http://regulome.stanford.edu/;
SAS version 9.3, http://www.sas.com/;
UCSC Genome Browser, http://genome.ucsc.edu/;
ONLINE METHODS
Study populations
The ABCC comprises 22,780 cases and 24,181 controls from 14 studies (Supplementary Table 1), including 15,483 Chinese women, 18,367 Korean women, and 13,111 Japanese women. Chinese participants came from six studies based in: Shanghai [n = 12,219, the Shanghai Breast Cancer Study (SBCS), the Shanghai Breast Cancer Survival Study (SBCSS), the Shanghai Endometrial Cancer Study (SECS, controls only), and the Shanghai Women’s Health Study (SWHS)]6, 26–28, Taiwan (n = 2,131)29, and Hong Kong (n = 1,133)30. Korean participants came from five studies: the Seoul Breast Cancer Study (SeBCS) (n = 6,179)11, the Hwasun Cancer Epidemiology Study-Breast (HCES-Br) (n = 6,573)31–33, the Korea Genome Epidemiology Study (KoGES; n = 3,209)34, the Korean Hereditary Breast Cancer study (KOHBRA; n = 1,397)35, and the Korean National Cancer Center (n = 1,009). Japanese participants came from three studies: the Biobank Japan Project (BBJ) (n = 11,021)36, the Nagoya Study (n = 1,288)37, and the Nagano Breast Cancer Study (n = 802)38 (Supplementary Table 1). Detailed descriptions of these participating studies are presented in the Supplementary Note. The protocols for all participating studies were approved by their relevant Institutional Review Boards, and all participants of participating studies provided written informed consent. We estimated that our study had a statistical power of >80% to identify an association with an OR of 1.09 or above at P <5×10−8 for SNPs with a MAF as low as 0.25.
Genotyping Methods
Stage 1 genotyping
Stage 1 included two GWAS, in which 5,285 Chinese women and 4,777 Korean women were scanned primarily using the Affymetrix Genome-Wide Human SNP Array 6.0. Genotyping protocols for Stage I have been described elsewhere6–9, 11. In the Chinese GWAS (SBCGS-1), the initial 300 samples were genotyped using the Affymetrix GeneChip Mapping 500K Array Set. The remaining 4,985 samples were genotyped using the Affymetrix Genome-Wide Human SNP Array 6.0. After QC exclusions, the final dataset included 2,867 cases and 2,285 controls for 677,157 markers. For the Korean GWAS (SeBCS1), the Affymetrix Genome-Wide Human SNP Array 6.0 Array was also used11. After QC exclusions, the final dataset included 2,246 cases and 2,052 controls for 555,117 markers. Detailed descriptions of QC and criteria for sample and marker exclusion are presented in the Supplementary Note.
Stage 2 genotyping
Genotyping assays for 3,944 cases and 3,980 controls (SBCGS-2) in Stage 2 were completed using Illumina Infinium assays as the add-on content to the Illumina HumanExome Beadchip (see URLs). Genotype calling was carried out by using Illumina’s GenTrain version 2.0 clustering algorithm in GenomeStudio version 2011.1. Cluster boundaries were determined using study samples. Further QC procedures were conducted using PLINK (see URLs). Detailed descriptions of QC and criteria for sample and marker exclusion are presented in the Supplementary Note. Of the 4,598 SNPs selected, assays for 4,071 SNPs were successfully designed using Illumina Infinium assays. A total of 3,850 SNPs were successfully genotyped, and 3,678 SNPs were included in the analyses of 3,472 breast cancer cases and 3,595 controls.
Stage 3 genotyping
Genotyping assays for the 50 SNPs in Stage 3 were completed at the Vanderbilt Molecular Epidemiology Laboratory using the iPLEX Sequenom MassArray platform for 19,423 samples from the Taiwan, Hong Kong, HCES-Br, KOHBRA/KoGES, SeBCS2, Korea-NCC, Nagoya, and Nagano studies. Detailed descriptions of QC and criteria for sample and marker exclusion are presented in the Supplementary Note. For the BBJ1 study, the SNP data needed for the study were extracted from either genotyped (n = 8) or imputed (n = 14, mean RSQR = 0.96) data generated using the OmniExpress BeadChip. Breast cancer cases included in the BBJ2 study were genotyped using multiplex-PCR Invader assays. SNP data for the BBJ2 controls were extracted from data generated using the OmniExpress BeadChip.
We also selected 16 SNPs that showed a promising trend in the other studies included in Stage 3 for additional genotyping assays among 2,021 cases and 1,958 controls included in a case-control study conducted in Malaysia and Singapore that used the iPLEX Sequenom MassArray platform at the Cancer Research Initiatives Foundation, Sime Darby Medical Centre, Malaysia. However, because of a potential concern about genetic admixture revealed in our previous study3 and an unusual pattern of associations observed in these studies (Supplementary Table 8), we did not include these samples in the final analysis.
Statistical analyses
PLINK version 1.07 (see URLs)39 was used to analyze the genome-wide data obtained in Stage 1. To evaluate the population structure in the Chinese GWAS (SBCGS-1), we performed principal component analyses using the EIGENSTRAT software40 in a set of approximately 6,000 independent SNPs that met the following criteria: (1) a neighboring distance > 200 kb from the SNP of interest, (2) a MAF > 0.2, (3) r2 < 0.1, and (4) a genotype call rate > 99%. The inflation factor (λ) was estimated to be 1.0426. Similar analyses were performed for the Korean GWAS (SeBCS1) and yielded a λ of 1.043111. We also rescaled the inflation statistic to an equivalent value for a study including 1,000 cases and 1,000 controls (λ1000) using the formula: λ1000 = 1 + 500 × (λ − 1) × (1/Ncases + 1/Ncontrols)41. The λ1000 was 1.02 for both studies included in Stage 1 and 1.01 in the meta-analysis of both studies. These data suggest that any population substructure, if present, should not have any appreciable effect on the results. ORs associated with each SNP and 95% confidence intervals (CIs) were estimated using logistic regression implemented in PLINK with adjustment for age and the first two principal components.
We used the program MACH 1.0 (see URLs)12 to impute genotypes for autosomal SNPs (n = 2,416,663) that were present in CHB (Han Chinese in Beijing, China) and JPT (Japanese in Tokyo, Japan) HapMap Phase II release 22 data for samples included the Chinese and Korean GWAS. Only SNPs with high imputation quality score (RSQR > 0.50) and a MAF > 0.05 in these two GWAS were included in the analyses. Dosage data for imputed SNPs for samples in each GWAS were analyzed using the program Mach2dat (see URLs)12. Associations between genotype dosage (0, 1, and 2) of the effect allele and breast cancer risk were assessed using logistic regression models after adjusting for age and the first two principal components. ORs associated with each SNP and 95% CIs were estimated under a log-additive model. We also used SAS version 9.3 (see URLs) to analyze genotype data, which yielded results virtually identical to those generated from dosage data using Mach2dat. We obtained summary ORs and 95% CIs of SNPs from the two GWAS by using METAL software (see URLs)13 to run a fixed-effects inverse-variance meta-analysis.
Individual data were obtained from all studies except for the two BioBank Japan studies (BBJ1 and BBJ2). Case-control differences in selected demographic characteristics and major risk factors were evaluated using t-tests (for continuous variables) and Chi-square tests (for categorical variables). Summary associations between SNPs and breast cancer risk were generated on the basis of a fixed-effects inverse-variance meta-analysis conducted using METAL software13. Analyses stratified by ethnicity and estrogen receptor (ER) status were also carried out. Heterogeneity across studies, among ethnicities, and according to ER status was assessed with a Cochran’s Q test. P values of < 5 × 10−8 in the combined analysis were considered statistically significant.
We assessed associations of breast cancer risk with the three newly identified risk variants among European-ancestry women in collaboration with the DRIVE GAME-ON Consortium (see URLs). Included in this analysis were data from 16,003 cases and 41,335 controls recruited in twelve studies. Genome-wide scan data from these studies were imputed and meta-analyzed, and summary data are presented herein.
We generated forest plots by using R version 3.0.0 (see URLs). Regional association plots were drawn using the website-based tool LocusZoom, version 1.1 (see URLs)42. LD matrices used in this study were reported based on HapMap release 22 data. All genomic references are based on NCBI Build 36.
Functional annotation
Functional annotation was performed using data from the ENCODE project (see URLs) accessed through the UCSC Genome Browser (see URLs). Enhancer and transcription elongation regions were predicted in HMEC by using ChromHMM annotation. DNase I hypersensitive areas, transcription factor binding sites, and miRNA binding sites were evaluated in all cell types, including breast cancer cells, included in ENCODE. RegulomeDB (see URLs)17, a database that annotates SNPs with known and predicted regulatory elements in the intergenic regions of the human genome using data from GEO, the ENCODE project, and published literature, was also used to predict the possibility of transcription factor binding sites and DNase I hypersensitivity. In addition, we performed functional annotation using HaploReg v2 (see URLs)18, a tool for exploring annotations of the noncoding genome at variants on haplotype blocks.
eQTL analysis
We used the TCGA breast cancer data (Supplementary Note) to perform an eQTL analysis for normal and tumor tissue samples separately. Detailed descriptions of eQTL analysis are presented in the Supplementary Note. We focused only on the SNPs and genes located within the 1Mb regions flanking the three newly identified risk loci to identify cis-eQTLs. A significance threshold P value of < 0.01 was used to determine candidate cis-eQTLs.
Differential gene expression analysis
To identify differentially expressed genes located in the three identified risk loci, we analyzed data from a total of 87 pairs of breast tumor-normal tissue samples included in TCGA (Supplementary Note). Detailed descriptions of differential gene expression analysis are presented in the Supplementary Note.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies. The authors wish to thank the study participants and research staff for their contributions and commitment to this project. We thank Regina Courtney, Jie Wu, Jing He, Hui Cai, Xingyi Guo, and Bethanie Rammer for their help with sample preparation and genotyping, statistical and bioinformatics analyses for the project, and editing and preparing the manuscript at Vanderbilt.
This research was supported in part by US National Institutes of Health grants R01CA124558, R01CA148667, and R37CA070867 (to W. Zheng); R01CA118229, R01CA092585, and R01CA064277 (to X.-O. Shu); R01CA122756 (to Q. Cai); and R01CA137013 (to J. Long), Department of Defense Idea Awards BC011118 (to X.-O. Shu) and BC050791 (to Q. Cai), and Ingram Professorship and Research Reward funds (to W. Zheng). Sample preparation and genotyping assays at Vanderbilt were conducted at the Survey and Biospecimen Shared Resources and Vanderbilt Microarray Shared Resource, which are supported in part by the Vanderbilt-Ingram Cancer Center (P30CA068485). The SeBCS was supported by the BRL (Basic Research Laboratory) program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (2011-0001564). KOHBRA/KOGES was supported by a grant from the National R&D Program for Cancer Control, Ministry for Health, Welfare and Family Affairs, Republic of Korea (#1020350).
Studies participating in the ABCC include (Principal Investigator, grant support): the Shanghai Breast Cancer Study (W. Zheng and X.-O. Shu, R01CA064277), the Shanghai Women’s Health Study (W. Zheng, R37CA070867), the Shanghai Breast Cancer Survival Study (X.-O. Shu, R01CA118229), the Shanghai Endometrial Cancer Study (X.-O. Shu, R01CA092585, controls only), the Seoul Breast Cancer Study [D. Kang, BRL (Basic Research Laboratory) program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (2012-0000347)], the BioBank Japan Project (S.-K. Low, the Ministry of Education, Culture, Sports, Sciences and Technology from the Japanese Government); the Hwasun Cancer Epidemiology Study-Breast (S.-S. Kweon, the National R&D Program for Cancer Control, Ministry of Health & Welfare, Republic of Korea, # 1020010), the Taiwan Study (C.-Y. Shen, Institute of Biomedical Sciences, Academia Sinica, Taiwan and National Science Council, DOH97-01), the Hong Kong Study (U.-S. Khoo, Research Grant Council, Hong Kong SAR, China, HKU 7520/05M and 76730M), the Korean-NCC Study (M.-K. Kim, grant-in-aid from the Korea National Cancer Center, NCC-0910310), the Nagano Breast Cancer Study (S. Tsugane, Grants-in-Aid for the Third Term Comprehensive Ten-Year Strategy for Cancer Control from the Ministry of Health, Labor and Welfare of Japan, and for Scientific Research on Priority Areas, 17015049 and for Scientific Research on Innovative Areas, 221S0001, from the Ministry of Education, Culture, Sports, Science, and Technology of Japan), the Hospital-based Epidemiologic Research Program at Aichi Cancer Center [K. Tajima, Grant-in-Aid for Scientific Research (C) (No.24590776) and Grant-in-Aid for Scientific Research on Priority Areas of Cancer (No. 17015018) and on Innovative Areas (No. 221S0001) from the Japanese Ministry of Education, Culture, Sports, Science and Technology], the Malaysian Breast Cancer Genetic Study [S.-H. Teo, the Malaysian Ministry of Science, Technology and Innovation, Malaysian Ministry of Higher Education (UM.C/HlR/MOHE/06) and Cancer Research Initiatives Foundation. Controls were recruited by the Malaysian More than a Mammo Programme, which was supported by a grant from Yayasan Sime Darby LPGA], the Singapore Breast Cancer Study [M. Hartman, the National Medical Research Council start-up Grant and Centre Grant (NMRC/CG/NCIS /2010). Additional controls were recruited by the Singapore Consortium of Cohort Studies-Multi-ethnic cohort (SCCS-MEC), which was funded by the Biomedical Research Council, grant number: 05/1/21/19/425]. The DRIVE GAME-ON consortium is funded by NIH grant U19CA148065 (D. Hunter).
Footnotes
Note: Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
W.Z. conceived and directed the Asia Breast Cancer Consortium (ABCC) and the Shanghai Breast Cancer Genetics Study. Q.C. and W.Z. wrote the paper with significant contributions from B.Z., J.S., J.L., R.J.D., B.L., and X.-O.S. Q.C., B.Z., J.L., and W.W. coordinated the project. Q.C. directed the laboratory operations. J.S. performed the genotyping experiments. B.Z., J.L., and W.W. managed the study data. B.Z., J.L., and W.W. performed the statistical analyses with significant contributions from B.L. and C.L. Q.C., J.L., R.J.D., Y.Z., and B.L. performed the bioinformatics analyses. H.S., S.K.L., S.S.K., L.W., J.Y.C., D.Y.N., C.Y.S., K.M., S.H.T., M.K.K., U.S.K., M.I., M.H., A.T., K.A., K.M., M.H.S., M.H.P., Y.Z., Y.B.X., B.T.J., S.K.P., P.E.W., C.N.H., H.I., Y.K., P.K., S.M., S.H.A., H.S.K., K.Y.K.C., E.P.S.M., H.I., S.T., H.M., J.L., Y.N., M.K., Y.T.G, X.O.S., D.K., and W.Z. contributed to the collection of the data and biological samples for the original studies. All authors have reviewed and approved the final manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Reference List
- 1.Mavaddat N, Antoniou AC, Easton DF, Garcia-Closas M. Genetic susceptibility to breast cancer. Mol Oncol. 2010;4:174–191. doi: 10.1016/j.molonc.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang B, Beeghly-Fadiel A, Long J, Zheng W. Genetic variants associated with breast-cancer risk: comprehensive research synopsis, meta-analysis, and epidemiological evidence. Lancet Oncol. 2011;12:477–488. doi: 10.1016/S1470-2045(11)70076-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zheng W, et al. Common genetic determinants of breast-cancer risk in East Asian women: a collaborative study of 23 637 breast cancer cases and 25 579 controls. Hum Mol Genet. 2013;22:2539–2550. doi: 10.1093/hmg/ddt089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Michailidou K, et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet. 2013;45:353–2. doi: 10.1038/ng.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garcia-Closas M, et al. Genome-wide association studies identify four ER negative-specific breast cancer risk loci. Nat Genet. 2013;45:392. doi: 10.1038/ng.2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng W, et al. Genome-wide association study identifies a new breast cancer susceptibility locus at 6q25.1. Nat Genet. 2009;41:324–328. doi: 10.1038/ng.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Long J, et al. Identification of a functional genetic variant at 16q12.1 for breast cancer risk: results from the Asia Breast Cancer Consortium. PLoS Genet. 2010;6:e1001002. doi: 10.1371/journal.pgen.1001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai Q, et al. Genome-wide association study identifies breast cancer risk variant at 10q21.2: results from the Asia Breast Cancer Consortium. Hum Mol Genet. 2011;20:4991–4999. doi: 10.1093/hmg/ddr405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Long J, et al. Genome-wide association study in east Asians identifies novel susceptibility loci for breast cancer. PLoS Genet. 2012;8:e1002532. doi: 10.1371/journal.pgen.1002532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haiman CA, et al. A common variant at the TERT-CLPTM1L locus is associated with estrogen receptor-negative breast cancer. Nat Genet. 2011;43:1210–1214. doi: 10.1038/ng.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim HC, et al. A genome-wide association study identifies a breast cancer risk variant in ERBB4 at 2q34: results from the Seoul Breast Cancer Study. Breast Cancer Res. 2012;14:R56. doi: 10.1186/bcr3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bernstein BE, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeller T, et al. Genetics and beyond--the transcriptome of human monocytes and disease susceptibility. PLoS One. 2010;5:e10693. doi: 10.1371/journal.pone.0010693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boyle AP, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–1797. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40:D930–D934. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adelaide J, et al. Integrated profiling of basal and luminal breast cancers. Cancer Res. 2007;67:11565–11575. doi: 10.1158/0008-5472.CAN-07-2536. [DOI] [PubMed] [Google Scholar]
- 20.Draheim KM, et al. ARRDC3 suppresses breast cancer progression by negatively regulating integrin beta4. Oncogene. 2010;29:5032–5047. doi: 10.1038/onc.2010.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rafiq S, et al. Identification of inherited genetic variations influencing prognosis in early-onset breast cancer. Cancer Res. 2013;73:1883–1891. doi: 10.1158/0008-5472.CAN-12-3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu C, Jiang W. Cell cycle-dependent translocation of PRC1 on the spindle by Kif4 is essential for midzone formation and cytokinesis. Proc Natl Acad Sci U S A. 2005;102:343–348. doi: 10.1073/pnas.0408438102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li C, Lin M, Liu J. Identification of PRC1 as the p53 target gene uncovers a novel function of p53 in the regulation of cytokinesis. Oncogene. 2004;23:9336–9347. doi: 10.1038/sj.onc.1208114. [DOI] [PubMed] [Google Scholar]
- 24.Mustacchi G, et al. Identification and validation of a new set of five genes for prediction of risk in early breast cancer. Int J Mol Sci. 2013;14:9686–9702. doi: 10.3390/ijms14059686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Markljung E, et al. ZBED6, a novel transcription factor derived from a domesticated DNA transposon regulates IGF2 expression and muscle growth. PLoS Biol. 2009;7:e1000256. doi: 10.1371/journal.pbio.1000256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao YT, et al. Association of menstrual and reproductive factors with breast cancer risk: results from the Shanghai Breast Cancer Study. Int J Cancer. 2000;87:295–300. doi: 10.1002/1097-0215(20000715)87:2<295::aid-ijc23>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 27.Shu X, et al. Soy food intake and breast cancer survival. JAMA: The Journal of the American Medical Association. 2009;302:2437–2443. doi: 10.1001/jama.2009.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng W, et al. The Shanghai Women’s Health Study: rationale, study design, and baseline characteristics. Am J Epidemiol. 2005;162:1123–1131. doi: 10.1093/aje/kwi322. [DOI] [PubMed] [Google Scholar]
- 29.Ding SL, et al. Genetic variants of BLM interact with RAD51 to increase breast cancer susceptibility. Carcinogenesis. 2009;30:43–49. doi: 10.1093/carcin/bgn233. [DOI] [PubMed] [Google Scholar]
- 30.Chan KY, et al. Functional polymorphisms in the BRCA1 promoter influence transcription and are associated with decreased risk for breast cancer in Chinese women. J Med Genet. 2009;46:32–39. doi: 10.1136/jmg.2007.057174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song HR, et al. Sex-specific differences in the association between ABO genotype and gastric cancer risk in a Korean population. Gastric Cancer. 2013;16:254–260. doi: 10.1007/s10120-012-0176-z. [DOI] [PubMed] [Google Scholar]
- 32.Cui LH, et al. Methylenetetrahydrofolate reductase C677T polymorphism in patients with gastric and colorectal cancer in a Korean population. BMC Cancer. 2010;10:236. doi: 10.1186/1471-2407-10-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kweon SS, et al. Cohort Profile: The Namwon Study and the Dong-gu Study. Int J Epidemiol. 2014;43:558–567. doi: 10.1093/ije/dys244. [DOI] [PubMed] [Google Scholar]
- 34.Cho YS, et al. A large-scale genome-wide association study of Asian populations uncovers genetic factors influencing eight quantitative traits. Nat Genet. 2009;41:527–534. doi: 10.1038/ng.357. [DOI] [PubMed] [Google Scholar]
- 35.Han SA, et al. The Korean Hereditary Breast Cancer (KOHBRA) Study: Protocols and Interim Report. Clin Oncol (R Coll Radiol ) 2011;23:434–441. doi: 10.1016/j.clon.2010.11.007. [DOI] [PubMed] [Google Scholar]
- 36.Elgazzar S, et al. A genome-wide association study identifies a genetic variant in the SIAH2 locus associated with hormonal receptor-positive breast cancer in Japanese. J Hum Genet. 2012;57:766–771. doi: 10.1038/jhg.2012.108. [DOI] [PubMed] [Google Scholar]
- 37.Hamajima N, et al. Gene-environment Interactions and Polymorphism Studies of Cancer Risk in the Hospital-based Epidemiologic Research Program at Aichi Cancer Center II (HERPACC-II) Asian Pac J Cancer Prev. 2001;2:99–107. [PubMed] [Google Scholar]
- 38.Itoh H, et al. Serum organochlorines and breast cancer risk in Japanese women: a case-control study. Cancer Causes Control. 2009;20:567–580. doi: 10.1007/s10552-008-9265-z. [DOI] [PubMed] [Google Scholar]
- 39.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 41.Freedman ML, et al. Assessing the impact of population stratification on genetic association studies. Nat Genet. 2004;36:388–393. doi: 10.1038/ng1333. [DOI] [PubMed] [Google Scholar]
- 42.Pruim RJ, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.