

Abstract
It has been reported that nicotine/alcohol alters epigenetic control and leads to abrogated DNA methylation and histone modifications, which could subsequently perturb transcriptional regulation critically important in cellular transformation. The aim of this study is to determine the molecular mechanisms of nicotine/alcohol-induced epigenetic alterations and their mechanistic roles in transcriptional regulation in human adult stem cells. We hypothesized that nicotine/alcohol induces deregulation of epigenetic machinery and leads to epigenetic alterations, which subsequently affect transcriptional regulation in oral epithelial stem cells. As an initiating step we have profiled transcriptomic alterations induced by the combinatory administration of EtOH and nicotine in primary normal human oral keratinocytes. Here we provide detailed experimental methods, analysis and information associated with our data deposited into Gene Expression Omnibus (GEO) under GSE57634. Our data provide comprehensive transcriptomic map describing molecular changes induced by EtOH and nicotine on normal human oral keratinocytes.
Keywords: Gene expression signatures, Alcohol, Nicotine, Normal human oral keratinocytes
| Specifications | |
|---|---|
| Organism/cell line/tissue | Human normal oral keratinocytes |
| Sex | N/A |
| Sequencer or array type | Affymetrix Human Genome Plus 2.0 |
| Data format | Raw and analyzed |
| Experimental factors | Normal oral keratinocytes treated with EtOH and/or nicotine |
| Experimental features | Time and dose dependency exposure experiment to compare molecular effects of EtOH and nicotine in normal human oral keratinocytes |
| Consent | N/A |
| Sample source location | N/A |
Direct link to deposited data
Experimental design, materials and methods
Cell culture
Primary normal human oral keratinocytes (NHOKs) were prepared from normal oral mucosal tissues according to methods described in elsewhere [1]. The isolation of primary cells was approved by the Institutional Review Board (IRB) under the protocol # IRB10-000222. Briefly, discarded normal human oral mucosal tissues from routine periodontal surgery were obtained and stored in MEM/Ca2 + free medium containing 3 × gentamycin (Invitrogen, Carlsbad, CA). Oral mucosal tissues were cut into small pieces and incubated in Dispase solution (Invitrogen) for 1 h in at 37 °C. Epithelial tissues were gently separated from the underlying connective tissues and minced into smaller pieces. Minced samples were then trypsinized in 37 °C for 3–5 min, and trypsinization is inactivated with the equal amount of fetal bovine serum (FBS; Invitrogen). Trypsinized keratinocytes were then collected, washed, seeded onto the dish, and maintained in EpiLife (Cascade Biologics, Portland, OR) supplemented with Human Keratinocytes Growth Supplement (HKGS) kit (Life Technologies, Grand Island, NY). Minimal passage number was maintained to prevent cell senescence. A maximum 60% confluence was maintained to prevent contact growth inhibition. The morphology of NHOK was confirmed under a 20 × inverted light microscope. NHOKs were transferred to 6 well tissue culture treated plates (34.8 mm diameter). NHOKs were treated with ethanol (0, 20 and 50 mM) and/or nicotine (0, 0.5 and 1 μM) in biological duplicates. After 24 h, media was removed and the cells were washed twice with PBS.
RNA isolation
Total RNA was isolated from NHOK treated with ethanol (0, 20, 50 mM) and/or nicotine (0, 0.5 and 1 μM) for 24 h. RNA was extracted using a RNeasy purification kit, following the manufacturer's instruction (Qiagen). Isolated RNA was further purified by DNAse treatment (Promega). RNA purity and concentration was determined by NanoDrop, ND-1000 spectrophotometer (Thermo) and microfluidics-based platform 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). RNA concentration ranged from 85 ng/ul to 438.5 ng/ul. RNA concentration >=50 ng/μl is recommended for the subsequent microarray analysis. A 260/280 ratio ranged from 2.03 to 2.1. The ideal 260/280 ratio for pure RNA is 2.0.
Gene expression microarray analysis
Biological duplicate samples were hybridized to Affymetrix Human Genome Plus 2.0 (Cat.# 900469). We set target intensity (TGT) at 500. The sensitivity of the system was measured by %P using the 3′ biased Affymetrix HG-U133A 2.0 arrays. %P ranged from 46.5 to 48.8% demonstrating the ability to detect a large number of transcripts across a wide range of abundance. All 18 arrays were assessed for recommended standard quality control metrics by Affymetrix including image quality, signal distribution and pairwise scatter plots and passed. mas5.CHP files were generated for each array by MAS 5.0 (Affymetrix) and combined to a final RESULTS.MAS5.TXT file.
Data analysis
Degradation plot was prepared with each curve corresponding to a single chip and visualizing the chip-averaged dependency between probe intensity and probe position (Fig. 1A). Raw data was initialized and analyzed for the quality of microarray analysis by log density estimates of the data across all arrays (Fig. 1B).
Fig. 1.

A. Degradation plot: Each curve corresponds to a single chip and visualizes the chip-averaged dependency between probe intensity and probe position. B. Log density estimates (histograms) of the data across arrays.
We performed background correction (Fig. 2), quantile normalization and log transformation with Robust Multi-array Average (RMA) approach on Affymetrix gene expression data using “Affy” R package (Fig. 3) [2].
Fig. 2.

Quality control statistics. Each array is presented by a separated line. The blue bar represents the region where all scale factor fall within 3-fold of the mean scale factor for all chips. The chips passed all the QC metrics, indicating good quality data.
Fig. 3.

Boxplot of intensity for each sample after normalization and log transformation by using (A) Mas5 method and (B) Robust Multi-array Average (RMA) method.
We removed probes with expression lower than the overall sample median; 27,061 out of 54,676 probes were kept for further analysis.
Further data analysis was performed according to the Weighted Gene Co-expression Network Analysis (WGCNA) package tutorial written by Peter Langfelder and Steve Horvath [3]. The complete tutorial including necessary codes to run the analysis is publically available from http://labs.genetics.ucla.edu/horvath/CoexpressionNetwork/Rpackages/WGCNA/Tutorials/. The latest R (R-3.0.0) was downloaded to iMAC 2.9 GHz quad-core Intel Core i5 with 8 GB 1600 MHz DDR3 memory. Package WGCNA 1.36 was installed in conjunction with dynamicTreeCut, cluster, flashClust, Hmisc, reshape, foreach, and doParallel. Before data can be loaded to WGCNA, pre-processing step is necessary. Generally, WGCNA associated files should be in CSV file format without spaces, special characters and/or empty cells in their file names, as well as in their columns and rows within the files themselves. WGCNA_matrix.CSV was made from the final RESULTS.MAS5.TXT file. An example of the first ten rows from the WGCNA_matrix.CGV is shown in Table 1.
Table 1.
Details of WGCNA_matrix.CSV (WGCNA input). The first row describes the experimental condition that NHOKs were exposed to. It contains information on different concentration of EtOH and/or nicotine used on NHOKs (0 = no treatment, 20 = 20 mM EtOH, 50 = 50 mM EtOH, 05 = 0.5 μM nicotine, 1 = 1.0 μM nicotine, _1 = 1 of 2 in biological duplicates, _2 = 2 of 2 in biological duplicates). The first column contains ProbeSetID representing a defined probe from affymetrix microarray. Only first ten rows are shown.
| ProbeSet ID | EtOHOnicotineO 1 | EtOHOnicotineO 2 | EtOHOnicotineOS 1 | EtOH50nicotinc05 2 | EtOHSOnicotinel 1 | EtOHSOnicotinel 2 |
|---|---|---|---|---|---|---|
| 1007_s_at | 5981.038 | 5889.953 | 6831.399 | 5330.862 | 5741.348 | 5483.501 |
| 1053_at | 660.922 | 900.0433 | 747.627 | 814.9226 | 1019.678 | 697.8356 |
| 117_at | 77.72807 | 46.67521 | 103.4658 | 99.7991 | 79.94872 | 51.02247 |
| 121_at | 528.5612 | 613.58 | 498.6766 | 486.1765 | 458.8339 | 429.0858 |
| 1255_g_at | 5.511828 | 4.936847 | 10.26788 | 2.371168 | 19.53059 | 4.99963 |
| 1294_at | 200.1463 | 199.8828 | 255.2035 | 166.7272 | 173.7267 | 165.8017 |
| 1316_at | 152.9277 | 134.7367 | 230.6803 | 103.8526 | 126.7582 | 98.9052 |
| 1320_at | 74.21634 | 45.85337 | 96.10732 | 82.3228 | 81.87113 | 67.86658 |
| 1405_i_at | 84.68176 | 56.01822 | 62.11277 | 68.74342 | 62.43927 | 64.13792 |
| 1431_at | 26.04898 | 22.96477 | 37.09872 | 37.06742 | 18.30075 | 9.857845 |
A sample_annotation.CSV was made using the corresponding transposed columns from 18 arrays including control and different combinations of ethanol and/or nicotine treatment (Table 2).
Table 2.
Details of sample_annotation.CSV (WGCNA input). The sample annotation file contains information on how the samples should be ordered and which samples should be compared. Second column should reflect the first ro w from WGCNA_matric.CSV exactly. Columns three to ten are user defined depending on the user's specific hypothesis. For example, if the user wishes to compare control and EtOH + nicotine, then a comparison analysis between column three and four would be appropriate. 0's and 1's in each cell reflect corresponding binary information (0 = negative and 1 = positive).
| OriginalOrder | ProbeSetID | Control | EtOH_and_Nicotine | EtOH_0 | EtOH_20 | EtOH_50 | Nicotine_0 | Nicotine_05 | Nicotine_l |
|---|---|---|---|---|---|---|---|---|---|
| 1 | EtOHOnicotineO_l | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| 2 | EtOHOnicotineO_2 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| 3 | EtOH0nicotine05_l | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| 4 | EtOH0nicotine05_2 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| 5 | EtOHOnicotinel_l | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| 6 | EtOHOnicotinel_2 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| 7 | EtOH20nicotine0_l | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| 8 | EtOH20nicotineO_2 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| 9 | EtOH20nicotine05_l | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| 10 | EtOH20nicotine05_2 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| 11 | EtOH20nicotinel_l | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| 12 | EtOH20nicotinel_2 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| 13 | EtOH50nicotine0_l | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 |
| 14 | EtOH50nicotineO_2 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 |
| 15 | EtOH50nicotine05_l | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 0 |
| 16 | EtOH50nicotine05_2 | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 0 |
| 17 | EtOH50nicotinel_l | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 |
| 18 | EtOH50nicotinel_2 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 |
The WGCNA_matrix.CSV and the sample_annotation.CSV were read by WGCNA and sample dendogram and trait heatmap was plotted based on their Euclidean distance (Fig. 4).
Fig. 4.

Sample dendrogram and trait heatmap (WGCNA output). This is the first output data using WGCNA. The sample dendrogram allows users to visualize how the samples are cluster and identify any obvious outliers. EtOH0nicotine05_1 falls into possible outlier category. Outlier can be removed by editing the original WGCNA_matrix.CSV manually or the user can choose to set a limit on height to remove the outlier.
The sample dendogram and trait heatmap allows the users to check the data for excessive missing values and visualize obvious outlier samples. Next, a soft threshold power beta value based on the scale free topology was calculated. This is a critical step since the success of subsequent network construction and identification of modules depends on choosing the most ideal soft threshold power beta value. WGCNA allows the users to choose an automated “convenient-1-step method” or more customizable step-by-step method. It is important to maximize scale-free topology model fit (R^2) while maintaining a high mean number of connections. Generally, R^2 should be close to 1, the mean connectivity should be high so that the network contains enough information. The slope of the regression line should be close to − 1. We chose the soft threshold power beta = 9 since this was where the curve reached a saturation point in the Soft Threshold (SFT) graph (Fig. 5).
Fig. 5.

Threshold beta value calculation (WGCNA output). The left panel shows the Scale-Free Topology (SFT) Index in y-axis as a function of the Soft Threshold in x-axis. The graph is reaching a saturation point at threshold beta value = 9. The right panel shows the Mean Connectivity in y-axis as a function of the Soft Threshold in x-axis. The slope of the regression line should be close to − 1.
Once the Soft threshold power beta value was chosen based on the criterion of approximate scale-free topology, we turned the adjacencies into Topological Overlap Matrix (TOM). The main objective of our study was to identify changes in biological function and pathway of normal oral keratinocytes due to EtOH and/or nicotine. Although it is possible to simply rank microarray expression data based on the fold change alone, the strength of WGCNA comes from its robustness and sensitivity to identify genes/proteins of interest and its ability to cluster genes of interest into a module based on their interconnectedness [4]. We set merging threshold value at 0.25, beta power value at 9, maximum block size at 10,000 and minimum module size at 1000. The rest of the “blockwiseModules” function was left at its default values. The result from automatic module detection via dynamic tree cutting is shown in Fig. 6.
Fig. 6.
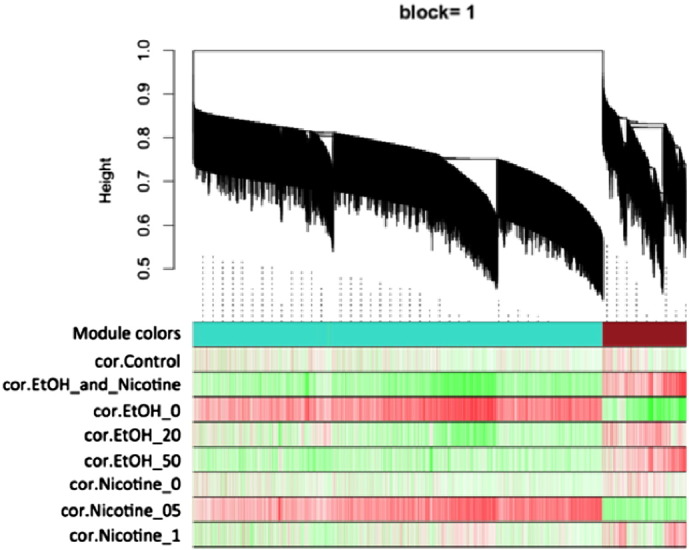
Module detection via dynamic tree cutting (WGCNA output). Each vertical line a.k.a. “leaf” represents a gene. A group of leaves form a “branch” which is densely interconnected co-expressing genes. Multiple branches converge into a tree which corresponds to a module. A random color is assigned to a module (i.e. turquoise and brown are shown here). Heatmaps show expression level of genes from corresponding experimental group (shown on left with starting with “cor.”) and denoted module (shown by “Module colors”). Heatmaps: Red — positive, green — negative.
In total, we found that genes from NHOKs treated with EtOH and/or nicotine were divided into six blocks and clustered into 14 distinctive modules using WGCNA (data not shown). Fig. 3 represents block 1 which contains two modules: turquoise and brown. Turquoise module had 15,174 genes. The 15,174 genes showed down-regulation by co-treatment of EtOH and nicotine compared to control. The turquoise module also contained genes that were downregulated upon EtOH treatment regardless of EtOH concentration. For nicotine, it seemed like the 0.5 μM nicotine treatment showed more significant change than the 1.0 μM concentration at least within the turquoise module.
In conclusion, we demonstrated that a complex dataset from microarray experiment can be analyzed effectively using WGCNA. Once the interesting modules are identified by WGCNA analysis, the modules can be related to external traits (i.e. EtOH/nicotine related cancers), used to find key drivers a.k.a. hub genes, and used for downstream functional annotation (i.e. DAVID analysis).
Acknowledgment
This work was supported by the UCLA Faculty Seed Grant and NIH/NIAAA R01 grant (R01AA21301) to Y.K.
References
- 1.Kim R.H., Lieberman M., Shin K.-H., Mehrazarin S., Park N.-H., Kang M. Bmi-1 extends the lifespan of normal human oral keratinocytes by inhibiting the TGF-β signaling. Exp. Cell Res. 2010;316:2600–2608. doi: 10.1016/j.yexcr.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gautier L., Cope L., Bolstad B.M., Irizarry R.A. affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 3.Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinforma. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yip A.M., Horvath S. Gene network interconnectedness and the generalized topological overlap measure. BMC Bioinforma. 2007;8:22. doi: 10.1186/1471-2105-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]