

Abstract
We report a 16-year-old boy who presented with weakness of lower limbs. He was diagnosed to have Wilson's disease, renal tubular acidosis and osteoporosis. Screening of siblings showed that his younger sister was also affected by the disease.
Keywords: Metabolic bone disease, renal tubular acidosis, Wilson's disease
Introduction
Wilson's disease is an autosomal recessive disorder caused by mutations in the ATP7B gene. There is failure of copper metabolism that leads to copper toxicity. Copper toxicity primarily affects the liver and the brain.[1]
Wilson's disease can cause distal renal tubular acidosis (RTA), which may lead to metabolic bone disease. Reports in this regard are scant. We report one such case with family analysis.
Case Report
A 16-year-old boy presented with history of fullness in left hypochondrium of the abdomen associated with early satiety and vague abdominal pain since the last one year. He had difficulty in walking since the last one year and difficulty in getting up from squatting position. He had hematemesis 6 months back, which lasted for a period of 1 week. It was small in amount and subsided spontaneously. There was no history of jaundice, weakness of upper limbs, sensory complaints in lower limbs or fever. His past medical history was unremarkable. He was born out of a consanguineous marriage and was the third of four siblings.
On examination, the patient was conscious, co-operative and oriented. He was of normal height for his age and gender. Gynecomastia was present on general examination [Figure 1]. On examination of the abdomen, spleen was enlarged 10 cm below the left costal margin and liver was enlarged 3 cm below the right costal margin. On central nervous system examination, higher function and cranial nerve examination was normal. Tone was normal in all the limbs. Power was decreased in both the lower limbs (Grade 4/5) and proximal muscles in both the lower limbs were more affected than distal muscles. Knee and ankle jerks were exaggerated bilaterally. Plantar response was flexor bilaterally. He had waddling type of gait on walking which may have been due to proximal muscle weakness. His left lower limb was shorter than the right lower limb by 2 cm. Length of his right lower limb was 81 cm while that of the left lower limb was 79 cm. The cause of shortening of the left lower limb was not known. However, clinical examination findings were not suggestive of rickets. His sensory system examination was normal and he had no signs of cerebellar involvement.
Figure 1.
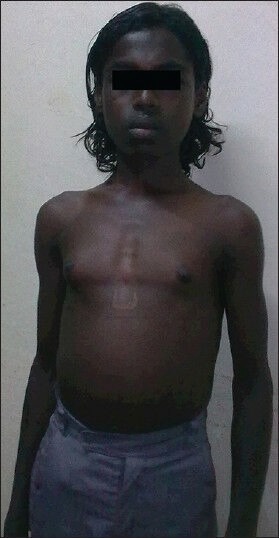
Gynecomastia in the patient (Patient consent obtained)
Investigations showed hemoglobin of 10.9 g/dl, total leucocyte count of 1900/mm3 with a differential count of 66 neutrophils, 30 lymphocytes, and 4 eosinophils. Platelet count was 33,000/mm3. Blood urea was 34 mg/dl and serum creatinine was 1.4 mg/dl. Serum sodium was 139 mEq/l and potassium was 3.4 mEq/l. Random blood glucose was 145 mg/dl. Liver function tests showed serum bilirubin to be 0.8 mg/dl, aspartate aminotransferase was 20 IU/l, alanine aminotransferase was 13 IU/l and raised alkaline phosphatase of 242 IU/l. Serum albumin was 3.7 g/dl and prothrombin time was 17.7 s with an INR of 1.47. Human immunodeficiency virus, hepatitis B surface antigen and anti-hepatitis C virus serologies were negative. His serum ceruloplasmin value was less than 15 mg/dl (normal range 30-50 mg/dl). Total serum calcium value was 8.9 mg/dl, serum phosphorous was 3.3 mg/dl and serum parathyroid hormone was 15 IU/l which was in normal range (normal range 12-95 IU/l). Vitamin D level (25-hydroxylation [OH] vitamin D) was 18.90 ng/ml, suggestive of vitamin D insufficiency. (normal value 30-100 ng/ml).
Urinalysis examination showed trace albumin, no glucose and 2-4 pus cells/high power fields on microscopy. Urine pH was 6.7 and analysis of 24 h urine electrolytes showed sodium of 106 mEq/l, potassium of 27 mEq/l, chloride of 129 mEq/L and urine anion gap was calculated to be 4 mEq/l. ([Na++ K+] − Cl−) The patient had hypercalciuria as calcium excretion was 329.8 mg/ day (normal range 100-250 mg/day) and phosphorous 387.6 mg/day (normal range 400-1300 mg/ day). There was no glucose or amino acids in the urine sample. His arterial blood gas (ABG) analysis was suggestive of hyperchloremic compensated metabolic acidosis (pH 7.4, pCO228.8 mm Hg, chloride 113 mEq/l, actual bicarbonate 17.3 mEq/l, and standard bicarbonate 20.5 mEq/l).
Slit lamp examination showed Kayser-Fleischer (KF) rings in both eyes [Figure 2]. Ultrasonography (USG) of abdomen showed coarsened echotexture of liver with a size of 13 cm, normal portal vein diameter and enlarged spleen with a size of 15.7 cm. Both kidneys were normal with no evidence of calculi on USG. Upper gastrointestinal endoscopy showed single Grade I varix in the lower esophagus and small erosion in the fundus of stomach. Digital X-rays of pelvis and lumbo-sacral spine showed osteopenia. X-ray of both wrist joints of the patient showed evidence of metaphyseal fraying and cupping. This could be a radiologic feature of rickets [Figure 3]. To confirm the radiologic findings, Dual-energy X-ray absorptiometry (DEXA) scan of lumbar spine (L2-L4) was done that revealed T score of −3.66 and Z score of −3.51 which was suggestive of severe osteoporosis. DEXA scan was done in this patient to confirm the radiologic findings and to document improvement after therapy. Nerve conduction velocity studies of lower limbs were normal. Electromyography of lower limb showed a myopathic pattern. Magnetic resonance imaging (MRI) studies of the brain showed bilaterally symmetrical pallidal hyperintensities on T1 sequence consistent with Wilson's disease. MRI of the spinal cord was normal.
Figure 2.
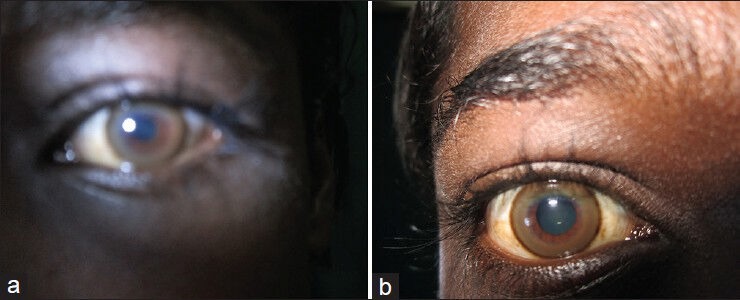
Kayser-Fleischer ring in (a) left eye and (b) right eye
Figure 3.
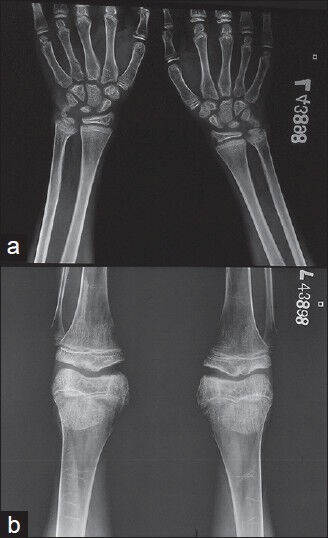
X-ray of both wrist joints of the patient
Screening of family members showed that the younger sister's ceruloplasmin value was 16.5 mg/dl and his elder brother's value was 31 mg/dl (normal range 30-50 mg/dl). KF ring was present on slit lamp examination in his sister and was absent in his brother. Eldest sister was not available for assessment, but historically was fine and was married with children.
Patient was started on oral zinc therapy (50 mg 3 times daily) and put on oral alkali (sodium bicarbonate) therapy for RTA. The patient was discharged on zinc tablets 3 times daily along with sodium bicarbonate two tablets 4 times a day. He was asked to come for follow-up. On follow-up after a period of 8 months, the patient reported subjective improvement in lower limb weakness. However, X-ray pelvis still showed osteopenia. His urine electrolytes could not be re-assessed due to financial constraints.
Discussion
Distal RTA is characterized by impaired ability of the distal renal tubules to excrete acid. It is identified by the kidneys' inability to acidify the urine to pH <5.5 in the presence of metabolic acidosis because of inability of the distal tubule to secrete hydrogen ions. In the absence of metabolic acidosis, distal RTA is diagnosed when hypercalciuria and hypocitraturia is present along with failure of the urine to reach pH <5.5 after an acid load. Hypercalciuria occurs due to release of calcium from bone that helps in the buffering of acid in urine and increased proximal citrate absorption results in hypocitraturia and both predispose to nephrocalcinosis and nephrolithiasis.
Metabolic acidosis in distal RTA causes reduction in bone carbonate content and leads to calcium loss from bone. This calcium loss from bone occurs as a result of physicochemical dissolution, inhibition of osteoblastic bone formation and stimulation of osteoclastic bone resorption. Metabolic acidosis also impairs the hydroxylation of 25(OH) vitamin D3 to 1, 25(OH) vitamin D3. This leads to decrease in both intestinal calcium absorption and renal tubular reabsorption of calcium. Renal loss of calcium (hypercalciuria) leads to nephrocalcinosis and nephrolithiasis in distal RTA.
Patients with Wilson's disease have the autosomal recessive form of distal RTA. Here, impaired function of distal renal tubules occurs due to gene mutations in chloride-bicarbonate exchanger in type A intercalated cells.
Hyperchloremia with low plasma bicarbonate and hypokalemia is seen with distal RTA, proximal RTA, diarrhea and chronic respiratory alkalosis. Diarrhea was ruled out by history while ABG analysis ruled out respiratory alkalosis. Urine pH <5.5 is a feature of proximal RTA while urine pH >6.0 is suggestive of distal RTA.[2] In this case, the positive urine anion gap (4 mEq/l) with low serum bicarbonate (17.3 mEq/l) and a urinary pH of 6.7 establishes the diagnosis of distal RTA. Hypercalciuria is also a feature of distal RTA and was present in this case.
The renal manifestations of Wilson's disease are nephrolithiasis and vitamin D-resistant rickets. Fanconi's syndrome may occur rarely.[3] This case was detected to have severe osteoporosis due to distal RTA. Wilson's disease was diagnosed to be the cause for distal RTA. Though Wilson's disease is known to be responsible for distal RTA, the presentation with severe osteoporosis and gait disturbance is relatively rare which was seen in this case.
To our knowledge, five cases of RTA (presenting as metabolic bone disease) have been previously reported in Wilson's disease. Out of the five cases, three had distal RTA on investigation and they have been reported from India.[4,5,6] Proximal RTA though less commonly seen in Wilson's disease was reported in one case.[7] In one case, the type of RTA was not exactly differentiated. Renal rickets occurring in Wilson's disease is reported to be refractory to treatment with vitamin D and calcium supplements.[8]
Treatment with zinc and oral alkali helps in clinical improvement of patients with Wilson's disease and RTA. Symptomatic and radiographic improvement of osteoporosis occurs gradually over a period of time.
The initial presentation of Wilson's disease as an osteomuscular syndrome is seen to be peculiar to the Indian subcontinent.[8] The late presentation of this patient to the physician may explain to some extent the reason why the prominent features were musculo-skeletal in this patient. This case had a similar presentation of Wilson's disease leading to metabolic bone disease. Bone disease in this patient is due to renal and hepatic involvement. The primary cause is metabolic acidosis and the contributing factors are vitamin D deficiency and chronic liver disease. Metabolic bone disease can occur as a result of RTA which impairs reabsorption of calcium. Metabolic bone disease due to RTA is a rare presenting feature of Wilson's disease.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Brewer GJ. Wilson's disease. In: Longo DL, Fauci AS, Kasper DL, Hauser SL, Jameson JL, Loscalzo J, editors. Harrison's Principles of Internal Medicine. 18th ed. Vol. 2. New York: The McGraw-Hill Companies; 2012. p. 3188. [Google Scholar]
- 2.Adrogue HJ, Madias NE. Renal tubular acidosis. In: Davison AM, Cameron JS, Grunfeld JP, Ponticelli C, Ritz E, Winearls CG, editors. Oxford Textbook of Clinical Nephrology. 3rd ed. Vol. 2. New York: Oxford University Press; 2005. p. 984.p. 987. [Google Scholar]
- 3.Cox DW, Roberts EA. Wilson disease. In: Feldman M, Friedman LS, Brandt LJ, editors. Sleisenger and Fordtran's Gastrointestinal and Liver Disease. 9th ed. Vol. 2. Philadelphia: Elsevier; 2010. p. 1252. [Google Scholar]
- 4.Palkar AV, Shrivastava MS, Padwal NJ, Padhiyar RN, Moulick N. Renal tubular acidosis due to Wilson's disease presenting as metabolic bone disease. BMJ Case Rep 2011. 2011 doi: 10.1136/bcr.04.2011.4121. pii: bcr0420114121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rais N, Dalal D, Bhandarkar SD, Donde UM. Wilson's disease (a case report) J Postgrad Med. 1980;26:149–51. [PubMed] [Google Scholar]
- 6.Kalra V, Mahajan S, Kesarwani PK. Rare presentation of Wilson's disease: A case report. Int Urol Nephrol. 2004;36:289–91. doi: 10.1023/b:urol.0000034630.73124.36. [DOI] [PubMed] [Google Scholar]
- 7.Kashyap AS, Kumar V. Short stature and rickets. Lancet. 2000;356:1894. doi: 10.1016/s0140-6736(00)03261-x. [DOI] [PubMed] [Google Scholar]
- 8.Dastur DK, Manghani DK, Wadia NH. Wilson's disease in India. I. Geographic, genetic, and clinical aspects in 16 families. Neurology. 1968;18:21–31. doi: 10.1212/wnl.18.1_part_1.21. [DOI] [PubMed] [Google Scholar]