

Abstract
β-Adrenergic receptor blockers (β-blockers) are commonly used to treat heart failure, but the biologic mechanisms governing their efficacy are still poorly understood. The complexity of β-adrenergic signaling coupled with the influence of receptor polymorphisms makes it difficult to intuit the effect of β-blockers on cardiac physiology. While some studies indicate that β-blockers are efficacious by inhibiting β-adrenergic signaling, other studies suggest that they work by maintaining β-adrenergic responsiveness. Here, we use a systems pharmacology approach to test the hypothesis that in ventricular myocytes, these two apparently conflicting mechanisms for β-blocker efficacy can occur concurrently. We extended a computational model of the β1-adrenergic pathway and excitation-contraction coupling to include detailed receptor interactions for 19 ligands. Model predictions, validated with Ca2+ and Förster resonance energy transfer imaging of adult rat ventricular myocytes, surprisingly suggest that β-blockers can both inhibit and maintain signaling depending on the magnitude of receptor stimulation. The balance of inhibition and maintenance of β1-adrenergic signaling is predicted to depend on the specific β-blocker (with greater responsiveness for metoprolol than carvedilol) and β1-adrenergic receptor Arg389Gly polymorphisms.
Introduction
β-Adrenergic receptor blockers (β-blockers) are front-line therapies for the treatment of heart failure, yet the biologic mechanism governing their success is still poorly understood (Krum, 2003; Tilley and Rockman, 2006; El-Armouche and Eschenhagen, 2009). The β1-adrenergic receptor pathway has a dominant role in the regulation of heart contractility (Saucerman and McCulloch, 2006). One of the hallmarks of heart failure is elevated catecholamine release, which desensitizes the β-adrenergic pathway, causing an inability to increase contractility and cardiac output in response to acute stress (Ungerer et al., 1994). Two apparently conflicting theories commonly postulated are that β-blockers are effective in heart failure by either inhibiting the harmful consequences of sustained adrenergic stimulation or maintaining the beneficial aspects of β1-adrenergic receptor pathway activation (Lohse et al., 2003). The inhibition hypothesis is supported by clinical and experimental evidence that β-blockers help prevent or reverse the cardiac remodeling that leads to heart failure (Lowes et al., 1999). Conversely, the maintenance hypothesis is given credence by clinical evidence that β-blockers increase β1-adrenergic receptor levels (Michel et al., 1988) and exercise tolerance (Engelmeier et al., 1985).
The ability of different β-blockers to either inhibit or maintain signaling is varied, causing controversy about which β-blocker is more effective in heart failure (Metra et al., 2006). Among the 17 US Food and Drug Administration–approved β-blockers, a variety of pharmacologic properties beyond receptor specificity alone may contribute to these differences (Mason et al., 2009). For example, some β-blockers are inverse agonists (Metra et al., 2006), reducing signaling below basal levels (Parra and Bond, 2007). Yet the importance of inverse agonism in determining clinical outcome during β-blocker treatment is unclear.
Genetic differences among patients also impact β-blocker efficacy (Krum, 2003). In vitro experiments in cell-expression systems show that the common β1AR-Arg389Gly single-nucleotide polymorphism has a higher fold increase in adenylyl cyclase activity after receptor stimulation but is more desensitized (Mason et al., 1999; Rathz et al., 2003). Carvedilol and metoprolol have similar affinities for both receptor variants in vitro (Joseph et al., 2004), but carvedilol has a larger effect on receptor conformation of the β1-Arg389 variant (Rochais et al., 2007). Thus, there may be compound-specific phenotypes for β1-adrenergic receptor polymorphisms (Dorn and Liggett, 2009).
The complexity of the β-adrenergic receptor pathway, coupled with the influence of receptor polymorphisms, makes it difficult to intuit the effect of β-blockers on observed cardiac physiology. Here we use a systems pharmacology approach (Sorger and Schoeberl, 2012), extending our previous computational models of β1-adrenergic signaling and excitation-contraction coupling (Saucerman et al., 2003, 2004) to investigate the apparently conflicting mechanisms by which β-blockers may inhibit or maintain β-adrenergic signaling. We tested the hypothesis that in normal ventricular myocytes, both proposed mechanisms for β-blocker efficacy can occur concurrently. To do this, a previous computational model of the β1-adrenergic receptor pathway was extended to include detailed receptor interactions for 19 ligands. Model predictions, validated with Ca2+ and Förster resonance energy transfer (FRET) imaging of isolated adult ventricular myocytes, surprisingly suggest that β-blockers can both inhibit and maintain signaling depending on the magnitude of receptor stimulation. In addition, the model predicted β-blocker-specific effects of receptor polymorphisms.
Materials and Methods
Computational Model of β-Blockers and β-Adrenergic Signaling.
A computational model was previously developed that integrates β1-adrenergic receptor signaling with excitation-contraction coupling in rat cardiac myocytes and is based on mass action kinetics (Saucerman et al., 2003). The receptor module was previously described by a ternary complex model (De Lean et al., 1980). To better model the inverse agonism of some β-blockers seen in in vitro experiments (Varma et al., 1999), the receptor module of our original β1-adrenergic receptor signaling model was replaced with the extended ternary complex model (ETCM) (Samama et al., 1993). The ETCM (Fig. 1) proposes two receptor states: active and inactive, and appropriately describes the constitutive activity of β-adrenergic receptors. The existence of these receptor states has been recently confirmed by determination of the crystal structure of the β2-adrenergic receptor (Rosenbaum et al., 2011). Parameters for the ETCM and detailed calibration procedures are described in the Supplemental Methods and Supplemental Table 1. The expanded model has 49 algebraic and differential equations and is constrained by 102 parameters. Sensitivity analysis was used to determine ETCM parameters with distinct effects on model prediction before sequential parameter estimation (Supplemental Figs. 1 and 2). In descriptions comparing model predictions and experimental data, the terms calibration and fitting are used to describe instances where model parameters were used to better fit those data, while the term validation is used to describe instances where model parameters were not adjusted to fit those data.
Fig. 1.

Extended ternary complex model of the β1-adrenergic receptor, coupled with the β1-adrenergic pathway and ventricular myocyte excitation-contraction coupling. KL, equilibrium dissociation constant of the agonist receptor complex; KR, propensity for switching between active and inactive receptor states; KG, dissociation constant for binding of G-protein to the receptor; α, differential affinity of the ligand for the inactive receptor; γ, differential affinity of the ligand-receptor complex for G-protein.
Isolation and Culture of Rat Cardiac Myocytes.
All procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health and approved by the University of Virginia Institutional Animal Care and Use Committee. Adult rat ventricular myocytes were isolated similar to Bers et al. (Bers et al., 1990) from adult male (200–250 g) Sprague-Dawley rats. Briefly, rats were anesthetized with ketamine/xylazine and hearts quickly excised before being Langendorff-perfused with collagenase (Cellutron Life Technologies, Baltimore, MD). Ventricular tissue was removed, mechanically dispersed, and filtered; and myocyte suspensions were rinsed and plated on 35-mm glass coverslips treated with 40 µg/ml laminin (Invitrogen, Carlsbad, CA) at a density of ∼3 x 106 cells per ml. Unattached myocytes were removed after 1 hour by replenishing media. Cells were then loaded with 1 µM fluo-4 acetoxymethyl (Gee et al., 2000) (Invitrogen) or infected with AKAR3 adenovirus (Vector Biolabs, Philadelphia, PA) following the manufacturer’s instructions in a solution of minimum Eagle’s medium (MEM) containing (in mM) NaHCO3 4.7, pyruvic acid 2, Na-HEPES 10, HEPES 10 and (in units/ml) insulin 0.4, and penicillin-streptomycin 50 (pH 7.35), Myocytes were then placed in an RC-21BRFS slotted bath chamber (Warner Instruments, Hamden, CT). The chamber was constantly perfused with Tyrode’s solution containing (in mM) NaCl 140, KCl 4, MgCl2 1, and HEPES 10 (pH 7.4) before cells were stimulated with isoproterenol (Tocris, Minneapolis, MN) and various β-blockers (propranolol, metoprolol, and carvedilol; Tocris). The flow rate of the perfusate was approximately 1–1.5 ml/min. Myocytes were field paced with the Myopacer (Ionoptix, Milton, MA) at a frequency of 1 Hz with bipolar pulse duration of 4 milliseconds at a voltage of 10 V. All measurements were performed at room temperature.
Camera-Based Ca2+ Imaging of Myocytes.
Ca2+ was measured using fluo-4 as described previously (Amanfu et al., 2011). Myocytes were imaged on an Olympus IX-81 inverted microscope (Olympus, Center Valley, PA) with a Hamamatsu C9300 charge-coupled device camera (Bridgewater, NJ) and automated stage (Prior Scientific, Rockland, MA) at a sampling frequency of 67 Hz using Metamorph (Molecular Devices, Sunnyvale, CA). To minimize photobleaching and phototoxicity, cells were imaged intermittently for 10 seconds after every minute. Automated cell segmentation using Otsu’s method identified regions of interest from which Ca2+ transients for each cell were extracted at each time point. Raw fluorescence values were background-subtracted and normalized to yield fold change in fluo-4 intensity:
 |
Average fluo-4-fold change was calculated by averaging five to seven consecutive transients at specific time points. All segmentation and feature extraction was implemented in MATLAB. Code for these analyses and example movies are freely available at http://bme.virginia.edu/saucerman.
FRET Imaging of Cardiac Myocytes.
Adenovirus was constructed from plasmid DNA of AKAR3 protein kinase–A reporter (Allen and Zhang, 2006). Myocytes were infected with adenovirus immediately after isolation in serum MEM media for 1 hour. Cells were then cultured for 24 hours in serum-free MEM media. Myocytes were preincubated in solutions of 0.1 µM isoproterenol with and without 0.1 µM propranolol. Cells were placed in a slotted bath with Tyrodes perfusate and paced at 10 Hz. Expressing myocytes were imaged on an Olympus IX-81 inverted microscope with a Hamamatsu C9300 charge-coupled device camera. A cocktail of 10 μM forskolin (Tocris) and 100 μM 3-isobutyl-1-methylxanthine (Sigma-Aldrich, St. Louis, MO) was used as positive control at the end of each experiment. Automated cell segmentation and FRET computation (using the precision FRET [PFRET] algorithm) (Chen and Periasamy, 2006) were performed in MATLAB. FRET response was normalized to positive control.
Results
Calibration and Validation of the β1-Adrenergic Model with the Extended Ternary Complex Receptor Model.
To quantitatively investigate how β-blockers modulate β-adrenergic signaling in cardiac myocytes, a computational model of the β1-adrenergic receptor pathway was developed that includes detailed interactions between ligand, receptor, and G-protein in the form of the extended ternary complex model (Fig. 1). The integrated model describes stimulation of the β1-adrenergic receptor, activation of receptor intermediates, production of cAMP, activation of protein kinase A (PKA), phosphorylation of downstream PKA targets, and the effect on Ca2+ transients. Receptor desensitization by both the β-adrenergic receptor kinase and PKA is also included.
Model predictions were compared with a range of experimental data from the literature and the current study (Fig. 2). The shift in agonist binding to the receptor in the presence of guanosine 5′-[β,γ-imido] triphosphate (which displaces the G protein) was validated (Fig. 2A). The model validates reasonably well against measured kinetics of cAMP (Fig. 2B) and PKA activity (Fig. 2D) in response to isoproterenol. The model is calibrated to have appropriate basal and maximally stimulated cAMP levels in cardiac myocytes, with validation of the sensitivity to isoproterenol (Fig. 2C). The EC50 of isoproterenol for phosphorylation of phospholamban by PKA is also accurately validated (Fig. 2E). The model was calibrated to have an appropriate EC50 of isoproterenol for Ca2+ transients to isoproterenol, as measured with fluo-4 by our group and others (Fig. 2F). In addition, we validated model predictions of Ca2+ transient responses to increasing propranolol concentration in the presence of 0.1 µM isoproterenol (Supplemental Fig. 4). A summary of all calibrations and validations is provided in Supplemental Table 2. These results indicate that the updated model is consistent with experimental data at multiple levels of the β1-adrenergic receptor pathway, providing confidence in the utility of the model for testing hypotheses regarding β-blocker efficacy.
Fig. 2.

Experimental validation of coupled β1-adrenergic signaling and excitation-contraction coupling model. (A) Model reproduces shift in agonist binding affinity in the presence of guanosine 5′-[β,γ-imido] triphosphate (GPP), which displaces Gs from the receptor. (B) Kinetics of [cAMP] in response to 10 nM isoproterenol (ISO) stimulation. (C) cAMP dose response to ISO. (D) PKA activity measured by FRET reporter AKAR3. (E) Phospholamban phosphorylation in response to ISO. (F) Ca2+ dose response to ISO. Results in (A)–(C), (E), and (F) show direct comparison with published experimental data (Mason et al., 1999), (Vila Petroff et al., 2001), (De Arcangelis et al., 2010), (Vittone et al., 1998), and (Collins and Rodrigo, 2010), whereas data in (D) and (F) were acquired in the current study.
Propranolol Inhibits and Maintains the β-Adrenergic Response Depending on the Magnitude of Receptor Stimulation.
While inhibition and maintenance of β-adrenergic responsiveness are typically thought to be incompatible explanations of β-blocker efficacy, we hypothesized that both could occur depending on the magnitude of receptor stimulation. To test this hypothesis in silico, we simulated low (0.1 µM) and then high (10 μM) levels of isoproterenol in the absence and presence of the first-generation β-blocker propranolol. We used 0.1 μM propranolol because this was the lowest dose that suppressed Ca2+ transients at 0.1 µM isoproterenol (Supplemental Fig. 3). Low and high doses of isoproterenol are analogous to chronically elevated levels of catecholamines in heart failure and acutely elevated levels in exercise, respectively. In the absence of propranolol, the model predicts that low receptor stimulation increases Ca2+ amplitude (Fig. 3A), with no further sensitivity to subsequent high levels of isoproterenol (Fig. 3B). In the presence of propranolol, responsiveness to low receptor stimulation is suppressed, but the pathway maintains sensitivity of Ca2+ transients to high isoproterenol (Fig. 3C). Independent experiments imaging Ca2+ dynamics in isolated adult rat ventricular myocytes qualitatively validated these model predictions (Fig. 3, D–F). These simulations and experiments indicate that the apparently conflicting roles of the β-blocker propranolol to inhibit signaling and maintain responsiveness are in fact compatible.
Fig. 3.

Propranolol both inhibits and maintains β1-adrenergic-mediated regulation of Ca2+ transients. (A) Model-predicted individual Ca2+ transients in response to increasing concentration of isoproterenol (ISO). (B) Ca2+ concentration increased in response to 0.1 µM ISO, with no further response to subsequent stimulation with 10 µM ISO. (C) The model predicted that propranolol (PRO) inhibits response to 0.1 μM ISO, but the responsiveness to 10 μM ISO is maintained (large sensitivity). (D) Individual Ca2+ transients as measured by fluo-4 from rat ventricular myocytes exposed to increasing [ISO]; scale bar 20 µm. (E) Similar to model predictions, myocytes were not responsive to further stimulation with 10 µM ISO. (F) PRO inhibited response to 0.1 μM ISO, but myocytes were responsive to further stimulation with 10 µM ISO. Sensitivity was quantified as the increase in Ca2+ transient magnitude when increasing from 0.1 µM ISO (analogous to chronically elevated catecholamines in heart failure) to 10 µM ISO (analogous to exercise).
To experimentally investigate whether these effects persist with chronic receptor stimulation, cells were pretreated with a low dose of isoproterenol for 24 hours before subsequent stimulation with high isoproterenol (Fig. 4). In the absence of propranolol, Ca2+ transient amplitude in pretreated cells was not further sensitive to high isoproterenol (Fig. 4C), as Ca2+ transients were already elevated. In contrast, cells pretreated with propranolol maintained sensitivity to high-dose isoproterenol in the presence of propranolol, similar to model predictions and the acute experiments (Fig. 3). Using a FRET reporter for PKA activity, we found that PKA (upstream of Ca2+ in the β1-adrenergic pathway) also maintains sensitivity to high isoproterenol in the presence of propranolol (Fig. 4B), again validating model predictions (Supplemental Fig. 8).
Fig. 4.

Propranolol both inhibits and maintains the β1-adrenergic-mediated Ca2+ and PKA response after 24-hour isoproterenol (ISO) pretreatment. (A) Expression and cytosolic distribution of PKA activity biosensor AKAR3 in rat adult ventricular myocytes (YFP emission); scale bar 40 µM. Following 24-hour pretreatment with both 0.1 µM ISO and 0.1 µM propranolol (PRO), both (B) PKA activity measured by AKAR3 and (C) Ca2+ response as measured by fluo-4 were still sensitive to a subsequent increase to 10 µM ISO.
β-Blockers Differ in Their Ability to Inhibit and Maintain β-Adrenergic Responsiveness.
To examine whether the dual role of propranolol in inhibiting and maintaining β-adrenergic responsiveness may be generalized to other β-blockers, we extended the model to 17 additional β-adrenergic receptor ligands. Two key ligand-specific properties: ligand dissociation constant (KL or KA) and inverse agonism (αL or αA), were calibrated using data on ligand binding and adenylyl cyclase activity for these 19 ligands in Chinese hamster ovary cells overexpressing human β1-adrenergic receptor (Hoffmann et al., 2004) (Supplemental Fig. 2).
We then performed an in silico screen of the 19 ligands for effects on β-adrenergic responsiveness (Fig. 5). Similar to simulations in Fig. 3, low and then high isoproterenol doses were simulated in the presence and absence of the indicated ligand at 1 µM. The model predicted substantial diversity in the ability of ligands to maintain cAMP sensitivity to subsequent high-dose isoproterenol (Fig. 5A). To understand this diversity, we examined correlations between cAMP sensitivity and the ligand-specific parameters KL and α. As shown in Fig. 5B, ligand binding affinity was predicted to influence cAMP sensitivity in a biphasic manner (e.g., metoprolol had the highest cAMP sensitivity with a moderate KL). In order for the initial binding event to occur, a β-blocker needs to have a low enough binding affinity to out-compete the receptor agonist. There is also a modest positive correlation between α and cAMP sensitivity, suggesting that ligands with high α (indicating a high degree of inverse agonism) preferentially increase cAMP sensitivity (Fig. 5B). This is due to such ligands keeping the receptor in an inactive state, preventing receptor desensitization (Supplemental Figs. 8–10). Neither ligand binding affinity nor inverse agonism is sufficient alone to predict cAMP sensitivity. Thus the ability of β-blockers such as metoprolol to maintain cAMP sensitivity was due to both inverse agonism and binding affinity.
Fig. 5.

Ligand-binding affinity and inverse agonism were both predicted to influence ligand cAMP sensitivity. (A) In silico screen of 19 β1-adrenergic ligands predicts differential cAMP sensitivity. (B) Effect of ligand dissociation constant (KL) on predicted cAMP sensitivity. (C) Effect of ligand inverse agonism (α) on predicted cAMP sensitivity. Propranolol (PRO), metoprolol, and carvedilol (highlighted in red) were predicted to have both distinct effects on cAMP sensitivity with distinct combinations of ligand dissociation constant and inverse agonism.
Metoprolol and Carvedilol Differ in Their Ability to Maintain β-Adrenergic Responsiveness.
An interesting prediction of the in silico screen is that carvedilol and metoprolol (two clinically prescribed β-blockers) differ significantly in their ability to enhance cAMP sensitivity. This difference indicates that the two drugs may have distinct effects (inhibition or maintenance) on the β1-adrenergic response. To experimentally validate model predictions for carvedilol and metoprolol, Ca2+ imaging experiments in adult rat ventricular myocytes were performed. As with propranolol, we empirically selected doses for carvedilol and metoprolol that were just sufficient to suppress responses to 0.1 µM isoproterenol (Supplemental Fig. 4). Sensitivity to high-dose isoproterenol was maintained in cardiac myocytes treated with 1 µM metoprolol (Fig. 6A) but suppressed with 1 µM carvedilol (Fig. 6B), qualitatively validating our model predictions (summarized in Fig. 6, C and D).
Fig. 6.
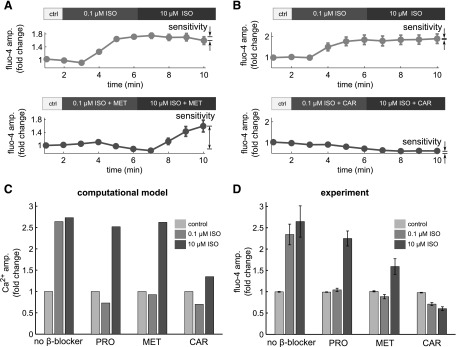
Metoprolol and carvedilol differentially influence β1-adrenergic responsiveness. (A) In adult ventricular myocytes, metoprolol (MET) blocked response to 0.1 μM isoproterenol (ISO), but the responsiveness to 10 μM ISO was maintained. (B) Carvedilol (CAR) blocked the response to both 0.1 μM ISO and 10 μM ISO. (C) Summary of model-predicted Ca2+ responses to 0.1 µM and 10 µM ISO in the presence of β-blockers. MET and propranolol (PRO) were both predicted to substantially enhance cAMP sensitivity to 10 µM ISO, but CAR was not. (D) Summary of experimental validations from adult ventricular myocytes for PRO, MET, and CAR.
To test the robustness of this result to β-blocker concentration, we further simulated how cAMP sensitivity is affected by propranolol, metoprolol, and carvedilol doses. The model predicted that propranolol and metoprolol robustly maintained β-adrenergic responsiveness, but that at doses lower than we had previously examined, carvedilol may also maintain β-adrenergic responsiveness (Supplemental Fig. 5). To test this prediction, we performed subsequent experiments with 0.3 µM carvedilol (Supplemental Fig. 6), which showed that lower carvedilol still suppressed the responsiveness to high-dose isoproterenol. The robust suppression seen in the carvedilol experiments can be explained by an alternative receptor model accounting for binding of carvedilol to an allosteric site on the β1-adrenergic receptor (Supplemental Fig. 7), as supported by previous experiments by Kindermann et al. (Kindermann et al., 2004).
Receptor Polymorphisms Are Differentially Modulated by Diverse β-Blockers.
Genetic differences among patients also impact β-blocker efficacy (Shin and Johnson, 2010). Patients with the β1-Arg389 variant have a better prognosis after β-blocker administration compared with patients with the β1-Gly389 polymorphism. Increased G-protein binding is observed experimentally for the β1-Arg389 variant, causing higher constitutive activity. This behavior was modeled by altering KG, the ETCM model parameter that affects binding of the active receptor to G-protein. KG in the β1-Arg389 model was manually calibrated to 0.7 µM to replicate the shift in agonist binding in the presence of guanosine 5′-[β,γ-imido] triphosphate (Fig. 7A) and the higher constitutive activity of the Arg389 variant (Fig. 7B). β1-Arg389 and β1-Gly389 polymorphisms were predicted to have varying responses to propranolol. Propranolol had more of an effect inhibiting the low-dose isoproterenol cAMP and Ca2+ responses in the Arg389 variant (Fig. 7C), but it also enhanced sensitivity to high-dose isoproterenol. The 19 ligands were predicted to have varying effects on cAMP sensitivity (Fig. 7D), similar to the β1-Gly389 variant (Fig. 5A). However, there are some significant differences in the responses to particular ligands between the receptor polymorphisms (Fig. 7E). For example, atenolol was predicted to be less effective at maintaining β-adrenergic responsiveness for the β1-Arg389 variant compared with the β1-Gly389 variant. This diversity of responses indicates that computational models may be useful for predicting pharmacogenetic interactions.
Fig. 7.

β1AR-Arg389 polymorphism responds differently to β-blockers. (A) Model reproduces the shift in agonist binding affinity in the presence of guanosine 5′-[β,γ-imido] triphosphate for Arg389. (B) Concentration dependence of adenylyl cyclase (AC) activity to isoproterenol for Gly389 and Arg389. (C) Arg389 is predicted to have higher cAMP sensitivity and Ca2+ response versus Gly 389 in cardiac myocytes. (D) In silico screen of 19 β1-adrenergic ligands against Arg389. (E) Differential cAMP sensitivity between Arg389 and Gly389 for different β1-adrenergic ligands predicted for cardiac myocytes. Experimental data in panels (A) and (B) are from (Mason et al., 1999; Mialet Perez et al., 2003).
Discussion
Mechanisms of β-Blocker Efficacy in Heart Failure.
A key feature of heart failure is the modest chronic elevation of circulating catecholamines (e.g., epinephrine) which desensitizes the β-adrenergic receptor signaling pathway, rendering patients incapable of increasing cardiac output in response to intense acute stress (e.g., exercise). Crucial alterations to the signaling pathway in this chronically activated state include reduced β1-adrenergic receptor density (Bristow et al., 1982) and Ca2+ (Harding et al., 1992) in response to adrenergic stimulation. Sustained stimulation has detrimental long-term consequences, including apoptosis and hypertrophy (Communal et al., 1998; Taimor et al., 2001). Maintenance of signaling in cardiomyopathy by adenylyl cyclase overexpression (Roth et al., 1999) or G-protein receptor kinase 2 inhibition (Reinkober et al., 2012) has improved cardiac function in in vitro murine models. Previous studies of mechanisms governing β-blocker efficacy have focused exclusively on one of two mechanisms: i.e., the inhibition (Lowes et al., 1999) or maintenance of β1-adrenergic receptor signaling (Engelmeier et al., 1985; Michel et al., 1988). With evidence supporting both theories, it is unclear how these two contradictory mechanisms can explain the same biologic phenomena or the appropriate context where one mechanism dominates. This study provides evidence that at least in normal isolated adult ventricular myocytes, both mechanisms can occur concurrently dependent on the magnitude of receptor stimulation.
Complexities at the receptor level and the influence of receptor polymorphisms complicate attempts to infer these mechanisms. Computational modeling is highly suited for this task by allowing the unbiased comparison of clinically available β-blockers. Previous computational models of the β1-adrenergic receptor pathway have used simplified receptor kinetic models (Saucerman et al., 2003, 2004). Although sufficient to describe the activation of the signaling pathway by agonists, these pathway models do not have the mechanistic detail of receptor kinetics needed to adequately model the inverse agonism of β-blockers. Detailed receptor models have been developed, but these models have been evaluated in isolation from downstream signaling pathways (Samama et al., 1993). To model β-blockers, detailed models of receptor kinetics were linked to the cardiac β1-adrenergic receptor pathway and excitation contraction coupling. Computational model simulations indicate that both inhibition and maintenance of signaling are compatible, dependent on the magnitude of receptor stimulation. Propranolol inhibited low-dose isoproterenol (analogous to chronic levels of catecholamine seen in heart failure) but enabled sensitivity to high-dose isoproterenol (analogous to acute catecholamine levels during exercise). Fluo-4 and FRET imaging of isolated cardiac myocytes confirmed this prediction.
Metoprolol and Carvedilol Have Distinct Mechanisms of Action in Isolated Ventricular Myocytes.
Separate clinical trials of the two β-blockers commonly used to treat heart failure show reduction in mortality. Results of the COMET trial, which aimed to compare both treatments, concluded that carvedilol had a larger effect on mortality (Poole-Wilson et al., 2003). Significant controversy surrounds this result with questions raised on the appropriate dose of each compound that merits fair comparison (Kveiborg et al., 2007). Another important clinical measure of heart failure treatment effectiveness is exercise tolerance. Studies have shown that metoprolol has a larger effect on exercise tolerance than carvedilol (Metra et al., 2000). Our computer simulations and Ca2+ imaging experiments confirm that metoprolol maintains β-adrenergic signaling in isolated ventricular myocytes due to its moderate binding affinity and high inverse agonism. Carvedilol, although also an inverse agonist, did not maintain isoproterenol sensitivity, due to its tight binding to the β1-adrenergic receptor and the potential contribution from allosteric binding (Kindermann et al., 2004).
Pharmacogenomic Targeted Treatment with β-Blockers.
Another factor complicating treatment of heart failure patients is the presence of β-adrenergic receptor polymorphisms. β1-Gly389 has been shown to couple less effectively to G-protein in expression cell systems, but the β1-Arg389 variant provides higher risk for heart failure and differential responses to β-blockers. A recent study has shown that carvedilol exhibits enhanced inverse agonism with the β1-Arg389 variant (Rochais et al., 2007), an example of the potential for personalized medicine. Understanding how genotype affects therapeutic response is expected to open a new era of pharmacogenomics and personalized medicine. One obstacle is that existing knowledge of β1-adrenergic receptor polymorphisms comes from cell lines that might function differently in healthy versus failing myocytes. We modeled β1-adrenergic receptor polymorphisms in the background of a ventricular myocyte. The model identified differences between the receptor polymorphisms’ cAMP sensitivity to high-dose isoproterenol in the presence of particular ligands. For example, atenolol was predicted to be less effective at maintaining β-adrenergic responsiveness in isolated ventricular myocytes expressing β1-Arg389 compared with the β1-Gly389 variant.
Limitations and Considerations
A critical decision in developing computational models is specifying the models’ scope. Uncertainty in parameters, and henceforth the ensuing predictions, becomes overwhelming as model scope increases. We have restricted our model to the β1-adrenergic receptor pathway and its effects on Ca2+ transients in isolated rat ventricular myocytes, because this pathway plays a central role in enhancing contractility after β-adrenergic stimulation. However, an alternative hypothesis is that other properties of β-blockers (i.e., binding to other adrenergic receptors and pharmacokinetic properties including half-life, lipid solubility, and nonspecific binding) may play a larger role than blockade of the β1-adrenergic receptors. Indeed our simulations suggest that binding of carvedilol to an allosteric site on the β1-adrenergic receptor influences its effect on β-adrenergic responsiveness. Our current computational model is not yet able to fully explore the consequence of this mechanism in vivo. Future work could couple the β1-adrenergic signaling model to whole-body pharmacokinetics or simulate crosstalk with other adrenergic receptors, including the β2-adrenergic receptor (Zamah et al., 2002).
Conclusions
Previous studies have suggested two seemingly conflicting mechanisms (inhibition or maintenance of the β-adrenergic receptor signaling pathway) to explain β-blocker efficacy. Here we show, both in pathway models and adult ventricular myocytes, that the β-blockers propranolol and metoprolol (but not carvedilol) not only block response to low isoproterenol (analogous to chronic stimulation in heart failure) but also maintain the β-adrenergic receptor response to subsequent high-dose isoproterenol (analogous to acute stimulation in exercise). Thus, both inhibition and maintenance of signaling can occur concurrently, dependent on the magnitude of receptor stimulation. Computational simulations indicate that these responses are modulated by particular receptor polymorphisms. Evaluating the mechanisms for these differences, with the help of computational models, is an important step toward designing personalized β-blocker therapies.
Supplementary Material
Acknowledgments
The authors thank Renata Polanowska-Grabowska for technical assistance.
Abbreviations
- β1-AR
β1-adrenergic receptor
- β-blockers
β-adrenergic receptor blockers
- ETCM
extended ternary complex model
- FRET
Förster resonance energy transfer
- IBMX
3-isobutyl-1-methylxanthine
- MEM
minimum Eagle’s medium
- PKA
protein kinase A
Authorship Contributions
Participated in research design: Amanfu, Saucerman.
Conducted experiments: Amanfu.
Contributed new reagents or analytic tools: Amanfu.
Performed data analysis: Amanfu.
Wrote or contributed to the writing of the manuscript: Amanfu, Saucerman.
Footnotes
This work was supported by the American Heart Association [Grant 0830470N] and the National Institutes of Health National Heart, Lung, and Blood Institute [Grants HL094476 and HL05242 (to J.J.S)].
 This article has supplemental material available
at molpharm.aspetjournals.org.
This article has supplemental material available
at molpharm.aspetjournals.org.
References
- Allen MD, Zhang J. (2006) Subcellular dynamics of protein kinase A activity visualized by FRET-based reporters. Biochem Biophys Res Commun 348:716–721 [DOI] [PubMed] [Google Scholar]
- Amanfu RK, Muller JB, and Saucerman JJ (2011). Automated image analysis of cardiac myocyte Ca2+ dynamics, in Proceedings of Engineering in Medicine and Biology Society, EMBC, 2011 Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Aug 30-Sept 3 2011, Boston, MA, pp. 4661–4664. [DOI] [PMC free article] [PubMed]
- De Arcangelis V, Liu S, Zhang D, Soto D, Xiang YK. (2010) Equilibrium between adenylyl cyclase and phosphodiesterase patterns adrenergic agonist dose-dependent spatiotemporal cAMP/protein kinase A activities in cardiomyocytes. Mol Pharmacol 78:340–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM, Lederer WJ, Berlin JR. (1990) Intracellular Ca transients in rat cardiac myocytes: role of Na-Ca exchange in excitation-contraction coupling. Am J Physiol 258:C944–C954 [DOI] [PubMed] [Google Scholar]
- Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, Billingham ME, Harrison DC, Stinson EB. (1982) Decreased catecholamine sensitivity and β-adrenergic-receptor density in failing human hearts. N Engl J Med 307:205–211 [DOI] [PubMed] [Google Scholar]
- Chen Y, Periasamy A. (2006) Intensity range based quantitative FRET data analysis to localize protein molecules in live cell nuclei. J Fluoresc 16:95–104 [DOI] [PubMed] [Google Scholar]
- Collins HE, Rodrigo GC. (2010). Inotropic response of cardiac ventricular myocytes to beta-adrenergic stimulation with isoproterenol exhibits diurnal variation: involvement of nitric oxide. Circ Res 106:1244–1252 [DOI] [PubMed] [Google Scholar]
- Communal C, Singh K, Pimentel DR, Colucci WS. (1998) Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the β-adrenergic pathway. Circulation 98:1329–1334 [DOI] [PubMed] [Google Scholar]
- Dorn GW, 2nd, Liggett SB. (2009) Mechanisms of pharmacogenomic effects of genetic variation within the cardiac adrenergic network in heart failure. Mol Pharmacol 76:466–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Armouche A, Eschenhagen T. (2009) β-adrenergic stimulation and myocardial function in the failing heart. Heart Fail Rev 14:225–241 [DOI] [PubMed] [Google Scholar]
- Engelmeier RS, O’Connell JB, Walsh R, Rad N, Scanlon PJ, Gunnar RM. (1985) Improvement in symptoms and exercise tolerance by metoprolol in patients with dilated cardiomyopathy: a double-blind, randomized, placebo-controlled trial. Circulation 72:536–546 [DOI] [PubMed] [Google Scholar]
- Gee KR, Brown KA, Chen W-NU, Bishop-Stewart J, Gray D, Johnson I. (2000) Chemical and physiological characterization of fluo-4 Ca(2+)-indicator dyes. Cell Calcium 27:97–106 [DOI] [PubMed] [Google Scholar]
- Harding SE, Jones SM, O’Gara P, del Monte F, Vescovo G, Poole-Wilson PA. (1992) Isolated ventricular myocytes from failing and non-failing human heart; the relation of age and clinical status of patients to isoproterenol response. J Mol Cell Cardiol 24:549–564 [DOI] [PubMed] [Google Scholar]
- Hoffmann C, Leitz MR, Oberdorf-Maass S, Lohse MJ, Klotz K-N. (2004) Comparative pharmacology of human beta-adrenergic receptor subtypes—characterization of stably transfected receptors in CHO cells. Naunyn Schmiedebergs Arch Pharmacol 369:151–159 [DOI] [PubMed] [Google Scholar]
- Joseph SS, Lynham JA, Grace AA, Colledge WH, Kaumann AJ. (2004) Markedly reduced effects of (-)-isoprenaline but not of (-)-CGP12177 and unchanged affinity of β-blockers at Gly389-β1-adrenoceptors compared to Arg389-β1-adrenoceptors. Br J Pharmacol 142:51–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindermann M, Maack C, Schaller S, Finkler N, Schmidt KI, Läer S, Wuttke H, Schäfers H-J, Böhm M. (2004) Carvedilol but not metoprolol reduces beta-adrenergic responsiveness after complete elimination from plasma in vivo. Circulation 109:3182–3190 [DOI] [PubMed] [Google Scholar]
- Krum H. (2003) Beta-blockers in chronic heart failure: what have we learned? What do we still need to know? Curr Opin Pharmacol 3:168–174 [DOI] [PubMed] [Google Scholar]
- Kveiborg B, Major-Petersen A, Christiansen B, Torp-Pedersen C. (2007) Carvedilol in the treatment of chronic heart failure: lessons from the Carvedilol Or Metoprolol European Trial. Vasc Health Risk Manag 3:31–37 [PMC free article] [PubMed] [Google Scholar]
- De Lean A, Stadel JM, Lefkowitz RJ. (1980) A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J Biol Chem 255:7108–7117 [PubMed] [Google Scholar]
- Lohse MJ, Engelhardt S, Eschenhagen T. (2003) What is the role of β-adrenergic signaling in heart failure? Circ Res 93:896–906 [DOI] [PubMed] [Google Scholar]
- Lowes BD, Gill EA, Abraham WT, Larrain JR, Robertson AD, Bristow MR, Gilbert EM. (1999) Effects of carvedilol on left ventricular mass, chamber geometry, and mitral regurgitation in chronic heart failure. Am J Cardiol 83:1201–1205 [DOI] [PubMed] [Google Scholar]
- Mason DA, Moore JD, Green SA, Liggett SB. (1999) A gain-of-function polymorphism in a G-protein coupling domain of the human β1-adrenergic receptor. J Biol Chem 274:12670–12674 [DOI] [PubMed] [Google Scholar]
- Mason RP, Giles TD, Sowers JR. (2009) Evolving mechanisms of action of beta blockers: focus on nebivolol. J Cardiovasc Pharmacol 54:123–128 [DOI] [PubMed] [Google Scholar]
- Metra M, Giubbini R, Nodari S, Boldi E, Modena MG, Dei Cas L. (2000) Differential effects of β-blockers in patients with heart failure: A prospective, randomized, double-blind comparison of the long-term effects of metoprolol versus carvedilol. Circulation 102:546–551 [DOI] [PubMed] [Google Scholar]
- Metra M, Dei Cas L, Cleland JGF. (2006) Pharmacokinetic and pharmacodynamic characteristics of beta-blockers: when differences may matter. J Card Fail 12:177–181 [DOI] [PubMed] [Google Scholar]
- Mialet Perez J, Rathz DA, Petrashevskaya NN, Hahn HS, Wagoner LE, Schwartz A, Dorn GW, Liggett SB. (2003) Beta 1-adrenergic receptor polymorphisms confer differential function and predisposition to heart failure. Nat Med 9:1300–1305 [DOI] [PubMed] [Google Scholar]
- Michel MC, Pingsmann A, Beckeringh JJ, Zerkowski HR, Doetsch N, Brodde OE. (1988) Selective regulation of beta 1- and beta 2-adrenoceptors in the human heart by chronic beta-adrenoceptor antagonist treatment. Br J Pharmacol 94:685–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra S, Bond RA. (2007) Inverse agonism: from curiosity to accepted dogma, but is it clinically relevant? Curr Opin Pharmacol 7:146–150 [DOI] [PubMed] [Google Scholar]
- Poole-Wilson PA, Swedberg K, Cleland JG, Di Lenarda A, Hanrath P, Komajda M, Lubsen J, Lutiger B, Metra M, Remme WJ, et al. Carvedilol Or Metoprolol European Trial Investigators (2003) Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the Carvedilol Or Metoprolol European Trial (COMET): randomised controlled trial. Lancet 362:7–13 [DOI] [PubMed] [Google Scholar]
- Rathz DA, Gregory KN, Fang Y, Brown KM, Liggett SB. (2003) Hierarchy of polymorphic variation and desensitization permutations relative to beta 1- and beta 2-adrenergic receptor signaling. J Biol Chem 278:10784–10789 [DOI] [PubMed] [Google Scholar]
- Reinkober J, Tscheschner H, Pleger ST, Most P, Katus HA, Koch WJ, Raake PWJ. (2012) Targeting GRK2 by gene therapy for heart failure: benefits above β-blockade. Gene Ther 19:686–693 [DOI] [PubMed] [Google Scholar]
- Rochais F, Vilardaga J-P, Nikolaev VO, Bünemann M, Lohse MJ, Engelhardt S. (2007) Real-time optical recording of β1-adrenergic receptor activation reveals supersensitivity of the Arg389 variant to carvedilol. J Clin Invest 117:229–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum DM, Zhang C, Lyons JA, Holl R, Aragao D, Arlow DH, Rasmussen SGF, Choi H-J, Devree BT, Sunahara RK, et al. (2011) Structure and function of an irreversible agonist-β(2) adrenoceptor complex. Nature 469:236–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth DM, Gao MH, Lai NC, Drumm J, Dalton N, Zhou JY, Zhu J, Entrikin D, Hammond HK. (1999) Cardiac-directed adenylyl cyclase expression improves heart function in murine cardiomyopathy. Circulation 99:3099–3102 [DOI] [PubMed] [Google Scholar]
- Samama P, Cotecchia S, Costa T, Lefkowitz RJ. (1993) A mutation-induced activated state of the β 2-adrenergic receptor. Extending the ternary complex model. J Biol Chem 268:4625–4636 [PubMed] [Google Scholar]
- Saucerman JJ, McCulloch AD. (2006) Cardiac β-adrenergic signaling: from subcellular microdomains to heart failure. Ann N Y Acad Sci 1080:348–361 [DOI] [PubMed] [Google Scholar]
- Saucerman JJ, Brunton LL, Michailova AP, McCulloch AD. (2003) Modeling β-adrenergic control of cardiac myocyte contractility in silico. J Biol Chem 278:47997–48003 [DOI] [PubMed] [Google Scholar]
- Saucerman JJ, Healy SN, Belik ME, Puglisi JL, McCulloch AD. (2004) Proarrhythmic consequences of a KCNQ1 AKAP-binding domain mutation: computational models of whole cells and heterogeneous tissue. Circ Res 95:1216–1224 [DOI] [PubMed] [Google Scholar]
- Shin J, Johnson JA. (2010) β-Blocker pharmacogenetics in heart failure. Heart Fail Rev 15:187–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorger PK, Schoeberl B. (2012) An expanding role for cell biologists in drug discovery and pharmacology. Mol Biol Cell 23:4162–4164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taimor G, Schlüter K, Piper HM. (2001) Hypertrophy-associated gene induction after β-adrenergic stimulation in adult cardiomyocytes. J Mol Cell Cardiol 33:503–511 [DOI] [PubMed] [Google Scholar]
- Tilley DG, Rockman HA. (2006) Role of beta-adrenergic receptor signaling and desensitization in heart failure: new concepts and prospects for treatment. Expert Rev Cardiovasc Ther 4:417–432 [DOI] [PubMed] [Google Scholar]
- Ungerer M, Parruti G, Böhm M, Puzicha M, DeBlasi A, Erdmann E, Lohse MJ. (1994) Expression of beta-arrestins and beta-adrenergic receptor kinases in the failing human heart. Circ Res 74:206–213 [DOI] [PubMed] [Google Scholar]
- Varma DR, Shen H, Deng XF, Peri KG, Chemtob S, Mulay S. (1999) Inverse agonist activities of β-adrenoceptor antagonists in rat myocardium. Br J Pharmacol 127:895–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vittone L., Mundina-Weilenmann C., Said M., Mattiazzi A. (1998) Mechanisms involved in the acidosis enhancement of the isoproterenol-induced phosphorylation of phospholamban in the intact heart. J Biol Chem 273:9804–11 [DOI] [PubMed] [Google Scholar]
- Vila Petroff M. G., Egan J. M., Wang X., Sollott S. J. (2001) Glucagon-like peptide-1 increases cAMP but fails to augment contraction in adult rat cardiac myocytes. Circ. Res. 89:445–452 [DOI] [PubMed] [Google Scholar]
- Zamah AM, Delahunty M, Luttrell LM, Lefkowitz RJ. (2002) Protein kinase A-mediated phosphorylation of the beta 2-adrenergic receptor regulates its coupling to Gs and Gi. Demonstration in a reconstituted system. J Biol Chem 277:31249–31256 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.