

Abstract
Obesity produces a chronic inflammatory state that contributes to the development of diabetes and atherosclerosis. In obese humans, fat depot adipocytes and macrophages produce inflammatory cytokines and other factors which exert unfavorable local and systemic immune responses. The expression of many cytokines is modulated at the post-transcriptional level by mRNA-binding proteins which recognize AU-rich elements (AREs) in the 3′-untranslated regions (3′-UTR) of these transcripts. One such protein, zinc finger protein 36 (Zfp36), is known to destabilize target mRNAs leading to decreased cytokine expression. Few regulators of Zfp36 expression in adipocytes have been described and mRNA targets of Zfp36 in adipocytes are largely unknown. We found that macrophage-derived inflammatory stimuli enhanced endogenous Zfp36 expression in 3T3-L1 adipocytes. Furthermore, the β-adrenergic receptor agonist isoproterenol (Iso) and the glucocorticoid dexamethasone (Dex) each enhanced Zfp36 expression in adipocytes, the former most likely via a cyclic adenosine monophosphate (cAMP)-dependent pathway. By contrast, Zfp36 expression in murine macrophages (RAW 264.7) was not enhanced by exposure to Dex but was stimulated by retinoic acid (RA). Zfp36 inhibited basal and lipopolysaccharide (LPS)-stimulated interleukin-6 (IL-6) expression in adipocytes. These data reveal important and cell type-specific modulators of Zfp36 expression in adipocytes and macrophages and identify Zfp36 as a potent repressor of adipocyte-derived IL-6. Furthermore, this work identifies new factors that stimulate adipocyte Zfp36 expression that are neither classically inflammatory nor mitogenic. Upregulating an mRNA-binding protein for therapeutic purposes may provide a novel mechanistic approach with which to treat diverse inflammatory disorders including common conditions associated with obesity.
INTRODUCTION
Roughly 66% of adults are overweight and ~33% of adults are clinically obese. Excess weight is a risk factor for numerous health conditions including cardiovascular disease, diabetes, dyslipidemia, hypertension, stroke, cancer, and osteoarthritis. Obesity produces a chronic, low-grade, systemic inflammatory state with increased adipose tissue infiltration by macrophages and increased inflammatory activation of these cells and their neighboring adipocytes (1,2). Adipose tissue macrophage numbers positively correlate with total bodyweight, BMI, and total body fat (1).
Adipose tissue expression of tumor necrosis factor-α (TNFα), monocyte chemotactic protein-1 (MCP-1), and plasminogen activator inhibitor-1 is elevated in human obesity and in mouse models of obesity (3,4). In humans, obesity also increases adipose tissue expression of interleukin-6 (IL-6), inducible nitric oxide synthase, matrix metalloproteinases, lipocalcin, and other factors (leptin, resistin etc.) that may contribute to atherosclerosis, insulin resistance, and conditions associated with obesity (1,2,5,6). Adipose tissue-expressed TNFα appears to originate predominantly from macrophages whereas adipocytes contribute to the systemic levels of MCP-1, IL-6, and leptin (7). Macrophage-adipocyte crosstalk is essential to the overall cytokine production from fat tissues and both cell types appear to contribute to the metabolic and health consequences of obesity. Fat depot-derived factors, here loosely referred to as “adipocytokines,” exert widespread physiologic effects.
IL-6 is secreted from adipocytes and adipose depot macrophages. Together, these cells produce ~15–35% of the systemic IL-6 (8). The effects of IL-6 on metabolism are complex, cell type-specific, and may vary depending upon the magnitude and duration of IL-6 exposure (9). Human obesity is correlated with elevated serum IL-6 which in turn correlates with the risk of insulin resistance and diabetes (10). In mice, chronically elevated IL-6 promotes hepatic and adipose tissue resistance to insulin (9,11). Adding to the complexity of IL-6 physiology, exercise promotes skeletal muscle secretion of IL-6 which in turn stimulates fatty acid oxidation and glucose uptake in the skeletal muscle (12). IL-6 regulates lipolysis, glucose disposal, and whole body metabolism through cell type-specific responses that are mediated by the Janus Kinase-Signal Transducer and Activator of Transcription and protein kinase B (PKB/Akt) pathways (9). IL-6 may also contribute to the pathogenesis of atherosclerosis (13).
The expression of many cytokines is regulated post-transcriptionally by factors that modulate mRNA transport, translation, and stability. Much of this regulation occurs by the binding and stabilizing, or destabilizing, of cytokine mRNAs by proteins that recognize adenosine and uridine-rich elements (AREs) in untranslated regions of target transcripts (14). In some cell types IL-6 transcripts are modulated at the level of mRNA stability (15,16) partly mediated by the ARE-binding protein (ARE-BP) AUF1/HNRNPD (17). It remains unknown which alternative ARE-BPs can similarly regulate IL-6 transcripts. Zinc finger protein 36 (Zfp36) has been reported to regulate the half-life of the mRNAs encoding important immune mediators such as TNFα, granulocyte macrophage colony-stimulating factor, IL-3, and cyclooxygenase-2 (18).
Recently, Zfp36 was demonstrated to physically interact with the p65 subunit of nuclear factor-κB leading to decreased nuclear import and diminished transcriptional activation mediated by nuclear factor-κB (19,20). Nuclear factor-κB is an important transcriptional activator of inflammatory genes including TNFA and MCP1. These data suggest two important mechanisms by which Zfp36 might suppress inflammatory responses: by regulating nuclear factor-κB-mediated transcriptional responses and by regulating cytokine mRNA stability.
Little is known about the function of Zfp36 in adipocytes. Zfp36 expression is enhanced by serum, lipopolysaccharide (LPS), insulin, and cinnamon extract in murine 3T3-L1 adipocytes (21-23). We hypothesized that enhancing adipocyte expression of Zfp36 would attenuate inflammatory adipocytokine expression to produce favorable metabolic effects. Our work addresses the regulation of Zfp36 expression in murine adipocytes and the role of this protein in modulating adipocyte-specific immune responses.
Glucocorticoids and β-adrenergic agonists (i.e., norepinephrine) are important activators of lipolysis in adipose tissue and participate in the stress response. We report that Zfp36 protein expression is enhanced by β-adrenergic receptor and glucocorticoid stimulation of 3T3-adipocytes. Exogenously expressed Zfp36 suppressed, and Zfp36-knockdown enhanced, IL-6 secretion from 3T3-L1 adipocytes. These findings have important implications for obesity-related inflammation and suggest that regulating Zfp36 expression in adipocytes might produce favorable health effects in obese individuals.
METHOD AND PROCEDURES
Cell Culture
3T3-L1 preadipocytes (American Type Culture Collection, Manassas, VA) were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% calf serum (Gibco), penicillin G, streptomycin, and l-glutamine (all Gibco, Carlsbad, CA), with a change of medium every 2–3 days. The standard method of differentiation to adipocytes was employed using fetal calf serum (FCS), dexamethasone (Dex), isobutylmethylxanthine, and insulin for 2 days followed by FCS and insulin for five additional days of culture. THP-1 cells were propagated in Eagles Minimum Essential Medium-α (Gibco) with 10% FCS, and penicillin G, streptomycin, and l-glutamine. THP-1 cells were induced to become adherent macrophage-like cells by treating with 50 ng/ml tetradecanoylphorbol acetate for 48 h, then tetradecanoylphorbol acetate was washed out with phosphate-buffered saline and cells were maintained in standard culture medium or changed to serum-free medium with 0.5% bovine serum albumin before treatments. RAW 264.7 cells (ATCC) were grown in RPMI 1640 medium (Gibco) with 10% FCS and penicillin G, streptomycin, and l-glutamine.
For all cell treatments and coculture experiments, cells were placed in serum-free medium supplemented with 0.5% bovine serum albumin for 24 h before treatments. Where indicated, treatments included vehicle control (100% EtOH and/or dimethyl sulfoxide and/or phosphate-buffered saline), TNFα (human recombinant, 20 ng/ml; Pierce, Rockford, IL), retinoic acid (RA, 1.0 μmol/l), LPS (10 ng/ml), Dex (1 μmol/l), isoproterenol (Iso, 10 μmol/l), SR59203A (5 μmol/l), forskolin (10 μmol/l), sulindac (100 μmol/l), or atorvastatin (5 μmol/l), all from Sigma, St. Louis, MO. Indirect coculture was performed using 0.4-μm pore size cell culture inserts from BD/Falcon, San Jose, CA. In experiments that required conditioned medium (CM), the duration of LPS exposure to the macrophages was 24 h. This medium was then sterile-filtered and placed on the adipocytes for 6 h. As such, the LPS from the macrophage cultures was still present during the adipocyte culture with the CM, and treatment effects should be compared to LPS-treated adipocytes.
Quantitative reverse transcriptase-PCR
Total RNA was purified from cell lysates using Trizol reagent (Invitrogen, Carlsbad, CA). Two micrograms of total RNA was used for reverse transcription using Anchored Oligo-(dT)23 (Sigma) as primer for 1st strand synthesis using the RT-AMV kit (Roche, Indianapolis, IN) per the manufacturer’s instructions. 1:100 dilutions of complementary DNA were used as template for quantitative-PCR using iQ-SYBR Green Master Mix (Biorad, Hercules, CA) in a Biorad Opticon 2 cycler. Quantitative reverse transcriptase-PCR values for target genes were normalized to reference gene expression (Rpl17) by calculating ΔΔCt values. Primer pairs for reverse transcriptase-PCR were Rpl17: 5′-TGCCAAGATGGTTCGCTACTCTCT, 3′-GGCTTTGCGGATATGCATTCCCTT, Mcp-1: 5′-TCACCTGCTGCTACTCATTCACCA, 3′-TACAGCTTCTTTGGGACACCTGCT, IL-6: 5′-ATCCAGTTGCCTTCTTGGGACTGA, 3′-TAAGCCTCCGACTTGTGAAGTGGT, Rantes: 5′-TCGTGCCCACGTCAAGGAGTATTT, 3′-TCTTCTCTGGGTTGGCACACACTT, and IL-1β: 5′-AA GGGCTGCTTCCAAACCTTTGAC, 3′-ATACTGCCTGCCTGAAGCTCTTGT.
Western blotting
Cells were washed with phosphate-buffered saline and cell lysates were collected in lysis buffer (Invitrogen) and quantified using the Micro BCA Protein Assay Kit (Pierce). Forty micrograms of total protein per well was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to polyvinylidene fluoride membranes, and probed with antibodies against β-actin (Sigma #A4700, used at 1:500 dilution) or Zfp36 (rabbit anti-mouse Zfp36 at 1:5,000 dilution (24)). Secondary goat anti-rabbit immunoglobulin G antibody (conjugated with horseradish peroxidase) was incubated at a dilution of 1:10,000 and blots were developed using Amersham ECL Plus Western Blotting Detection Reagents (GE Healthcare, Piscataway, NJ). Western blot data were quantitated using ImageJ software to measure Zfp36 band intensities and data were normalized to each respective Actin band measured in parallel. These data are presented relative to Zfp36 expression in the control (vehicle-treated) cells.
Enzyme-linked immunosorbent assays
Enzyme-linked immunosorbent assay (ELISA) for mouse IL-6 (RayBiotech, Norcross, GA) was performed using conditioned media per the manufacturer’s instructions. Although there is inter-experimental variability in adipocyte density and consequent IL-6 secretion, all enzyme-linked immunosorbent assay data were normalized to percent lipid in each well as assayed by AdipoRed reagent (Lonza, Basel, Switzerland) per the manufacturer’s instructions. Similar results were observed when IL-6 data were normalized to total protein in each well (data not shown). Furthermore, lentiviral transduction was performed using replicate wells of fully differentiated adipocytes, eliminating concerns regarding possible effects of high or low Zfp36 on the effciency of preadipocyte-to-adipocyte differentiation.
Cloning of genomic human ZFP36
PCR amplification of the genomic locus of ZFP36 from human endometrial stromal cell genomic DNA was performed after DNA extraction using the Genomic DNA Extraction kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. The PCR product containing the complete coding sequence for ZFP36 (for consistency, referred to in this manuscript as Zfp36) along with the first intron was ligated with pCDH-CMV-EF1-puro (System Biosciences, Mountain View, CA). All constructs employed in this work were confirmed by DNA sequencing. Primers used for genomic locus amplification were 5′-tatcccgggCCACTCTCGGCCGACACCCCTCATG and 3′-atacggccgtttatttGAGACGTGGCTCCCCGCTGAGATCCAGCTGATC.
Lentiviral expression in adipocytes
MISSION Lentiviral Transduction Vectors targeting mouse Zfp36 transcripts for repression/knockdown via the intracellular production of short hairpin RNAs (shRNAs) were obtained from Sigma. These vectors, along with genomic Zfp36 coding sequence (in pCDH-CMV-EF1-puro vector) were used to produce lentiviral particles as previously described (25). Briefly, the target vector pCDH-Zfp36, or pLK0.1-Zfp36-shRNA (Sigma), or a green-fluorescent protein-expressing empty control vector (Sigma), is co-transfected into HEK293T cells with the plasmids delta 8.9 (expressing gag, pol, and rev), and vesicular stomatitis virus G expression plasmid (expressing env) to produce replication-incompetent but highly infective viral particles. The packaged, unconcentrated virus is collected over a period of 5 days post-transfection and then concentrated using ultracentrifugation. Lentiviral particles were used at titers of 6 × 107 transducing units/ml of culture media.
Statistical analyses
Comparisons between two groups were made using a two-tailed Student’s t-test with P values indicated. The threshold for statistical significance was taken to be P < 0.05.
RESULTS
Proinflammatory stimuli enhance Zfp36 expression in 3T3-L1 adipocytes
Zfp36 expression is enhanced by mitogens, growth factors, and proinflammatory stimuli in macrophages and embryonic fibroblast cells (24,26). In murine RAW 264.7 monocyte/macrophage cells, LPS is a potent inducer of Zfp36 expression (24). We observed that LPS acutely (6 h) stimulated Zpf36 protein expression in 3T3-L1 adipocytes (Figure 1a). Because of variable phosphorylation at multiple residues, Zfp36 migrates as a broad-staining band at ~37–44 kDa molecular weight on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (24,26,27). Longer (24 h) exposure of adipocytes to LPS did not produce a sustained increase in Zfp36 expression (Figure 1a) consistent with a role for Zfp36 as an acute phase responder.
Figure 1.
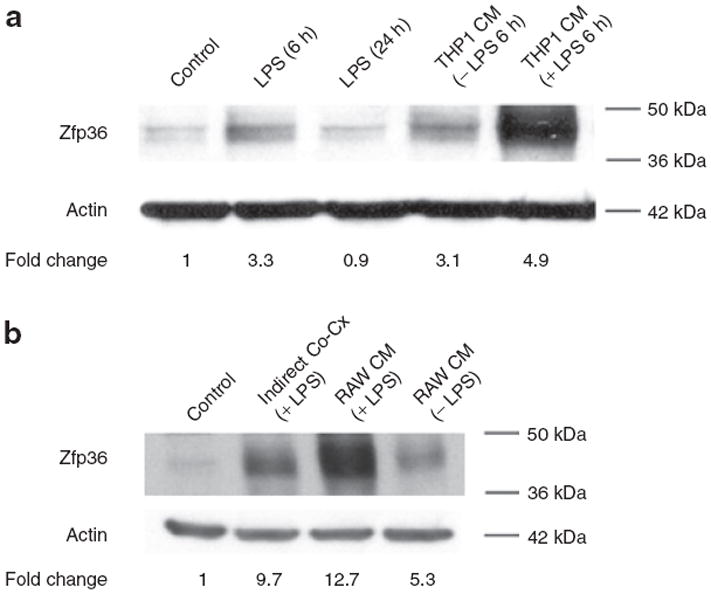
Lipopolysaccharide (LPS) and macrophage-conditioned medium (CM) enhance zinc finger protein 36 (Zfp36) expression in 3T3-L1 adipocytes. Western blot analysis of adipocyte Zfp36. (a) 3T3-L1 adipocytes were serum starved overnight and treated with LPS (10 ng/ml) for the indicated time in serum-free culture medium. Zfp36 expression was enhanced with short (6 h) exposure to LPS but returned to baseline after 24 h exposure to LPS. Serum starved 3T3-L1 adipocytes were also cultured for 6 h with CM from THP-1 macrophage cells grown in serum-free medium. THP-1 cells were pretreated with vehicle or LPS (10 ng/ml) for 24 h before harvesting of medium. THP-1 CM enhanced Zfp36 expression in 3T3-L1 adipocytes and the effect was more pronounced in LPS-stimulated THP-1 cells. (b) Indirect coculture of 3T3-L1 adipocytes with RAW 264.7 macrophages similarly enhances Zfp36 expression in the adipocytes. 3T3-L1 adipocytes were serum starved overnight and then cultured with RAW 264.7 macrophages grown on cell-impermeable inserts (macrophages in the upper chamber, adipocytes in the lower chamber). Coculture was performed in the presence of LPS (10 ng/ml) for 6 h. In addition, adipocytes were treated for 6 h with RAW 264.7 CM that was pretreated with vehicle or LPS for 24 h before harvesting of medium. Quantitative image analysis indicating Zfp36 expression relative to control cells, normalized to actin, is shown. Data in a and b are representative of three independent experiments.
Because obesity is correlated with increased numbers of adipose depot macrophages and increased fat depot inflammation (1,2), we tested whether macrophage CM can regulate adipocyte expression of Zfp36. Serum-free CM from human THP-1 macrophages stimulated Zfp36 expression in 3T3-L1 adipocytes and this effect was strongly enhanced using LPS-stimulated THP-1 CM (Figure 1a). Similar results were observed when using CM from murine RAW 264.7 macrophage cells (Figure 1b). These data indicated that inflammatory stimuli, including factors derived from macrophages, enhance adipocyte expression of Zfp36.
In order to more closely model the macrophage-rich environment of adipose tissues in obese individuals we performed indirect coculture of THP-1 macrophages with 3T3-L1 adipocytes: 0.4-μm cell culture inserts physically separated the THP-1 and 3T3-L1 cells while permitting free exchange of paracrine factors. Indirect coculture of THP-1 macrophages with 3T3-L1 adipocytes, with and without LPS treatment, produced enhanced adipocyte mRNA expression of the proinflmmatory factors IL-6, Mcp-1, and regulated upon activation, normal T-cell expressed, and secreted (Rantes) (Figure 2). These data confirm the proinflammatory paracrine effects of macrophages on adipocytes in our in vitro model system. These effects recapitulate the inflammatory macrophage-adipocyte interactions that are observed in mammalian adipose tissue in obese individuals in vivo (3). Indirect coculture of RAW 264.7 cells (or THP-1 macrophages, date not shown) with 3T3-L1 adipocytes also enhanced Zfp36 protein expression in the adipocytes (Figure 1b). Our observations indicate that adipocyte Zfp36 expression is acutely regulated by inflammatory stimuli (i.e., LPS) and by diffusible factors secreted by macrophages under basal and inflammatory (LPS-stimulated) conditions.
Figure 2.
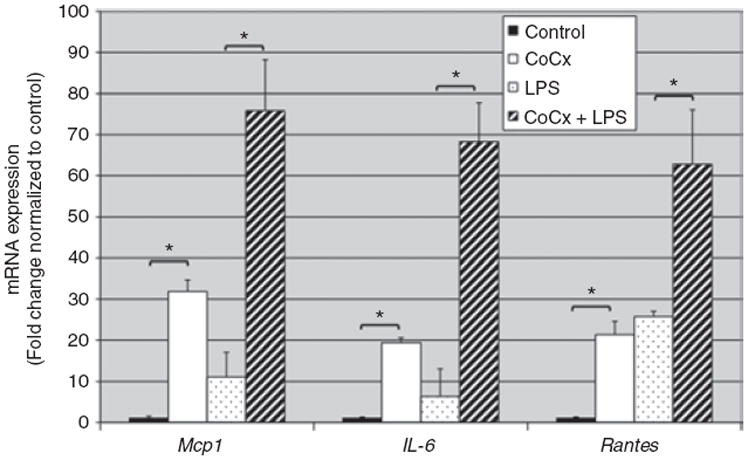
Indirect coculture of macrophages with adipocytes promotes adipocyte mRNA expression of inflammatory factors. Quantitative reverse transcriptase (RT)-PCR was performed using adipocyte mRNA. Adipocyte mRNA was isolated with and without prior indirect coculture with THP-1 macrophages, with and without costimulation using lipopolysaccharide (LPS) (10 ng/ml). mRNA expression of 3T3-L1-expressed IL-6, Mcp-1, and Rantes was enhanced with macrophage coculture and further enhanced by costimulation with LPS. Values represent the means of at least three independent experiments with s.e.m. *P < 0.05.
Dexamethsone, TNFα, and forskolin stimulate Zfp36 expression in 3T3-L1 adipocytes
We tested rational candidate molecules and immune modulators for the ability to regulate Zfp36 expression in 3T3-L1 adipocytes. Macrophage lineage cells may be induced secrete TNFα and/or RA which, in turn, regulate important autocrine and paracrine immune functions. Recombinant TNFα, but not RA, stimulated Zfp36 expression in 3T3-L1 adipocytes (Figure 3). In addition, FCS, forskolin (a stimulator of intracellular cyclic adenosine monophosphate (cAMP)), and the glucocorticoid Dex each stimulated Zfp36 expression in 3T3-L1 adipocytes. These data indicate that both proinflammatory stimuli (i.e., LPS, TNFα, and macrophage-conditioned media) and anti-inflammatory stimuli (i.e., pharmacologic doses of Dex) can enhance adipocyte expression of Zfp36.
Figure 3.
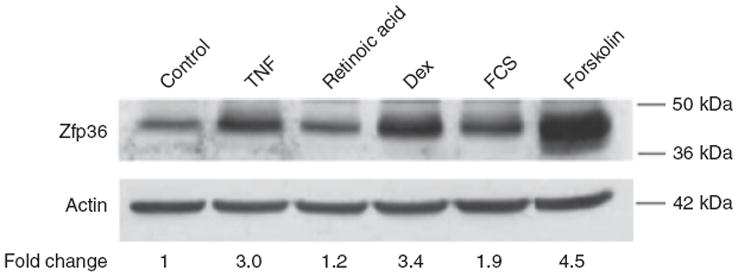
Dexamethsone, tumor necrosis factor-α (TNFα), and forskolin stimulate zinc finger protein 36 (Zfp36) expression in 3T3-L1 adipocytes. Western blot analysis of Zfp36 expression in 3T3-L1 adipocytes grown in serum-free medium with the following 6 h treatments: vehicle control, TNFα (20 ng/ml), retinoic acid (RA, 1.0 μmol/l), dexamethasone (Dex, 1 μmol/l), fetal calf serum (FCS, 10%), and forskolin (10 μmol/l). Quantitative image analysis indicating Zfp36 expression relative to control cells, normalized to actin, is shown. Data are representative of three independent experiments.
Nonsteroidal anti-inflammatory drugs (NSAIDs) and the cholesterol-lowering statin class of drugs demonstrate anti-inflammatory properties. In contrast to the Zfp36-stimulating effects of Dex, we found that neither the nonsteroidal anti-inflammatory drugs sulindac nor the statin atorvastatin stimulated adipocyte expression of Zfp36 (Figure 4). These data suggest that the anti-inflammatory properties of these agents do not occur through direct enhancement of adipocyte Zfp36 expression. Similarly, we did not observe any effect of sulindac or atorvastatin treatments on macrophage expression of Zfp36 (data not shown).
Figure 4.
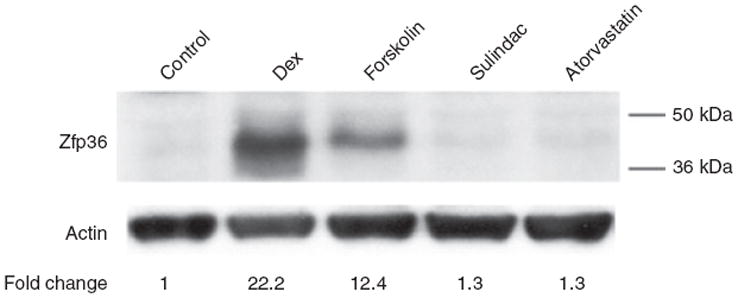
Sulindac and atorvastatin do not enhance zinc finger protein 36 (Zfp36) expression in 3T3-L1 adipocytes. Western blot analysis of Zfp36 expression in 3T3-L1 adipocytes grown in serum-free medium with the following 6 h treatments: vehicle control, dexamethasone (Dex, 1 μmol/l), forskolin (10 μmol/l), the nonsteroidal anti-inflammatory drug (NSAID) sulindac (100 μmol/l), and the lipid-lowering statin drug atorvastatin (5 μmol/l). Quantitative image analysis indicating Zfp36 expression relative to control cells, normalized to actin, is shown. Data are representative of three independent experiments.
The β-adrenergic receptor agonist Iso stimulates Zfp36 expression in 3T3-L1 adipocytes
β-Adrenergic receptors (β-AR) are important modulators of adipocyte metabolism acting via the generation of intracellular cAMP. Human and mouse adipocytes express multiple subtypes of the β-AR. In response to sympathetic neuroendocrine stimuli, principally catecholamines, β-AR subtypes trigger increased lipolysis and energy mobilization by the adipocytes. Having observed that forskolin treatment enhanced Zfp36 expression in adipocytes, we tested whether β-AR stimulation could similarly modulate Zfp36 expression in these cells. We found that the β-AR agonist Iso stimulated Zfp36 expression in 3T3-L1 adipocytes and that this effect could be blocked with the β3-AR antagonist SR 59230A (Figure 5). Similarly, forskolin (Figure 6) and Iso (data not shown) stimulated Zfp36 expression in RAW 264.7 macrophages, suggesting a role for adrenergic tone in the immune-regulation of these cells. These data indicate a new modulator of adipocyte and macrophage Zfp36 expression that is neither a classical immune factor nor a mitogen.
Figure 5.
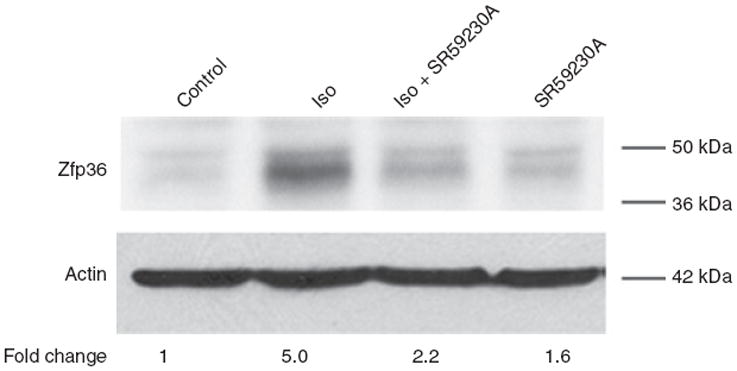
The β-adrenergic receptor agonist isoproterenol stimulates zinc finger protein 36 (Zfp36) expression in 3T3-L1 adipocytes. Western blot analysis of Zfp36 expression in 3T3-L1 adipocytes grown in serum-free medium with the following 6 h treatments: vehicle control, β-adrenergic agonist isoproterenol (Iso, 10 μmol/l), β3-adrenergic antagonist SR59230A (5 μmol/l), or Iso plus SR59230A (same concentrations). Quantitative image analysis indicating Zfp36 expression relative to control cells, normalized to actin, is shown. Data are representative of three independent experiments.
Figure 6.
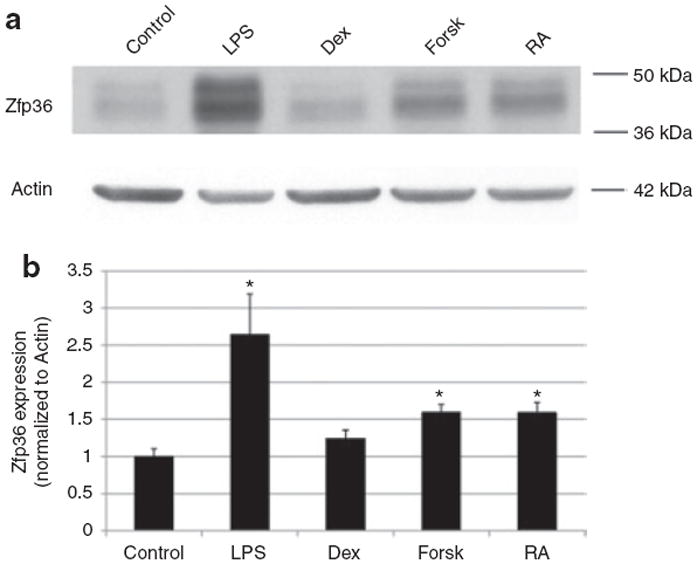
Forskolin and retinoic acid (RA), but not dexamethasone, rapidly enhance zinc finger protein 36 (Zfp36) expression in RAW 264.7 macrophages. (a) Western blot analysis of Zfp36 expression in RAW 264.7 cells grown in serum-free medium with the following 6 h treatments: vehicle control, lipopolysaccharide (LPS) (10 ng/ml), dexamethasone (Dex, 1 μmol/l), forskolin (Forsk, 10 μmol/l), and retinoic acid (RA, 1.0 μmol/l). Actin loading control is shown. Data are representative of three independent experiments. (b) Quantitative image analysis of western blots showing Zfp36 expression in RAW 264.7 cells after the same treatments described above. Band intensities were measured and normalized to respective Actin band intensities for three independent experiments. Data are shown relative to control cells. *P< 0.05 compared with control cells.
RA and forskolin, but not Dex, stimulate Zfp36 expression in macrophages
Given the set of signals that stimulate Zfp36 expression in adipocytes, we explored whether these same signals have similar function in macrophages. The glucocorticoid Dex was previously described to inhibit Zfp36 expression in activated J774 murine macrophages in a glucocorticoid receptor-dependent fashion. We tested RAW 264.7 macrophages and similarly observed that Dex did not increase Zfp36 expression whereas forskolin and RA rapidly enhanced Zfp36 expression in these cells (Figure 6). These data indicate cell type-specific regulation of Zfp36 expression by Dex and RA in adipocytes and macrophages, respectively. Collectively, our data indicate new modulators of Zfp36 expression in adipocytes and macrophages. The data also indicate that Zfp36 expression can be enhanced by diverse immune modulators in a cell type-specific fashion.
Zfp36 suppresses adipocyte expression of IL-6
We employed lentiviral particles to stably modulate Zfp36 expression in 3T3-L1 adipocytes. Full-length human ZFP36 cDNA (for consistency, referred to in this manuscript as Zfp36) or anti-Zfp36-shRNA expressing lentiviral particles were produced and tested in 3T3-L1 adipocytes (Figure 7a). While minimal endogenous Zfp36 could be detected in resting 3T3-L1 adipocytes, LPS-stimulated adipocytes demonstrated enhanced expression of Zfp36 and this expression was significantly reduced by expression of the anti-Zfp36-shRNA (i.e., Zfp36-knockdown/KD, Figure 7a). Cytomegalovirus promoter-driven overexpression of Zfp36 in transduced adipocytes was also observed (Figure 7a).
Figure 7.
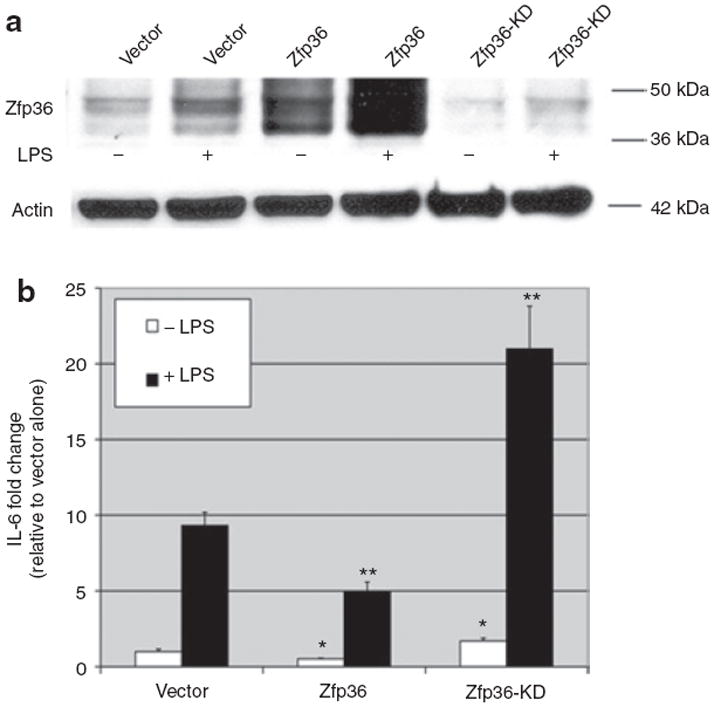
Zfp36 represses interleukin (IL)-6 expression in basal and LPS-stimulated 3T3-L1 adipoctyes. (a) Lentivirus-mediated zinc finger protein 36 (Zfp36) overexpression and Zfp36-knockdown in 3T3-L1 adipocytes. Western blot analysis of Zfp36 expression in 3T3-L1 adipocytes. Cells were transduced with empty vector-containing lentiviral particles (vector) or human Zfp36-expressing lentiviral particles (Zfp36) or anti-Zfp36-shRNA-expressing lentiviral particles (Zfp36-KD), with and without lipopolysaccharide (LPS) (10 ng/ml) stimulation for 6 h. Actin loading control is also shown and data are representative of three independent experiments. (b) IL-6 was measured in the culture medium of 3T3-L1 adipocytes engineered to over- or under-express Zfp36. Cells were transduced with empty vector-containing lentiviral particles (vector) or human Zfp36-expressing lentiviral particles (Zfp36) or anti-Zfp36-shRNA-expressing lentiviral particles (Zfp36-KD) (see Figure 7a). Cells were grown in serum-free medium for 24 h and then treated with vehicle or LPS (10 ng/ml) for 24 h. Culture medium was collected and IL-6 protein quantitated using enzyme-linked immunosorbent assay (ELISA). IL-6 data were normalized to percent lipid in each well. IL-6 levels are expressed as fold change relative to vector control. Shown are means with s.e.m. of at least three independent experiments performed in triplicate. *P < 0.05 compared with vector-transduced cells. **P < 0.05 compared with LPS-treated vector-transduced cells. shRNA, short hairpin RNA.
Zfp36 expression decreased, and Zfp36-knockdown increased, both basal and LPS-stimulated secretion of IL-6 from 3T3-L1 adipocytes (Figure 7b). In unstimulated adipocytes, Zfp36 overexpression reduced IL-6 secretion by nearly half and Zfp36-knockdown enhanced IL-6 expression by ~71% (Figure 7b, P < 0.05 for both effects relative to vector-transduced cells). In LPS-stimulated cells, Zfp36 overexpression reduced IL-6 secretion by ~47% and Zfp36-knockdown enhanced IL-6 secretion greater than twofold (Figure 7b, P < 0.05 for both effects compared with vector-transduced cells). In a typical experiment, the average basal secretion of IL-6 was ~400–500 pg/ml/day. Our data indicate that adipocyte-expressed Zfp36 negatively regulates an important adipocytokine, IL-6, in 3T3-L1 adipocytes.
Zfp36 regulates mRNA expression both transcriptionally (19,28) and post-transcriptionally (18). We studied the effects of enhanced adipocyte expression of Zfp36 on the expression of IL-6, Mcp-1, and IL-1β transcripts. Enhanced expression of Zfp36 reduced the mRNA expression of IL-6, Mcp-1, and IL-1β compared to lentiviral vector-treated adipocytes (Figure 8). These data indicate that Zfp36 regulates these targets at the mRNA level (either transcriptionally or post-transcriptionally, or both) and not at the level of mRNA translation (as is observed for other ARE-binding proteins such as the translational silencer TIA1 cytotoxic granule-associated RNA-binding protein-like 1, or TIAL-1). Our gene expression data are consistent with a model in which Zfp36 regulates IL-6 mRNA production and/or stability.
Figure 8.
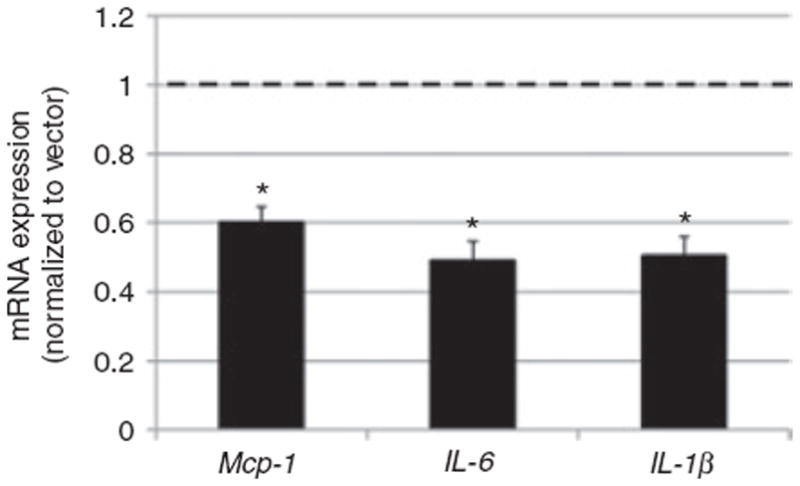
Zinc finger protein 36 (Zfp36) reduces adipokine mRNA expression in 3T3-L1 adipocytes. Cells were transduced with empty vector-containing lentiviral particles (vector) or human Zfp36-expressing lentiviral particles as described in Figure 7. Quantitative reverse transcriptase (RT)-PCR was performed and expression was normalized to vector-treated cells (dashed line). mRNA expression of IL-6, Mcp-1, and IL-1β was reduced in Zfp36-expressing adipocytes (black bars). Values represent the means of three independent experiments with s.e.m. *P < 0.05 relative to vector control.
DISCUSSION
We have identified pro- and anti-inflammatory factors that enhance Zfp36 expression in 3T3-L1 adipocytes. Macrophage-derived stimuli, TNFα, and bacterial LPS enhance endogenous Zfp36 expression in adipocytes. Furthermore, the β-adrenergic receptor agonist Iso and the anti-inflammatory glucocorticoid Dex each stimulates Zfp36 expression in adipocytes. By contrast, Zfp36 expression in murine and human macrophage-like cells is not enhanced by exposure to Dex but is stimulated by RA. Finally, high Zfp36 expression inhibits, and low Zfp36 enhances, basal and LPS-stimulated adipocyte production of IL-6 protein.
The markedly inflamed phenotype of the Zfp36 null mouse indicates that this protein serves important and nonredundant functions that are principally anti-inflammatory (29,30). We studied the regulation of Zfp36 in an adipocyte model system in preparation for studies of enhanced adipocyte expression of Zfp36 in vivo, in a transgenic mouse model system. Our data confirm a role for Zfp36 in responding to inflammatory stimuli to reduce adipocytokine expression. In particular, we have shown that forced expression of Zfp36 reduces basal and LPS-stimulated IL-6 expression in adipocytes. Our observations are consistent with a model in which inflammation is tightly regulated by compensatory increases in anti-inflammatory proteins such as Zfp36 (27,31). A negative feedback loop in which inflammatory factors enhance expression of the anti-inflammatory Zfp36 is reminiscent of the regulation of other immune processes: T-cell receptor signaling leads to activation of proteins (i.e., IκB and Calcipressin) which ultimately terminate the T-cell response (32), toll-like receptor (TLR) signaling in macrophages is followed by inactivation of MyD88 leading to inhibition of further TLR signaling (33), and a multitude of inflammatory stimuli also induce expression of suppressor of cytokine signaling (SOCS) proteins which feedback to inhibit additional cytokine signaling (34).
More encouraging, from a therapeutic perspective, is the identification of new modulators of Zfp36 expression in adi-poctyes; namely glucocorticoids and a β-adrenergic receptor agonists. Our data also indicate that there is cell type-specific enhancement of Zfp36 expression by retinoids and glucocorticoids in macrophages and adipocytes, respectively. These findings suggest the potential to develop cell type-specific modulators of Zfp36 expression for targeted therapy.
Diet-induced obesity increases local and systemic inflammatory adipocytokines in humans and in rodents; these factors contribute to adverse health outcomes (35). Factors that might mitigate against this inflammatory response have remained elusive. Orosomucoid (ORM1) was recently identified as an acute phase, adipocyte-expressed protein that may be anti-inflammatory (36). Interestingly, recent single-nucleotide polymorphism analysis revealed Zfp36 to be a candidate gene for influencing the metabolic syndrome in humans (37). Additional work suggested that high Zfp36 expression in adipose tissue may offer protection against the development of insulin resistance and the metabolic syndrome (38). Our data provide justification for future studies to assess whether enhanced Zfp36 expression in adipocytes in vivo will favorably affect the adverse metabolic and health consequences of obesity. Studies to characterize additional adipocytokine and noncytokine mRNA targets of adipocyte Zfp36 are ongoing.
The fat depot comprises diverse cell types including adipocytes, preadipocytes, leukocytes (including monocytes and macrophages), fibroblasts, endothelial and stromal cells. Adipose depot macrophages can polarize along both proinflammatory (M1) and anti-inflammatory (M2) phenotypes: obesity favoring M1 (classical) and leanness favoring M2 (alternative) polarization of fat depot macrophages (5,35,39). The phenotypic switch between M2 and M1 polarized macrophages in the setting of obesity is incompletely understood but correlates with fat-expressed adipocytokine production. Whether adipocyte- or macrophage-expressed Zfp36, by limiting inflammatory adipocytokine expression, plays a role in directing macrophage polarization in fat tissues remains to be explored. Our data suggest that enhanced macrophage infiltration of the adipose depot in obese individuals may modulate the expression of Zfp36 in neighboring adipocytes. In vivo studies are underway to address the net effects of adipose tissue-expressed Zfp36 in lean and obese mice.
Fat depots are innervated by the sympathetic nervous system and regulated by neuroendocrine mediators including catecholamines secreted by the adrenal medulla and sympathetic nerves. We observed that a β-adrenergic agonist which triggers lipolysis in fat cells also enhances adipocyte expression of Zfp36. Enhanced lipolysis is part of the normal stress response to catecholamine exposure. Recent data indicate that lipolysis and circulating lipids can trigger macrophage recruitment to the fat depot (40). This observation supports a relationship between lipolysis and the immune environment of the adipose depot. As such, stress hormone-stimulated expression of Zfp36 in adipocytes, whether by catecholamines or glucocorticoids, may reffect a shared compensatory response of adipocytes to noncytokine/nonmitogen stimuli. While intriguing to consider in light of recent reports linking Zfp36 to the metabolic syndrome (37,38), a broader role for adipocyte-expressed Zfp36 in directly modulating metabolism, lipolysis, or glucose homeostasis in the fat depot remains to be established.
Our data indicate the potential for metabolic and immune consequences of altered Zfp36 expression through changes in adipocyte secretion of IL-6 (8). The 3′-UTR of the human IL-6 transcript has at least six putative AU-rich elements (AREs) that may be bound and regulated by Zfp36. When the IL-6 3′-UTR was fused to a heterologous luciferase reporter it regulated reporter activity in MCF-7 breast cancer cells, consistent with a role for the IL-6 3′-UTR in regulating mRNA stability (data not shown). 3T3-L1 adipocytes are notoriously difficult to transiently transfect, a problem which has hindered our ability to demonstrate that Zfp36 directly binds and regulates IL-6 transcripts in these cells using heterologous luciferase reporter systems.
IL-6 regulates lipolysis, glucose disposal, and metabolism (9). Obese humans have elevated circulating IL-6, which is correlated with increased risk of insulin resistance and diabetes (10). Chronically elevated IL-6 promotes hepatic and adipose tissue resistance to insulin in rodents and may contribute to the development of atherosclerosis (9,11). The targeted manipulation of mRNA-binding proteins such as Zfp36 may one day represent a therapeutic means with which to reduce obesity-associated inflammation.
Acknowledgments
The authors are grateful to Drs Friedman and Taylor for scientific discussions, to Dr Ressler (and grant NS055077) for assistance in developing the lentiviral expression systems, and to Drs Blackshear and Kennington for sharing their Zfp36 antibody. This work was supported by NIH grants R01-HD055379 (N.S.) and R01-CA129424-01A1 (N.S.) and the RSDP/UCSF-K12-HD000849 award (C.B.K.).
Footnotes
DISCLOSURE
The authors declared no conflict of interest.
References
- 1.Ferrante AW., Jr Obesity-induced inflammation: a metabolic dialogue in the language of inflammation. J Intern Med. 2007;262:408–414. doi: 10.1111/j.1365-2796.2007.01852.x. [DOI] [PubMed] [Google Scholar]
- 2.Weisberg SP, McCann D, Desai M, et al. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sartipy P, Loskutoff DJ. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc Natl Acad Sci USA. 2003;100:7265–7270. doi: 10.1073/pnas.1133870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 5.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-α function. Nature. 1997;389:610–614. doi: 10.1038/39335. [DOI] [PubMed] [Google Scholar]
- 7.Fain JN, Buehrer B, Bahouth SW, Tichansky DS, Madan AK. Comparison of messenger RNA distribution for 60 proteins in fat cells vs the nonfat cells of human omental adipose tissue. Metab Clin Exp. 2008;57:1005–1015. doi: 10.1016/j.metabol.2008.02.019. [DOI] [PubMed] [Google Scholar]
- 8.Fried SK, Bunkin DA, Greenberg AS. Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: depot difference and regulation by glucocorticoid. J Clin Endocrinol Metab. 1998;83:847–850. doi: 10.1210/jcem.83.3.4660. [DOI] [PubMed] [Google Scholar]
- 9.Glund S, Krook A. Role of interleukin-6 signalling in glucose and lipid metabolism. Acta Physiol (Oxf) 2008;192:37–48. doi: 10.1111/j.1748-1716.2007.01779.x. [DOI] [PubMed] [Google Scholar]
- 10.Fernandez-Real JM, Vayreda M, Richart C, et al. Circulating interleukin 6 levels, blood pressure, and insulin sensitivity in apparently healthy men and women. J Clin Endocrinol Metab. 2001;86:1154–1159. doi: 10.1210/jcem.86.3.7305. [DOI] [PubMed] [Google Scholar]
- 11.Klover PJ, Clementi AH, Mooney RA. Interleukin-6 depletion selectively improves hepatic insulin action in obesity. Endocrinology. 2005;146:3417–3427. doi: 10.1210/en.2004-1468. [DOI] [PubMed] [Google Scholar]
- 12.Carey AL, Steinberg GR, Macaulay SL, et al. Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP-activated protein kinase. Diabetes. 2006;55:2688–2697. doi: 10.2337/db05-1404. [DOI] [PubMed] [Google Scholar]
- 13.Pai JK, Pischon T, Ma J, et al. Inflammatory markers and the risk of coronary heart disease in men and women. N Engl J Med. 2004;351:2599–2610. doi: 10.1056/NEJMoa040967. [DOI] [PubMed] [Google Scholar]
- 14.Garneau NL, Wilusz J, Wilusz CJ. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol. 2007;8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- 15.Ng SB, Tan YH, Guy GR. Differential induction of the interleukin-6 gene by tumor necrosis factor and interleukin-1. J Biol Chem. 1994;269:19021–19027. [PubMed] [Google Scholar]
- 16.Elias JA, Lentz V. IL-1 and tumor necrosis factor synergistically stimulate fibroblast IL-6 production and stabilize IL-6 messenger RNA. J Immunol. 1990;145:161–166. [PubMed] [Google Scholar]
- 17.Paschoud S, Dogar AM, Kuntz C, et al. Destabilization of interleukin-6 mRNA requires a putative RNA stem-loop structure, an AU-rich element, and the RNA-binding protein AUF1. Mol Cell Biol. 2006;26:8228–8241. doi: 10.1128/MCB.01155-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blackshear PJ. Tristetraprolin and other CCCH tandem zinc-finger proteins in the regulation of mRNA turnover. Biochem Soc Trans. 2002;30:945–952. doi: 10.1042/bst0300945. [DOI] [PubMed] [Google Scholar]
- 19.Schichl YM, Resch U, Hofer-Warbinek R, de Martin R. Tristetraprolin impairs NF- κB/p65 nuclear translocation. J Biol Chem. 2009;284:29571–29581. doi: 10.1074/jbc.M109.031237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang J, Lei T, Song Y, et al. RNA-destabilizing factor tristetraprolin negatively regulates NF- κB signaling. J Biol Chem. 2009;284:29383–29390. doi: 10.1074/jbc.M109.024745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao H, Urban JF, Jr, Anderson RA. Insulin increases tristetraprolin and decreases VEGF gene expression in mouse 3T3-L1 adipocytes. Obesity (Silver Spring) 2008;16:1208–1218. doi: 10.1038/oby.2008.65. [DOI] [PubMed] [Google Scholar]
- 22.Lin NY, Lin CT, Chen YL, Chang CJ. Regulation of tristetraprolin during differentiation of 3T3-L1 preadipocytes. FEBS J. 2007;274:867–878. doi: 10.1111/j.1742-4658.2007.05632.x. [DOI] [PubMed] [Google Scholar]
- 23.Lai WS, Stumpo DJ, Blackshear PJ. Rapid insulin-stimulated accumulation of an mRNA encoding a proline-rich protein. J Biol Chem. 1990;265:16556–16563. [PubMed] [Google Scholar]
- 24.Cao H, Tuttle JS, Blackshear PJ. Immunological characterization of tristetraprolin as a low abundance, inducible, stable cytosolic protein. J Biol Chem. 2004;279:21489–21499. doi: 10.1074/jbc.M400900200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chhatwal JP, Hammack SE, Jasnow AM, Rainnie DG, Ressler KJ. Identification of cell-type-specific promoters within the brain using lentiviral vectors. Gene Ther. 2007;14:575–583. doi: 10.1038/sj.gt.3302898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taylor GA, Thompson MJ, Lai WS, Blackshear PJ. Phosphorylation of tristetraprolin, a potential zinc finger transcription factor, by mitogen stimulation in intact cells and by mitogen-activated protein kinase in vitro. J Biol Chem. 1995;270:13341–13347. doi: 10.1074/jbc.270.22.13341. [DOI] [PubMed] [Google Scholar]
- 27.Carballo E, Lai WS, Blackshear PJ. Feedback inhibition of macrophage tumor necrosis factor-α production by tristetraprolin. Science. 1998;281:1001–1005. doi: 10.1126/science.281.5379.1001. [DOI] [PubMed] [Google Scholar]
- 28.Liang J, Lei T, Song Y, Yanes N, Qi Y, Fu M. RNA-destabilizing factor tristetraprolin negatively regulates NF-κB signaling. J Biol Chem. 2009;284:29383–29390. doi: 10.1074/jbc.M109.024745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor GA, Carballo E, Lee DM, et al. A pathogenetic role for TNF α in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity. 1996;4:445–454. doi: 10.1016/s1074-7613(00)80411-2. [DOI] [PubMed] [Google Scholar]
- 30.Carballo E, Blackshear PJ. Roles of tumor necrosis factor-α receptor subtypes in the pathogenesis of the tristetraprolin-deficiency syndrome. Blood. 2001;98:2389–2395. doi: 10.1182/blood.v98.8.2389. [DOI] [PubMed] [Google Scholar]
- 31.Sauer I, Schaljo B, Vogl C, et al. Interferons limit inflammatory responses by induction of tristetraprolin. Blood. 2006;107:4790–4797. doi: 10.1182/blood-2005-07-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu JO. The yins of T cell activation. Sci STKE. 2005;2005:re1. doi: 10.1126/stke.2652005re1. [DOI] [PubMed] [Google Scholar]
- 33.Han C, Jin J, Xu S, et al. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat Immunol. 2010;11:734–742. doi: 10.1038/ni.1908. [DOI] [PubMed] [Google Scholar]
- 34.Starr R, Willson TA, Viney EM, et al. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;387:917–921. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- 35.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee YS, Choi JW, Hwang I, et al. Adipocytokine orosomucoid integrates inflammatory and metabolic signals to preserve energy homeostasis by resolving immoderate inflammation. J Biol Chem. 2010;285:22174–22185. doi: 10.1074/jbc.M109.085464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bouchard L, Tchernof A, Deshaies Y, et al. ZFP36: a promising candidate gene for obesity-related metabolic complications identified by converging genomics. Obes Surg. 2007;17:372–382. doi: 10.1007/s11695-007-9067-5. [DOI] [PubMed] [Google Scholar]
- 38.Bouchard L, Vohl MC, Deshaies Y, et al. Visceral adipose tissue zinc finger protein 36 mRNA levels are correlated with insulin, insulin resistance index, and adiponectinemia in women. Eur J Endocrinol. 2007;157:451–457. doi: 10.1530/EJE-07-0073. [DOI] [PubMed] [Google Scholar]
- 39.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 40.Kosteli A, Sugaru E, Haemmerle G, et al. Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. J Clin Invest. 2010;120:3466–3479. doi: 10.1172/JCI42845. [DOI] [PMC free article] [PubMed] [Google Scholar]