

Abstract
Purpose
Acute kidney injury (AKI) is a common and serious complication of severe sepsis. No targeted therapies exist for sepsis-associated AKI, suggesting a pressing need for elucidation of the underlying pathogenic mechanisms.
Recent findings
Emerging studies of human and experimental septic AKI kidneys have affirmed the longstanding observation that cell death in the tubule is uncommon despite often severe impairment of filtration. Rather than cell death, there appears to be widespread sublethal injury to tubular epithelial mitochondria. These organelles efficiently harness energy through controlled oxidation of metabolic fuels, they house pro-apoptotic proteins, and they produce reactive oxygen species. Derangement in one or more of these functions may contribute to the large reduction in renal function in septic AKI despite only scant cell death. In experimental septic AKI, molecular markers of mitochondrial biogenesis and function – whose renal expression dips during injury – rebound to normal levels as kidney function improves. Results from knockout mice suggest that restoration of mitochondrial function within the nephron may be critical to functional recovery.
Summary
Recent findings from human and experimental septic AKI studies strongly implicate the mitochondrion as an important target for sublethal kidney injury. Stimulating the natural pathways through which mitochondrial function is normally recovered following sepsis represents a promising strategy for the development of novel therapies.
Keywords: biogenesis, kidney, metabolism, mitochondria, sepsis
Introduction
The concept of the kidney as a highly metabolic organ dates back nearly a century to studies relating urea extraction to renal oxygen consumption [1]. In the 1960s, the Nobel-winning physician-scientist Hans Krebs was keenly interested in the substrates that serve as fuels for the kidney [2]. Contemporaneously, development of the transmission electron microscopy technique revealed the kidney epithelium to be rich in mitochondria, particularly in the proximal tubule and medullary thick ascending limb [3]. Electron microscopy of kidney specimens taken from young adults dying of shock and acute kidney injury (AKI) showed clear-cut mitochondrial swelling within tubular cells [4]. Sepsis, the body's inflammatory response to severe infection, is not only a common cause of shock, but also a top-10 cause of mortality in American adults. When AKI compounds severe sepsis, the mortality of sepsis is doubled. Although the availability of renal replacement therapy implies that septic AKI is not per se deadly, understanding why and how sepsis frequently affects the kidney has major implications for understanding organ dysfunction in sepsis, for illuminating novel factors that influence the balance between renal health and disease, and most tangibly, for developing new therapeutic strategies against AKI in sepsis. This review will focus on recent advances that proffer the mitochondrion as a compelling therapeutic target in septic AKI.
The Mitochondrion in Health and Sepsis
The body's major fuel-burning organs include the central nervous system, heart, liver, kidney, and skeletal muscles. Cells that comprise each of these organs are rich in mitochondria. Although the structure and oxidative capacities of mitochondria vary between cells of different organs [5], the constant demand for efficient ATP production requires their healthy function, a fact substantiated by the involvement of these organs in genetic disorders of the mitochondrion [6].
The electron transport chain progressively oxidizes metabolic intermediates to CO2 while pumping protons from the innermost mitochondrial matrix into the intermembrane space, resulting in a large electrochemical gradient that provides the power to phosphorylate ADP to ATP. Several derangements observed in sepsis are thought to contribute to the impairment of electron transport and ATP production in sepsis [7], including the following: poor oxygen delivery arising from macro-vascular and/or micro-vascular flow perturbations; nongenomic actions of inflammatory signaling pathways [8]; and free radical oxidants (e.g., ROS) directly damaging protein and/or lipid components of the mitochondrion [9].
Through mechanisms that are incompletely understood, damaged mitochondria can undergo fission and clearance through autophagy (discussed below). If the stressors are severe enough, mitochondria can rapidly swell. Hunter et al. [10], first described this abrupt increase in the unselective permeability of the mitochondrial inner membrane to small solutes in calcium-treated isolated mitochondria from bovine hearts. Proximal tubular cells of the septic kidney develop these same swollen mitochondria (Fig. 1) [11]. Mitochondrial swelling can lead to the escape of pro-apoptotic mitochondrial proteins into the cytoplasm. Moreover, mitochondrial swelling is also associated with increased production and decreased detoxification of dangerous oxidants. Thus, mitochondria are not only a target of injury from the septic milieu, but through at least three distinct putative mechanisms – reduced ATP, release of pro-apoptotic proteins, and increased oxidants – can, in turn, propagate and amplify cellular damage (Fig. 2).
Figure 1.
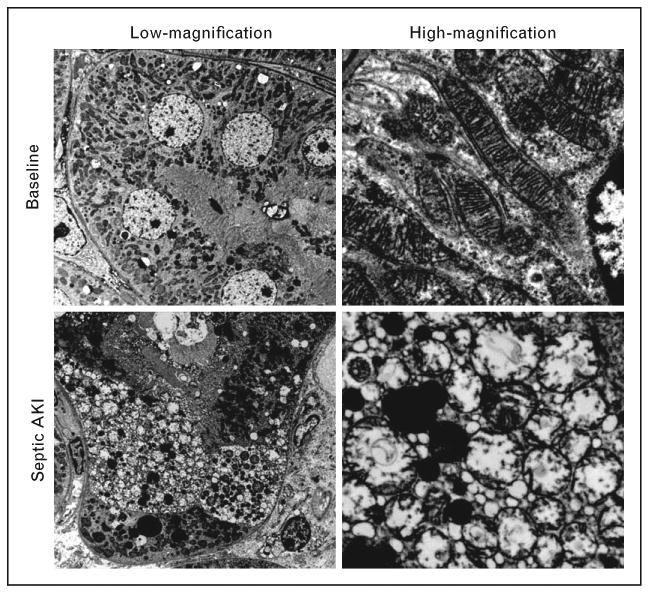
Swollen mitochondria in septic AKI. During experimental sepsis, individual proximal tubular cells exhibit accumulation of small vacuolar structures as observed by electron microscopy (left column). Higher-power examination reveals these structures to be swollen mitochondria. Adapted from Ref. [11].
Figure 2.
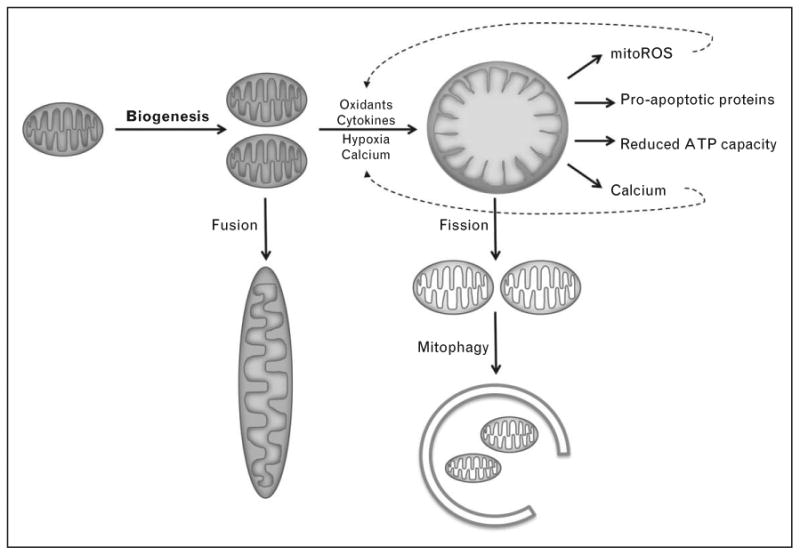
Mitochondrial biogenesis and factors in the septic milieu that promote injury. During sepsis, multiple factors conspire to damage mitochondria. In turn, damaged mitochondria can amplify the dysfunction of cells through the production of oxidants, the release of pro-apoptotic proteins, the reduction of ATP synthetic capacity, and the release of calcium ions. The long fusiform mitochondria observed in renal tubules likely arise from fusion. Damaged mitochondria may undergo fission and ultimately be cleared from the cell through mitophagy. Adapted from Ref. [5].
Acute Kidney Injury in Sepsis: A Case of Severe ‘Injury’ Without Massive Cell Death
Dysfunction of the septic kidney typically arises in the setting of shock, and so is considered by some a variant of ‘prerenal’ azotemia with no actual ‘injury’ taking place in the kidney. Indeed, efforts to document histopathological changes in septic AKI have yielded a paucity of findings [12], a paradox mirrored by primate and rodent models of septic AKI [13, 14]. Yet, functional and molecular changes do occur within the septic kidney. From clinical experience, it is clear that aggressive resuscitation of the macrocirculation, that is, the correction of the prerenal state, may be insufficient to thwart the development of persistently reduced filtration function.
Schrier's group showed that endotoxemia in rodents leads to transiently enhanced sodium avidity (consistent with the prerenal state) followed by a more sustained period of decreased sodium avidity (consistent with intrinsic tubular injury) [15]. Using a combination of knockout mice and renal transplantation prior to endotoxin exposure, Quigg's group showed that local, rather than systemic, inflammatory signaling through TNF-receptors and Toll-like receptors is critical to the septic AKI phenotype [16, 17]. Tran et al. [11], reported substantial genome-wide expression changes in the endotoxemic kidney that resolve back to a normal, preseptic pattern of expression as renal function resolves.
If injury in the septic kidney is not manifest as necrotic tubules and apoptotic cells, is there evidence of sublethal injury? Vacuolization within proximal tubular cells has been observed in both human and animal models of septic AKI. In some cases, such ‘vacuoles’ may actually be swollen mitochondria [11, 18▪▪]. Even in human renal ischemiareperfusion, mitochondrial swelling may be the dominant structural derangement [19▪▪]. In our studies of septic mice, mitochondrial swelling affects individual proximal tubular cells in an apparently stochastic fashion and only involves 5–10% of that cell population in vivo [11]. These observations emphasize the need for electron microscopy to evaluate tubule pathology in AKI. Molecular assays of mitochondrial electron transport chain activity consistently show a drastic reduction of critical enzymes affecting an entire anatomical segment of the kidney, the corticomedullary junction and the outer medulla. As kidney function resolves in the experimental models, expression of electron transport chain components returns to normal levels. Although speculative, we hypothesize that an early widespread reduction in electron transport chain expression during sepsis – that occurs through unknown mechanisms – activates a cascade of events that culminates in mitochondrial swelling. Therefore, the most widespread change we observe in the septic kidney is molecular rather than ultrastructural or histopathological (Fig. 3).
Figure 3.
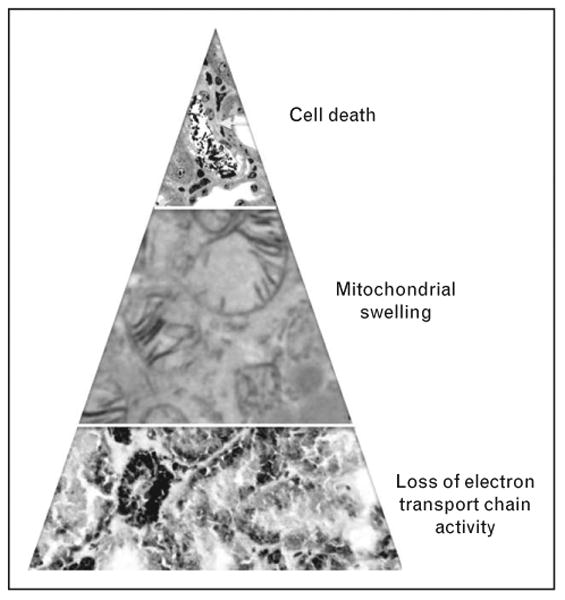
Mitochondrial injury may contribute to the predominantly sublethal injury associated with septic AKI. During sepsis, tubular cells at the corticomedullary junction exhibit marked reduction of electron transport chain enzyme activity (bottom panel, scattered brown staining indicative of small pockets where cytochrome c oxidase activity is preserved). A fraction of these cells progress to more overt mitochondrial damage, as observed by the swelling of mitochondria (middle panel). Finally, a much smaller subfraction of tubular cells die (top panel, yellow arrow).
Therapeutic Targeting of Mitochondria in Septic Acute Kidney Injury
Early observations from the laboratories of Joel Weinberg and Richard Zager studying rodent AKI models and isolated proximal tubules have led the field toward considering the role of tubular energy metabolism in AKI [20,21]. Work from several labs now suggests that mitochondrial injury may be an early pathogenic event in multiple forms of AKI, including toxic injury and ischemia-reperfusion injury (IRI). Rational therapeutic strategies in AKI based on this mitochondrial perspective broadly focus on preventing mitochondrial injury, enhancing the disposal of injured mitochondria through mitophagy, or replacing one or more injured components of the mitochondrion through biogenesis pathways.
Prevention of mitochondrial injury: fission and antioxidants
Dong's group has shown that mitochondrial fission (the division of long fusiform mitochondria into smaller shorter organelles) may contribute to experimental AKI following cisplatin exposure or IRI. They also demonstrated that prevention of fission through genetic or pharmacological means confers renoprotection [22,23]. These results suggest that fission is not solely a consequence of mitochondrial damage, but rather, may itself be a proximate cause or an amplifer mechanism of mitochondrial injury Portilla's laboratory has shown that induction of the metabolic transcription factor, PPARα, within the nephron prevents AKI following cisplatin and IRI [24,25]. They have attributed these beneficial effects to the restoration of fatty acid metabolism that is otherwise impaired during toxic and ischemic AKI. Notably, they have observed reduction of AKI-associated lipid peroxidation when PPARα is induced. Oxidative stress arising from mitochondrial injury may be an important means by which cisplatin impacts renal function, as mitochondrially targeted antioxidants dose-dependently ameliorate the AKI without attenuating the antitumor effect [26▪▪,27]. Related compounds are in clinical trials for several indications [28].
Stimulating clearance of injured mitochondria: mitophagy
Mitophagy, a subset of the broader cellular process known as autophagy, is the selective sequestration and degradation of injured mitochondria. This process may prevent cell death from mitochondrial oxidant stress and pro-apoptotic proteins. Jiang et al. [29▪], showed that acceleration of this clearance mechanism by administrating rapamycin attenuated cisplatin nephrotoxicity whereas inhibition of autophagy with chloroquine or a proximal tubule knockout of autophagy-gene-related 7 (Atg7) exacerbated the cisplatin AKI model in mice. More recently, Ishihara et al. [30] have utilized the IRI model to identify Sestrin-2 and BNIP3 as upstream regulators of autophagy and mitophagy in AKI.
Mitochondrial biogenesis
Synthesis of new mitochondrial mass may be an important mechanism for recovery from septic AKI. Tran et al. [11], studied this question using a fluid-resuscitated mouse endotoxemia model in which renal function nadirs ∼24 h after lipopolysaccharide exposure and recovers ∼24 h thereafter. They found that the recovery phase of this model was marked by a highly coordinated re-induction of genes encoding mitochondrial proteins. The peroxisome proliferator γ co-activator-1-α (PGC1α) is a transcriptional co-activator that binds to several transcription factors to drive mitochondrial biogenesis) [31,32]. PGC1α is highly expressed in the kidney in a pattern that overlaps the distribution of mitochondria [11,31].
In the resuscitated endotoxemia model, the temporal profile of renal PGC1α expression mirrored the initial fall in GFR followed by its subsequent recovery. In fact, throughout the time course of this model, the expression of PGC1α was highly correlated to the levels of downstream gene targets involved in electron transport, fatty acid oxidation, antioxidant enzymes, and other mitochondrial processes. These observations raised the question of whether the changes in renal PGC1α were contributing to or were downstream of the change in GFR. To address this, Tran et al., studied the endotoxemia model in global and proximal tubule-specific PGC1α knockouts. In both genetically engineered strains, baseline renal function was indistinguishable from controls. However, both loss-of-function mutants suffered similar and more sustained loss of GFR in the endotoxemia model compared to wild-type controls, arguing for an important role for PGC1α in renal recovery after septic AKI. Studies in other models of AKI from Schnellmann's laboratory independently support the concept that PGC1α-mediated mitochondrial biogenesis is a key factor for recovery of tubular cells from oxidative and ischemic stressors [33–35].
Efforts to stimulate the PGC1α program have focused on unbiased discovery approaches and pharmacological regulators of signaling pathways upstream of PGC1α [36–40]. As the renoprotective effects of such candidates, including resveratrol and AICAR, may be through several mechanisms, no clear consensus has emerged on the relative contribution of PGC1α induction in these settings [41,42].
Future Studies
Along each of the above-described rational approaches toward mitochondrial targeting in septic AKI, more mechanistic studies remain to be completed. First, combining both loss-of-function and gain-of-function genetically engineered mice in models of sepsis will help to narrow the field of intriguing molecular candidates. Second, as tissue biorepositories become more robust, these resources must be utilized for early human validation of molecules that emerge from preclinical studies. Third, as mentioned above, mitochondrial damage in the septic kidney likely mirrors the impact of inflammation and ischemia on other metabolically active organs throughout the body. Thus, a successful mitochondria-based drug may, in fact, ameliorate multiorgan dysfunction in sepsis. The inverse may also be true, namely that successful adjunctive strategies against sepsis are likely to ameliorate AKI as well. Finally, mitochondrial injury has also been observed in chronic kidney diseases, most prominently diabetic nephropathy [43,44▪▪]. Studies remain to be completed to address whether longstanding mitochondrial injury contributes to the well recognized AKI susceptibility among those suffering chronic kidney disease.
Conclusion
The kidney is a highly metabolically active organ whose cells rely on ATP generated by mitochondria to support their transport functions. Sepsis is both a major public health burden as well as a model condition of mitochondrial injury in the kidney and several other organs, an observation affirmed by numerous human and experimental studies. Relatively little is known about the behavior and molecular regulation of mitochondria during the course of septic AKI. However, promising data from multiple independent investigators suggest that the mitochondrion is not only critically affected by the septic milieu, but is also amenable to several pharmacologic strategies.
Key Points.
In septic AKI, both the human specimens and the experimental models demonstrate that cell injury and death is disproportionately low compared with the severity of renal dysfunction.
Ultrastructural, biochemical, and bioinformatics studies suggest that mitochondria may be a new and important target of sub-cellular injury within the tubular epithelium during sepsis.
Mitochondria within tubular cells may also be a target of toxic and ischemic forms of AKI.
Promising strategies for the amelioration of septic AKI include prevention of mitochondrial oxidative stress, removal of damaged mitochondria, and replacement of mitochondrial mass.
Acknowledgments
S.M.P. is supported by the National Institutes of Health (R01-HL093234 and R01-DK095072) and the American Diabetes Association (1-13-BS-141). S.M.P. would like to thank Dr Isaac Stillman for critical review of the article.
Footnotes
Conflicts of interest: None declared.
References and Recommended Reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
- 1.Van Slyke DD, Rhoads CP, Hiller A, Alving AS. Relationships between urea extraction, renal blood flow, renal oxygen consumption, and diuresis. The mechanism of urea excretion. Am J Physiol. 1934;109:336–374. [Google Scholar]
- 2.Weidemann MJ, Krebs HA. The fuel of respiration of rat kidney cortex. Biochem J. 1969;112:149–166. doi: 10.1042/bj1120149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sjostrand F, Rhodin J. The ultrastructure of the proximal convoluted tubules of the mouse kidney as revealed by high resolution electron microscopy. Exp Cell Res. 1953;4:426–456. [Google Scholar]
- 4.Trump BF, Valigorsky JM, Jones RT, et al. The application of electron microscopy and cellular biochemistry to the autopsy. Observations on cellular changes in human shock. Hum Pathol. 1975;6:499–516. doi: 10.1016/s0046-8177(75)80068-2. [DOI] [PubMed] [Google Scholar]
- 5.Hill BG, Benavides GA, Lancaster JR, Jr, et al. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol Chem. 2012;393:1485–1512. doi: 10.1515/hsz-2012-0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vafai SB, Mootha VK. Mitochondrial disorders as windows into an ancient organelle. Nature. 2012;491:374–383. doi: 10.1038/nature11707. [DOI] [PubMed] [Google Scholar]
- 7.Singer M, De Santis V, Vitale D, Jeffcoate W. Multiorgan failure is an adaptive, endocrine-mediated, metabolic response to overwhelming systemic inflammation. Lancet. 2004;364:545–548. doi: 10.1016/S0140-6736(04)16815-3. [DOI] [PubMed] [Google Scholar]
- 8.West AP, Brodsky IE, Rahner C, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472:476–480. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Piantadosi CA, Carraway MS, Haden DW, Suliman HB. Protecting the permeability pore and mitochondrial biogenesis. Novartis Foundation symposium. 2007;280:266–276. doi: 10.1002/9780470059593.ch18. discussion 76-80. [DOI] [PubMed] [Google Scholar]
- 10.Hunter DR, Haworth RA, Southard JH. Relationship between configuration, function, and permeability in calcium-treated mitochondria. J Biol Chem. 1976;251:5069–5077. [PubMed] [Google Scholar]
- 11.Tran M, Tam D, Bardia A, et al. PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest. 2011;121:4003–4014. doi: 10.1172/JCI58662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Langenberg C, Bagshaw SM, May CN, Bellomo R. The histopathology of septic acute kidney injury: a systematic review. Crit Care. 2008;12:R38. doi: 10.1186/cc6823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carraway MS, Welty-Wolf KE, Miller DL, et al. Blockade of tissue factor: treatment for organ injury in established sepsis. Am J Respir Crit Care Med. 2003;167:1200–1209. doi: 10.1164/rccm.200204-287OC. [DOI] [PubMed] [Google Scholar]
- 14.Miyaji T, Hu X, Yuen PS, et al. Ethyl pyruvate decreases sepsis-induced acute renal failure and multiple organ damage in aged mice. Kidney Int. 2003;64:1620–1631. doi: 10.1046/j.1523-1755.2003.00268.x. [DOI] [PubMed] [Google Scholar]
- 15.Schrier RW, Wang W, Poole B, Mitra A. Acute renal failure: definitions, diagnosis, pathogenesis, and therapy. J Clin Invest. 2004;114:5–14. doi: 10.1172/JCI22353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cunningham PN, Dyanov HM, Park P, et al. Acute renal failure in endotoxemia is caused by TNF acting directly on TNF receptor-1 in kidney. J Immunol. 2002;168:5817–5823. doi: 10.4049/jimmunol.168.11.5817. [DOI] [PubMed] [Google Scholar]
- 17.Cunningham PN, Wang Y, Guo R, et al. Role of Toll-like receptor 4 in endotoxin-induced acute renal failure. J Immunol. 2004;172:2629–2635. doi: 10.4049/jimmunol.172.4.2629. [DOI] [PubMed] [Google Scholar]
- 18▪▪.Takasu O, Gaut JP, Watanabe E, et al. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am J Respir Crit Care Med. 2013;187:509–517. doi: 10.1164/rccm.201211-1983OC. In rapidly obtained postmortem renal and cardiac specimens from 44 septic patients and 28 controls, the authors observed that most renal tubular cells appear normal by light and electron microscopy, and that the degree of cell injury and death does not account for the severity of renal dysfunction. They noted increased ‘hydropic’ mitochondria and autophagosomes in septic specimens. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19▪▪.Parekh DJ, Weinberg JM, Ercole B, et al. Tolerance of the human kidney to isolated controlled ischemia. J Am Soc Nephrol. 2013;24:506–517. doi: 10.1681/ASN.2012080786. The authors examined the renal response to clamp ischemia and subsequent reperfusion among patients undergoing partial nephrectomy. They found that that 30-60 min of clamp ischemia resulted in only mild structural changes and no acute functional loss. The most prominent structural changes were loss of apical brush-border and swelling of mitochondria within tubular cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weinberg JM, Venkatachalam MA, Roeser NF, Nissim I. Mitochondrial dysfunction during hypoxia/reoxygenation and its correction by anaerobic metabolism of citric acid cycle intermediates. Proc Natl Acad Sci U S A. 2000;97:2826–2831. doi: 10.1073/pnas.97.6.2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zager RA, Johnson AC, Hanson SY. Renal tubular triglyercide accumulation following endotoxic, toxic, and ischemic injury. Kidney Int. 2005;67:111–121. doi: 10.1111/j.1523-1755.2005.00061.x. [DOI] [PubMed] [Google Scholar]
- 22.Brooks C, Cho SG, Wang CY, et al. Fragmented mitochondria are sensitized to Bax insertion and activation during apoptosis. Am J Physiol Cell Physiol. 2011;300:C447–C455. doi: 10.1152/ajpcell.00402.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest. 2009;119:1275–1285. doi: 10.1172/JCI37829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li S, Basnakian A, Bhatt R, et al. PPAR-alpha ligand ameliorates acute renal failure by reducing cisplatin-induced increased expression of renal endonuclease G. Am J Physiol Renal Physiol. 2004;287:F990–F998. doi: 10.1152/ajprenal.00206.2004. [DOI] [PubMed] [Google Scholar]
- 25.Li S, Nagothu KK, Desai V, et al. Transgenic expression of proximal tubule peroxisome proliferator-activated receptor-alpha in mice confers protection during acute kidney injury. Kidney Int. 2009;76:1049–1062. doi: 10.1038/ki.2009.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26▪▪.Mukhopadhyay P, Horvath B, Zsengeller Z, et al. Mitochondrial-targeted antioxidants represent a promising approach for prevention of cisplatin-induced nephropathy. Free Radic Biol Med. 2012;52:497–506. doi: 10.1016/j.freeradbiomed.2011.11.001. Using a rodent cisplatin nephrotoxicity model, this study demonstrated two important findings that proximal tubular cells' mitochondria are selectively damaged when compared with distal tubular cells' mitochondria, correlating with the clinical presentation of this toxicity; targeting antioxidants to the mitochondria dose-dependently ameliorates renal dysfunction, suggesting that cisplatin-induced mitochondrial oxidative stress within the proximal tubule is an important mechanism for renal injury. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zsengeller ZK, Ellezian L, Brown D, et al. Cisplatin nephrotoxicity involves mitochondrial injury with impaired tubular mitochondrial enzyme activity. J Histochem Cytochem. 2012;60:521–529. doi: 10.1369/0022155412446227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith RA, Murphy MP. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann New York Acad Sci. 2010;1201:96–103. doi: 10.1111/j.1749-6632.2010.05627.x. [DOI] [PubMed] [Google Scholar]
- 29▪.Jiang M, Wei Q, Dong G, et al. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012;82:1271–1283. doi: 10.1038/ki.2012.261. The authors created mice that exhibited impaired autophagy specifically within the proximal tubule throughout a conditional knockout of ATG7. Although these mice showed no baseline impairment in renal function, they were markedly more sensitive to cisplatin-induced nephrotoxicity than wild-type counterparts. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishihara M, Urushido M, Hamada K, et al. Sestrin2 and BNIP3 (Bcl-2/adenovirus E1B 19kDa-interacting protein3) regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am J Physiol Renal Physiol. 2013 doi: 10.1152/ajprenal.00642.2012. [DOI] [PubMed] [Google Scholar]
- 31.Puigserver P, Wu Z, Park CW, et al. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 32.Wu Z, Puigserver P, Andersson U, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 33.Funk JA, Schnellmann RG. Persistent Disruption of Mitochondrial Homeostasis after Acute Kidney Injury. Am J Physiol Renal Physiol. 2011 doi: 10.1152/ajprenal.00035.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rasbach KA, Schnellmann RG. PGC-1alpha over-expression promotes recovery from mitochondrial dysfunction and cell injury. Biochem Biophys Res Commun. 2007;355:734–739. doi: 10.1016/j.bbrc.2007.02.023. [DOI] [PubMed] [Google Scholar]
- 35.Rasbach KA, Schnellmann RG. Signaling of mitochondrial biogenesis following oxidant injury. J Biol Chem. 2007;282:2355–2362. doi: 10.1074/jbc.M608009200. [DOI] [PubMed] [Google Scholar]
- 36.Arany Z, Wagner BK, Ma Y, et al. Gene expression-based screening identifies microtubule inhibitors as inducers of PGC-1alpha and oxidative phosphorylation. Proc Natl Acad Sci U S A. 2008;105:4721–4726. doi: 10.1073/pnas.0800979105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holthoff JH, Wang Z, Seely KA, et al. Resveratrol improves renal microcirculation, protects the tubular epithelium, and prolongs survival in a mouse model of sepsis-induced acute kidney injury. Kidney Int. 2012;81:370–378. doi: 10.1038/ki.2011.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kolgazi M, Sener G, Cetinel S, et al. Resveratrol reduces renal and lung injury caused by sepsis in rats. J Surg Res. 2006;134:315–321. doi: 10.1016/j.jss.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 39.Lempiainen J, Finckenberg P, Levijoki J, Mervaala E. AMPK activator AICAR ameliorates ischemia reperfusion injury in the rat kidney. Br J Pharmacol. 2012 doi: 10.1111/j.1476-5381.2012.01895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin A, Sekhon C, Sekhon B, et al. Attenuation of ischemia-reperfusion injury in a canine model of autologous renal transplantation. Transplantation. 2004;78:654–659. doi: 10.1097/01.tp.0000131664.18670.17. [DOI] [PubMed] [Google Scholar]
- 41.Lagouge M, Argmann C, Gerhart-Hines Z, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 42.Canto C, Gerhart-Hines Z, Feige JN, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brownlee M, Cerami A. The biochemistry of the complications of diabetes mellitus. Annu Rev Biochem. 1981;50:385–432. doi: 10.1146/annurev.bi.50.070181.002125. [DOI] [PubMed] [Google Scholar]
- 44▪▪.Wang W, Wang Y, Long J, et al. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012;15:186–200. doi: 10.1016/j.cmet.2012.01.009. Mitochondrial dysfunction has been implicated in the microvascular complications of diabetes. The authors utilize several genetically engineered mice in models of diabetes to show that hyperglycemia triggers a cellular kinase called ROCK1 to induce mitochondrial fission in podocytes and endothelium, suggesting ROCK1 as a new candidate mediator of mitochondrial dysfunction in diabetic nephropathy. [DOI] [PMC free article] [PubMed] [Google Scholar]