

Abstract
Trypanosomal S-adenoyl-L-homocysteine hydrolase (Tc-SAHH), considered as a target for treatment of Chagas disease, has the same catalytic mechanism as human SAHH (Hs-SAHH) and both enzymes have very similar X-ray structures. Efforts toward the design of selective inhibitors against Tc-SAHH targeting the substrate binding site have not to date shown any significant promise. Systematic kinetic and thermodynamic studies on association and dissociation of cofactor NAD/H for Tc-SAHH and Hs-SAHH provide a rationale for the design of anti-parasitic drugs directed toward cofactor-binding sites. Analogues of NAD and their reduced forms show significant selective inactivation of Tc-SAHH, confirming that this design approach is rational.
1. Introduction
Professor Morris J. Robins has made many meritorious contributions to the field of modified nucleosides and we wish in this review to discuss work carried out under his intellectual influence.
S-Adenosyl-L-homocysteine (AdoHcy) hydrolase (SAHH; EC 3.3.1.1.) is the only known enzyme responsible for the reversible conversion of AdoHcy to adenosine (Ado) and homocysteine (Hcy) (Scheme 1.1.). SAHH occurs downstream of the S-adenosylmethionine (AdoMet)-dependent transmethylation enzymes,1, 2 which are involved in a wide variety of important biological functions. The methyl acceptors in this pathway include macromolecules such as proteins, nucleic acids, and polysaccharides, and small molecules such as histamines and phospholipids3. AdoHcy is a powerful product inhibitor which regulates the AdoMet-dependent methyl transfers, so that SAHH plays a crucial role in the AdoMet-dependent transmethylation pathway through controlling the intracellular levels of AdoHcy1. The catabolic reaction of AdoHcy favors the hydrolytic direction under physiological conditions1. Thus, inhibition of SAHH in mammalian cells will result in increased cellular levels of AdoHcy and further blocking of the methyl cycle.
Scheme 1.1.
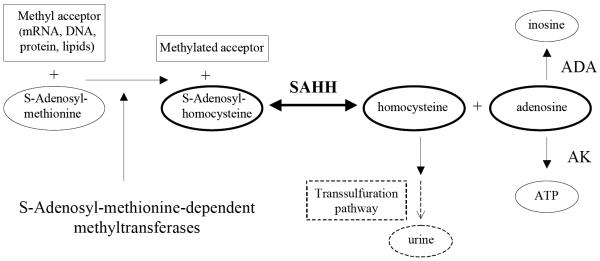
The role of SAHH in methyl transfer and metabolic pathways. Abbreviations: ADA, adenosine deaminase; AK, adenosine kinase.
SAHH has been considered an antiviral target in chemotherapy for decades, the 5'-terminus of viral mRNA being an AdoMet-dependent transmethylase substrate4, 5. Moreover, an overactive malfunction of SAHH will result in abnormally high blood levels of Hcy, which increases the risk of cardiovascular disease6–8 and possibly amyloid diseases9, and so SAHH has been considered as a target for treatment of these diseases as well. Recently, a new pharmacological interest has developed in inhibitors of SAHH as anti-parasitic agents10.
2. The importance of selective inhibitors against Tc-SAHH as therapeutics in Chagas disease
Besides mammalian organisms, parasites such as Trypanosoma cruzi11, Plasmodium falciparum12 and Leishmania donovani10, 13 also encode their own SAHHs (Tc-SAHH, Pf-SAHH and Ld-SAHH, respectively) as well as AdoMet-dependent methyltransferases14. As in mammalian organisms, parasitic AdoMet-dependent methyltransferases serve to methylate the 5'-terminus of mRNAs, which is important for growth14. Thus, inactivation of parasitic SAHHs will result in the blocking of methyl transfer and will suppress the growth of parasites. Earlier studies10, 11 have shown that there are differences in some kinetic and thermodynamic parameters between human and parasite enzymes. Parasite enzymes have weaker binding affinities for NAD+ and/or lower catalytic activities than those of the human enzyme10, 11. These differences provide the possibility of designing selective inhibitors of parasite SAHHs that will not inhibit human SAHH (Hs-SAHH).
In this review we are specifically interested in Trypanosoma cruzi. There currently are 8–11 million people infected with Chagas disease (also known as American trypanosomiasis) by the protozoan parasite Trypanosoma cruzi (Center for Disease Control and Prevention, Chagas Disease Detailed Fact Sheet, March 2009). This disease is transmitted by triatomine insects when they feed on the blood of human hosts15. Chagas disease mainly resides in rural areas of Mexico, Central America and South America and seriously threatens the public health there15. Because of increased population movement, it has been reported that there are rare cases in southern parts of the USA (Center for Disease Control and Prevention, Chagas Disease Detailed Fact Sheet, March 2009). A study of the distinguishing structural and enzymological features between Hs-SAHH and Tc-SAHH could lead to development of compounds with clinical potential as anti-parasitic agents and eventually to improved treatments of Chagas disease.
3. Structural similarities between Hs-SAHH and Tc-SAHH
So far there are fifteen available X-ray structures of SAHH from different sources (Protein Data Bank files: 1A7A, 1KY4, 1KY5, 1LI4, 1D4F, 1XWF, 1K0U, 2H5L, 1XBE, 1V8B, 2ZIZ, 2ZJ0, 2ZJ1, 3CE6, 3DHY) including wild-type enzymes from human placenta, rat liver, Plasmodium falciparum, Trypanosoma cruzi (unpublished data), and Mycobacterium tuberculosis as well as various mutated forms. The alignment of SAHH amino acid sequences from different sources shows the primary structure is highly conserved1.
3.1. The structure of Hs-SAHH
Hs-SAHH is a homotetramer (Figure 1) composed of four ~ 47.5 kDa monomers1, 16. Each monomer has 432 residues and can be divided into three parts: one substrate-binding domain, one cofactor-binding domain (complexed to a molecule of NAD+/NADH) and a small C-terminal extension or “tail”1, 16. Both structure and normal mode analysis identify a hinge region [residues 182–196 (hinge region 1) and 352–356 (hinge region 2)] which connects the substrate-binding domain (SBD, residues 5–180 and 357–374) with the cofactor-binding domain (CBD, residues 191–356)1, 17, 18. This hinge regulates a ~ 19° reorientation of the SBD relative to the CBD in the direction of the open-to-closed transition and enables a conformational change in SAHH, which plays an important role in substrate capture and in the subsequent series of catalytic reactions16, 17.
Figure 1.

The tetrameric structure of SAHH containing cofactor NAD+ (light ball-and-stick) (pdb code: 1KY4). Four cofactor-binding domains (central dark regions) form the core at the center and four substrate-binding domains (corners of the figure) are located outside.
Between the SBD and the CBD there is a deep cleft which works as a channel for substrates entering the active site and also for products being released into solution1. In terms of quaternary structure, the tetramer is composed of two dimers, formed by monomers A and B, and monomers C and D, respectively. The two dimers bind each other tightly and build a “dimer of dimers”1. The short tail (residues 380–432) of monomer A extends into monomer B and helps to form the cofactor-binding site of monomer B, and vice versa1. This dual interpenetration also occurs between monomers C and D1. Moreover, the four cofactor-binding domains from the four monomers bind each other tightly to form a central core with the four substrate-binding domains facing outside1.
The available X-ray structures show that there are two different conformations of SAHH: the open form and the closed form. Since rat SAHH shares 97% sequence identity with the human enzyme, the entire protein structure of rat SAHH is assumed to be very similar to that of the human enzyme19. The open conformation comes from the structure of the rat enzyme (PDB code: 1KY4)20 while the closed conformation is from the structure of the human placental enzyme [PDB code: 1A7A1 or 1LI416]. It is worth emphasizing that the X-ray structure (PDB:1A7A) of SAHH which contains the inhibitor DHCeA shows a twisted-closed conformation: a 14° reorientation of two halves of the homotetramer seem to seal the enzyme core1. Comparison of the open and closed conformations, Figure 2, shows a 19° rotation of the substrate binding domain toward the cofactor binding domain, which leads from the open to the closed conformation. The open-to-closed conformational transition is related to the catalytic reactions involving the substrate and cofactor NAD(H) in the active site. The open conformation of SAHH enables substrate binding and the closed conformation provides a closer contact between substrate and cofactor in the active site, as is needed for catalysis. The enzyme converts back to the open conformation upon product release21.
Figure 2.

Open conformation (pdb: 1KY4, rat source) and closed conformation (pdb: 1LI4, human source) of the SAHH monomer (created by Molecular Operation Environment). The top domain is the cofactor-binding domain; the bottom domain is the substrate-binding domain; the loop at the right is the C-terminal extension. SAHH in the open conformation contains cofactor NAD+ (ball-and-stick) and SAHH in closed conformation contains cofactor NADH (ball-and-stick) and oxidized inhibitor-NepA (ball-and-stick). From the open conformation, the substrate-binding domain rotates ~ 19° toward to the cofactor binding domain to form an active site for catalysis in the closed conformation.
Besides the X-ray structures, there is other evidence for SAHH domain motions. Computationally, normal mode analysis of low-frequency collective motions of SAHH reveals the different mechanical properties of the open and closed forms of SAHH. The hinge-bending motion in the direction of the open-to-closed conformational transition is unique to the open form of SAHH and occurs independently of other protein vibrations17. In the closed form, normal mode calculations show the amplitude of the hinge-bending motion in each subunit of SAHH is smaller than that in the open form, and the hinge bending motions of individual subunits are strongly coupled to each other and other low frequency vibrations, including subunit reorientations17. This mechanical coupling, a characteristic of the closed form, may transmit the information of any structural changes related to the catalytic reactions in one of the four active sites to the other active sites. Normal mode analysis also suggested that the 20-ps hinge-bending vibrations of subunits reach an amplitude of motion of ~ 1° which is much smaller than the amplitude of 19° in the global structural transition, so the 19° SBD vs. CBD reorientation cannot be described as a simple elastic deformation17.
Experimentally, time-resolved fluorescence anisotropy measurements were carried out on native SAHH and three catalytically active mutants, M351P, H353A and P354A, all labeled with the fluorescent probe PMal at residues C113 and C42118. Data analysis was focused on the time constants for reorientation motions. The data for wild type SAHH with/without ligand provides important information on the effect of ligand binding and oxidation on hinge-bending motions. Wild type SAHH without substrate exhibits three rotational correlation times: a time constant of 0.5 ns reflecting local chromophore reorientations and protein vibrations; one of 14 ns involving domain reorientation; and a third of 82 ns manifesting the overall protein tumbling in solution18. Wild type SAHH bound with 3'-deoxyAdo/NAD+ or 3'-keto-Ado/NADH (Ado/NAD+) shows a similar three-component anisotropy decay, but for wild type SAHH bound with 3'-keto-NepA/NADH, the 10-20 ns domain reorientation is not detected18. These results indicate that there is an equilibrium between open and closed structures of SAHH. This equilibrium is shifted toward a more mobile open form in the substrate-free enzyme (E-NAD+), as well as in complexes with intermediates formed early in the catalytic cycle after substrate binding or formed late prior to product release (E-NAD+/ligand). This equilibrium is shifted toward the more rigid closed form in SAHH complexes of analogues of the central catalytic intermediate (E-NADH/ligand)18. In addition, fluorescence anisotropy decay studies of three mutant enzymes with/without ligand illuminated the roles of the mutated residues in the hinge region 2 (residues 352–356) in hinge bending motions. Mutants M351P and P354A show a similar pattern of behavior to wild type SAHH, which indicates that residues M351 and P354 have no significant effects on domain motion18. For the H353A mutant, the 10-20 ns domain motion was not detected either with or without ligand, and the longest component was shortened to 55–71 ns for all four ligation states of mutant H353A. This indicates mutant H353A changes its domain motion dynamics independent of the presence of ligand or oxidation state of cofactor18. Since the H353A mutant remains catalytically active, this mutant may slow down its hinge bending motions to a longer time scale similar to that of overall protein tumbling or longer, or exhibit a modified shape of the protein18. If the hinge bending motions slow down to the time scale of enzyme turnover (the order of a second), this would be out of the detectable range for fluorescence anisotropy decay (0.1 – 200 ns)18. Overall, hinge region 2 makes no direct contribution to catalytic activity. In contrast, hinge region 1 contains residues N181, K186, N190, N191 which are in the active site, which are involved in binding the substrate and thus are directly able to affect the activity18.
To get a direct and clear picture of the domain motions of SAHH, a 15 ns dynamics simulation of the open form of SAHH with explicit solvent was performed using Amber7 software. Very similar to the X-ray structure, the trajectories show that at the tetramer level the four cofactor binding domains form a central core which remains relatively rigid, and that the four substrate binding domains, located at the protein exterior, exhibit flexible reorientations of large amplitude. Interestingly, the fluctuations of domains between open and closed conformations within each subunit constitute only ~ 20% of the trajectory domain motions. At the tetramer level, the remaining ~ 80% of the domain motions are perpendicular to the direction of the open-to-closed structural transition, and may be described as a “breathing-type” motion with substrate-binding domains moving to and from the tetrameric core of SAHH. Furthermore, the domain reorientations in solution can be represented as a combination of a faster process with 20–50 ps rotational correlation times and 3–4° amplitude, and a slower process with 8–23 ns correlation times and 14–22° amplitude. The time scale of the faster process is very close to that of the hinge-bending vibrations which were found in the normal mode analysis while the slow process well matches the fluorescence anisotropy decay measurements, which detected the 10-20 ns domain motion with ca. amplitude of 26° in SAHH without substrate. Therefore, the slow process is assigned to the rotational diffusion of the domains within a cone with 10-20° half-angle. Overall, the results of the simulation agree with the previous data and help us to better understand SAHH domain motions in solution22.
3.2. The structure of Tc-SAHH
The SAHH of Trypanosoma cruzi (437 aa and ~ 48 kDa per subunit) was cloned and purified in this laboratory11. The sequence of Tc-SAHH shares 74% identity with Hs-SAHH11. The X-ray structure of Tc-SAHH treated with the inhibitor Neplanocin A has been recently determined (unpublished data of Drs. Q.-S. Li and W. Huang). Comparison of the X-ray structures of Hs-SAHH and Tc-SAHH shows that there is no significant difference between them at the structural levels of the tetramer, subunit, CBD, SBD and C-terminal extension. The residues directly interacting with the substrate and the residues directly interacting with NAD+ are all conserved. However it was reported that parasitic enzymes such as Tc-SAHH11, Ld-SAHH10 and Pf-SAHH (unpublished data, S. Cai) bound cofactor NAD+ less tightly than Hs-SAHH, which suggests that there ought to be some structural or dynamic factors causing the differences in properties between Hs-SAHH and parasite enzymes.
4. The catalytic mechanism of SAHH
SAHH is a hydrolase for which the catalytic mechanism was the topic of intense earlier studies. Briefly, the catalytic reaction of SAHH for human and parasite is the reversible hydrolysis and synthesis of AdoHcy to/from Ado and Hcy.
This equilibrium favors the hydrolytic direction in vivo because of rapid removal of products - Ado catalyzed by Ado deaminase and Hcy by the transsulfuration pathway. In contrast, thermodynamic equilibrium favors the synthetic direction in the biochemical assays in vitro1.
The total catalytic process can be divided into two parts: a reduction/oxidization portion and an elimination/addition part, which is shown as a cycle in Scheme 1.2.16, 23, 24. The redox part utilizes a tightly bound NAD+/NADH as a cofactor for hydrogen transfer. In the hydrolysis direction, the 3'-CH center of substrate AdoHcy transfers a hydride equivalent to NAD+, forming 3'-keto-AdoHcy and NADH. In the 3'-keto-AdoHcy, the 4'-CH bond susceptible to deprotonation and thus activates elimination of Hcy across the 4'–5' bond. The elimination is followed by Michael addition of water to form 3'-keto-Ado. Tightly bound NADH now returns a hydride equivalent to 3'-keto-Ado and the catalytic process is completed with release of Ado from SAHH, which contains the cofactor again in the NAD+ state.
Scheme 1.2.

Catalytic cycle of SAHH in both hydrolytic and synthetic directions with redox partial reactions and elimination/addition partial reactions. B represents the enzymic residue that accepts and returns the proton at the 4' position. SAHH with bound NAD+ exists in an open conformation while SAHH with bound NADH exists in a closed conformation for a series of catalytic reactions.
The catalytic cycle involves two enzyme conformational states (Protein Data Bank code: 1KY420 and 1A7A1, 21): an open-closed interconversion of each monomer, which regulates substrate binding and product release, and a ~14° rotation of one dimer relative to the second dimer, which leads to a reduction in the volume of the tetramer and may serve to “seal” the closed form. The “sealed” active sites help to prevent contact with the environment of intermediates among a series of transition states21, 25.
The kinetics of individual steps in the SAHH catalytic cycle have been measured and a model suggested that integrates the kinetics, the structural and dynamic results, and the findings from site-directed mutagenesis16. The model emphasizes the roles of avoidance of abortive reactions and stabilization of transition states in achieving efficient levels of catalysis16, 26–28. There are three 3'-keto intermediates in the catalytic cycle and none of them could survive exposure to the aqueous buffer environment16.
Any release and/or exposure of intermediates will result in abortive products and an uncompleted catalytic cycle. This is hindered by a tight closure of the active site, which builds a barrier of 110 kJ/mol for abortive release of intermediates. This barrier is ~ 50 kJ/mol higher than a diffusional barrier and decreases the rate of abortive release by 108–109 fold16. In addition, the central intermediate 4', 5'-didehydro-5'-deoxy-3'-keto-adenosine also needs to be protected from the nearby cofactor NADH to avoid premature reduction of the 3'-keto group of the intermediate16. The 3'-keto structure of the central intermediate is crucial for activation of the next step of Michael addition of water/Hcy to the 4', 5'-double bond. To achieve the necessary protection, residue His 301 shifts its position so that its side chain buttresses the cofactor NADH at a greater separation from the intermediate. The result is an apparent increase in distance of about 0.4 Å between C-4' of the cofactor NADH and C-3' of the substrate16. The barrier for the abortive reduction of the central intermediate is 86–89 kJ/mol, which is 22–24 kJ/mol higher than the barrier for productive reduction (65–67 kJ/mol). This difference in barrier heights results in a rate of productive reduction that is faster by 103–104 fold than the rate of abortive reduction16. The catalysis of most steps in the reaction depends on acid-base catalysis for transition-state stabilization, so that the potentially needed exchange of protons with the medium would seem to be difficult in the closed conformation. However, SAHH appears to depend on a chain of water molecules to deliver protons between the active site and the buffer environment to maintain the suitable protonation states of functional residues during the catalytic cycle16.
Many attempts have been made to design mechanism-based inhibitors for Hs-SAHH targeting the substrate binding site and utilizing its catalytic potentialities, and some progress has been achieved1, 2, 29. The partial “oxidative” reaction (reduction/oxidation) and partial “hydrolytic” (elimination/addition) reaction provide two design targets29. Type I inactivators, which are oxidized to the 3'-keto form with SAHH catalysis and conversion of NAD+ to NADH, target the oxidative partial reaction30. Neplanocin A (NepA) with a Ki of 8.4 nM is an example of type I inactivators and its catalytic reaction stops after oxidation at the 3' position produces a material that is structurally an analogue of the central intermediate 4', 5'-didehydro-5'-deoxy-3'-keto-adenosine1, 31. On the other hand, type II inactivators are not only oxidized in the 3'-position by SAHH but also form covalent bonds with enzyme following reactions analogous to the elimination/addition partial reaction30. For example, (E)-5', 6'-didehydro-6'-deoxy-6'fluorohomoadenosine (EDDFHA) is a type II inactivator by generating electrophiles at the active site and forming a covalent bond to irreversibly inactivate the enzyme32. Figure 3 shows the structures of the above two types of inhibitors and their inactivation mechanisms.
Figure 3.

Type I inhibitor (NepA) and Type II inhibitor (EDDFHA) structure and their inactivation mechanisms (adopted from Elrod, et al. 200237).
5. Substrate binding site is not a suitable target for designing anti-parasitic drugs
A good antiparasitic drug needs to show selective inhibition of parasitic SAHHs (such as Tc-SAHH) without significant inactivation of Hs-SAHH. The available inhibitors against Hs-SAHH such as NepA or DHceA are all substrate (Ado or AdoHcy) analogues. So the substrate binding site was firstly considered as the target for designing anti-parasitic selective inhibitors. But Type I and Type II inhibitors of Hs-SAHH are obviously unsuitable as antiparasitic agents. Furthermore, Drs. Morris Robins and Stanislaw Wnuk kindly provided a total of 122 substrate analogs (unpublished work) for a screen against both Hs-SAHH and Tc-SAHH. There are only 12 compounds showing some weak selective inhibition of Tc-SAHH while most of the remainder, 89 compounds, show no inhibition of either enzyme. Only 10 compounds show roughly equivalent inhibition of both enzymes, while 11 compounds show stronger inhibition of Hs-SAHH. In addition, ribavirin (1, 2, 4-triazole-3-carboxamide riboside) was reported to show some time-dependent selective inactivation of Tc-SAHH over Hs-SAHH38. But the KI values of ribavirin for Hs-SAHH (266uM) and Tc-SAHH (194uM) are similar and the weak selectivity results from the differential slow inactivation rate where ribavirin reacts with Tc-SAHH five times faster than with Hs-SAHH.
Overall, we concluded that it was very unlikely that selective inhibitors targeted toward the substrate binding site of Tc-SAHH would be identified.
6. The cofactor binding site is a promising target for designing selective inhibitors against Tc-SAHH
As mentioned in section 3.2, Tc-SAHH and other parasitic SAHHs show a weaker affinity for the cofactor NAD+ than does Hs-SAHH. Such enzymological differences between Hs-SAHH and parasitic SAHHs may provide a clue or rationale for designing selective inhibitors against parasitic SAHHs. Therefore, the comparative kinetics of cofactor association and dissociation for Hs-SAHH and Tc-SAHH have been studied systemically33, 34.
6.1. The basic features of the cofactor association and dissociation process33
The equilibrium and kinetic properties of the association and dissociation of the cofactor NAD+ from Hs-SAHH and Tc-SAHH form a very complex picture. In a word, those properties of Hs-SAHH and Tc-SAHH are qualitatively similar but quantitatively distinct. All data suggest that the four cofactor binding sites of the homotetrameric apoenzyme fall into two numerically equal classes, one class denoted fast-binding sites and the other class denoted slow-binding sites. Fast-binding sites possess a weak cofactor binding affinity but occupation by cofactor generates enzymatic activity rapidly (< 1 min). Slow-binding sites possess a relatively strong cofactor binding affinity but occupation by cofactor generates no catalytic activity initially. Instead, it initiates a process in which the site is slowly (> 30 mins) transformed from a non-catalytic status to a catalytic-active status. This transformation of the slow-binding site may involve a series of conformational changes induced by cofactor binding, resulting in a delay of generation of catalytic activity. In addition, the transformation of slow-binding sites must affect the fast-binding sites: the cofactor binding affinity and catalytic activity of all four sites of the holoenzyme are same. Thereafter, Tc-SAHH persistently exhibits a cofactor binding affinity at micromolar levels. The slow-binding phase of Hs-SAHH terminates with a cofactor binding affinity also in the micromolar range. Yet over a period of some 30 mins, Hs-SAHH develops a nanomolar level of affinity for NAD+, due entirely to a decreased dissociation rate constant. The structural reason for this increased cofactor affinity of Hs-SAHH is still unknown.
The kinetics of cofactor association with both human and trypanosomal apoenzymes create saturation and exponential curves as a function of cofactor concentration. But the maximum cofactor-binding rate constant of Hs-SAHH is ten times larger than that of Tc-SAHH. Furthermore, the cofactor-association rate constant exhibits a complex temperature-dependence such that NAD+ binds faster to Hs-SAHH than to Tc-SAHH above ~16 °C.
Compared to the cofactor-association process, the cofactor-dissociation process is relatively simple. Cofactor NAD+ dissociates from all four binding sites in a single first-order reaction for both Hs-SAHH and Tc-SAHH. However, cofactor always dissociates from Tc-SAHH much more rapidly than from Hs-SAHH. For example, the cofactor-dissociation rate constant of Tc-SAHH is 80-fold larger than that of Hs-SAHH at 37 °C. The differential cofactor-dissociation rate constants provide a potential opportunity for inhibitors to bind selectively to Tc-SAHH, which supports the idea that it is feasible to design selective inhibitors targeted toward the cofactor binding site and not toward the substrate-binding site, as with previous efforts.
6.2. Structure elements responsible for differential cofactor binding properties (34 and unpublished data)
Hs-SAHH and Tc-SAHH thus exhibit distinct kinetic and thermodynamic properties of cofactor association and dissociation, which should be based on a structural distinction. Although the X-ray structures show no significant differences between two enzymes, some local elements near the cofactor-binding site may be important to NAD+ binding. The C-terminal extension of SAHH is a flexible structure. The extension includes a short helix-18, located 8 residues ahead of Lys, a final residue for Hs-SAHH and for three parasitic SAHHs (Tc-SAHH, Ld-SAHH and Pf-SAHH). The residues Lys426 and Tyr430 in the C-terminal loop of Hs-SAHH are found near NAD+ in X-ray structures, and may form hydrogen bonds with NAD+. A stable helix-18 may help to locate residues Lys426 and Tyr430 at suitable positions in the NAD+ binding pocket. Therefore, the relative stability of helix-18 should be important for determining the differences in cofactor-association-dissociation and affinity between human and parasitic enzymes. Each residue has its helix-propensity value, so the helix-propensity value of helix-18 can be easily calculated according to its seven-residue sequence. A high propensity value means a more stable helix structure. The hypothesis is that the SAHH with a more stable helix-18 will bind the cofactor NAD+ more strongly. The calculated helix-propensity values of helix-18 fall in a decreasing order Hs-SAHH (8.96) > Tc-SAHH (8.35) > Ld-SAHH (7.73) > Pf-SAHH (7.31), consistent with the order of their cofactor binding affinities as previously observed.
A mutagenesis approach was taken to create a “parasitized” human enzyme mutant, with helix-18 of Hs-SAHH replaced by the less stable helix-18 of Pf-SAHH (Hs-18Pf-SAHH). A “humanized” trypanosomal mutant was also constructed, with helix-18 of Tc-SAHH replaced by the more stable helix-18 from Hs-SAHH (Tc-18Hs-SAHH). Both mutants exhibit biphasic association kinetics (fast and slow binding) similar to that of wild type Hs-SAHH and Tc-SAHH. The temperature dependence of the rate constant for NAD+ association shows a thermal transition for all SAHHs. The thermal-transition temperatures follow the order Hs-SAHH (35 °C) > Hs-18Pf-SAHH (33 °C) > Tc-18Hs-SAHH (30 °C) > Tc-SAHH (15 °C). These transitions are considered to arise from local structure changes because the global unfolding temperatures of all four apo-SAHHs are greater than 40 °C. The thermal-transition order for the four enzymes clearly indicates the influence of helix-18, but doubtless reflects other properties of the “host enzyme” as well. The NAD+ dissociation rate constants of all four enzymes also exhibit thermal transitions. The thermal-transition temperatures decrease in the order Hs-SAHH (41 °C) > Hs-18Pf-SAHH (38 °C) > Tc-18Hs-SAHH (36 °C) > Tc-SAHH (29 °C). This order matches the expected stability order for helix-18. Here too it may reflect the contribution of other properties of the “host enzyme”. Circular dichroism and differential scanning calorimetry provided the global unfolding temperatures of all fully reconstituted holoenzymes which are around 63 °C and much higher than the thermal-transition temperatures seen in the temperature dependence of the NAD+ dissociation rate constants. This fact indicates that local structural alterations are the origin of the thermal transitions.
Besides the structure element helix-18 of the C-terminal extension, the β sheet of Rossmann motif has also been identified as playing a role in NAD+ binding (unpublished data). The Rossmann motif can be found in most classical NAD+ binding proteins35. Its βαβαβ unit often associated with an additional β strand, forms a “core”, the minimum secondary structure necessary for binding the cofactor36. In addition, the first 30–35 amino acids of the “core” are a fingerprint region for the identification of dinucleotide binding. There are several conserved characteristics within this fingerprint sequence: 1) a six-residue glycine-rich sequence (GXGXXG) involved in phosphate binding; 2) six conserved positions containing only hydrophobic amino acids; 3) a conserved, negatively charged residue and 4) a conserved, positively charged residue35. Specifically, the six conserved hydrophobic residues are located on the first β-sheet, the first α-helix and the second β-sheet, which form a hydrophobic core and are crucial to pack the β-sheets against the α-helix in crucial secondary-structure interactions36. Moreover, a second repeated βαβαβ unit, related to the first one by a two-fold rotation, can usually be found in NAD(P)+ binding proteins36.
There are two classical Rossmann motifs seen in each subunit of SAHH: one in the substrate-binding domain and the other in the cofactor-binding domain. The Rossmann motif in the NAD+ binding domain has a second repeated βαβαβ unit. Alignment of the fingerprint sequences (first 30–35 amino acids) of the Rossmann motif in the NAD+-binding domain of human and parasitic SAHHs reveals that the two small conserved hydrophobic residues on the first β-sheet of the Rossmann motif are conserved in Hs-SAHH and Pf-SAHH, but replaced by two hydrophilic residues in Tc-SAHH and Ld-SAHH. The replacement of the hydrophobic residues by two hydrophilic residues could weaken hydrophobic interactions and increase the flexibility of the hydrophobic core formed by the six hydrophobic residues and other residues. Such difference could influence NAD+ association, dissociation, and affinity.
The principle of our study of the first β sheet of the Rossmann motif is similar to that of our study of the helix-18. The β-sheet propensity estimates of the first β sheet of the Rossmann motif predict a greater stability of this sheet in Hs-SAHH than in parasitic SAHHs. The enzymes with more stable β-sheets are expected to bind NAD+ more tightly. Parasitized Hs-SAHH and humanized Tc-SAHH were created by mutations in the first β-sheet of the Rossmann motif. Systematic kinetic and thermodynamic studies were performed on these mutants. As expected, parasitized Hs-SAHH shows weaker cofactor binding affinity than Hs-SAHH while humanized Tc-SAHH shows stronger cofactor binding affinity than Tc-SAHH. The affinity changes indicate the role of this β-sheet in differential properties of the human and parasitic enzymes.
These results confirm that structural distinctions between Hs-SAHH and Tc-SAHH cause the observed differential kinetic properties and thermodynamic properties. Thus it seems a promising avenue for anti-parasitic therapy to design inhibitors targeted on the cofactor binding site of SAHH.
6.3. Applications of NAD(/H) analogues as selective inhibitors of Hs-SAHH and Tc-SAHH (See article by Li et al in this issue)
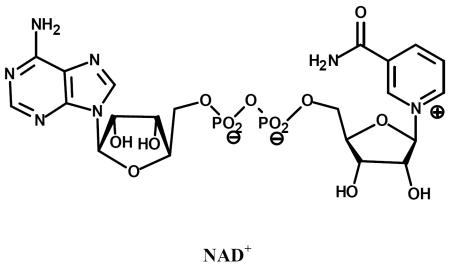
Analogues of NAD+ and its reduced form NADH can be divided into two categories: (a) modifications in the nicotinamide part; (b) modifications in the adenine part. S-NAD and S-NADH are the thione analogues of natural cofactors, modified in the nicotinamide part. These two analogues have been reported to exhibit time-dependent inactivation of both Tc-SAHH and Hs-SAHH but they show significant inactivation on Tc-SAHH during time periods where they reduce the activity of Hs-SAHH very little. Under the competition of natural cofactor NAD+ (50 μM) at 37 °C, S-NADH reaches an apparently complete inactivation of Tc-SAHH within 20 mins. In contrast, S-NAD+ reaches and maintains an apparent equilibrium of inactivation at 50% activity of Tc-SAHH. Under the same conditions, Hs-SAHH loses only 10% of its activity in the presence of S-NAD or S-NADH within 20 mins and reaches an apparent equilibrium inhibition only after twelve hours of incubation.
So far seven analogues of NAD and their reduced forms have been tested on Tc-SAHH and Hs-SAHH using the same approach (competition against 50 μM NAD+). H-NAD/H, C-NAD/H, O-NAD are NAD/H analogues modified in the nicotinamide part. NGD, NHD/H and ethno-NAD are NAD/H analogues modified in the adenine part. All structures are given in Table 1. Investigations of these analogues of Hs-SAHH and Tc-SAHH were carried out at 37 °C in the presence of 50 μM NAD+ to simulate in vivo concentrations. None of these analogues, either in oxidized form or reduced form, can inactivate Hs-SAHH by more than 4% within 6 mins. However, because cofactor NAD+ dissociates 90-fold faster from Tc-SAHH than from Hs-SAHH, the analogues enjoy a greater probability of entrance to a vacant cofactor binding site with Tc-SAHH, thus resulting in significant selective inactivation. S-NADH and H-NADH are the best of the seven analogues, leading to more than 90% inactivation of Tc-SAHH.
Table 1.
NAD+ and its analoguesa.
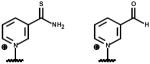
| Name | Structure |
|---|---|
| left: Thionicotinamide adenine dinucleotide (S-NAD) right: 3-Pyridinealdehyde adenine dinucleotide (H-NAD) |
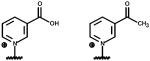
|
| left: Nicotinic acid adenine dinucleotide (O-NAD) right: 3-Acetylpyridine adenine dinucleotide (C-NAD) |
|
| left: Nicotinamide hypoxanthine dinucleotide (NHD) right: Nicotinamide guanine dinucleotide (NGD) |
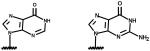
|
| Nicotinamide 1, N6-ethenoadenine dinucleotide (etheno-NAD) |
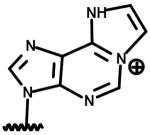
|
Reproduced from the paper by Li et al. in this issue.
In summary, several analogues of NAD and NADH achieve selective inhibition under conditions of competition with NAD+. NADH analogues are better candidates for selective inhibition of Tc-SAHH because SAHH binds NADH more strongly than NAD+. Analogues with modifications in the nicotinamide group do not exhibit cofactor function but, except for O-NAD, they are strong competitive inhibitors. Analogues with modifications in the adenine part show partial cofactor function, but are more weakly bound than NAD+ to Hs-SAHH by four orders of magnitude. They are more weakly bound than NAD+ to Tc-SAHH by two orders of magnitude. These observations suggest that the adenine group of NAD plays a negligible role in cofactor function but makes a contribution to the cofactor-binding interaction.
The successful in-vitro inhibition by NAD/H analogues of Tc-SAHH, but not Hs-SAHH, support the design of selective inhibitors targeted on the cofactor binding site of SAHH. Rational modifications of the structures of NAD and NADH may be capable of generating candidate anti-parasitic drugs, especially for the treatment of Chagas disease.
Acknowledgement
Research on this subject at The University of Kansas has been supported by Grant GM-29332 from the National Institute of General Medical Sciences and by the K-INBRE Bioinformatics Core, NIH Grant P20 RR016475.
7. References
- 1.Turner MA, Yang X, Yin D, Kuczera K, Borchardt RT, Howell PL. Structure and function of S-adenosylhomocysteine hydrolase. Cell Biochem Biophys. 2000;33(2):101–25. doi: 10.1385/CBB:33:2:101. [DOI] [PubMed] [Google Scholar]
- 2.Yin D, Yang X, Yuan C-S, Borchardt RT. Mechanism-based S-adenosyl-L-homocysteine hydrolase inhibitors in the search for broad-spectrum antiviral agents. In: Torrence PF, editor. Biomedical Chemistry: Applying Chemical Principles to the Understanding and Treatment of Disease. John Wiley & Sons, Inc; New York: 2000. pp. 41–71. [Google Scholar]
- 3.Chiang PK. Biological effects of inhibitors of S-adenosylhomocysteine hydrolase. Pharmacol Ther. 1998;77(2):115–34. doi: 10.1016/s0163-7258(97)00089-2. [DOI] [PubMed] [Google Scholar]
- 4.De Clercq E. Carbocyclic adenosine analogues as S-adenosylhomocysteine hydrolase inhibitors and antiviral agents: recent advances. Nucleosides Nucleotides. 1998;17(1–3):625–34. doi: 10.1080/07328319808005205. [DOI] [PubMed] [Google Scholar]
- 5.Robins MJ, Wnuk SF, Yang X, Yuan CS, Borchardt RT, Balzarini J, De Clercq E. Inactivation of S-adenosyl-L-homocysteine hydrolase and antiviral activity with 5',5',6',6'-tetradehydro-6'-deoxy-6'-halohomoadenosine analogues (4'-haloacetylene analogues derived from adenosine) J Med Chem. 1998;41(20):3857–64. doi: 10.1021/jm980163m. [DOI] [PubMed] [Google Scholar]
- 6.Schnyder G, Roffi M, Pin R, Flammer Y, Lange H, Eberli FR, Meier B, Turi ZG, Hess OM. Decreased rate of coronary restenosis after lowering of plasma homocysteine levels. N Engl J Med. 2001;345(22):1593–600. doi: 10.1056/NEJMoa011364. [DOI] [PubMed] [Google Scholar]
- 7.Ashfield-Watt PA, Moat SJ, Doshi SN, McDowell IF. Folate, homocysteine, endothelial function and cardiovascular disease. What is the link? Biomed Pharmacother. 2001;55(8):425–33. doi: 10.1016/s0753-3322(01)00125-1. [DOI] [PubMed] [Google Scholar]
- 8.Ueland PM, Refsum H, Brattstrom L. Plasma homocysteine and cardicascular disease. In: Francis RB Jr., editor. Arteriosclerotic Cardiovascular Disease, Hemostasis and Endothelial Function. Marcel Dekker; New York: 1992. pp. 183–196. [Google Scholar]
- 9.Kruman II, Kumaravel TS, Lohani A, Pedersen WA, Cutler RG, Kruman Y, Haughey N, Lee J, Evans M, Mattson MP. Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer's disease. J Neurosci. 2002;22(5):1752–62. doi: 10.1523/JNEUROSCI.22-05-01752.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang X, Borchardt RT. Overexpression, purification, and characterization of S-adenosylhomocysteine hydrolase from Leishmania donovani. Arch Biochem Biophys. 2000;383(2):272–80. doi: 10.1006/abbi.2000.2087. [DOI] [PubMed] [Google Scholar]
- 11.Parker NB, Yang X, Hanke J, Mason KA, Schowen RL, Borchardt RT, Yin DH. Trypanosoma cruzi: molecular cloning and characterization of the S-adenosylhomocysteine hydrolase. Exp Parasitol. 2003;105(2):149–58. doi: 10.1016/j.exppara.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 12.Nakanishi M, Yabe S, Tanaka N, Ito Y, Nakamura KT, Kitade Y. Mutational analyses of Plasmodium falciparum and human S-adenosylhomocysteine hydrolases. Mol Biochem Parasitol. 2005;143:146–151. doi: 10.1016/j.molbiopara.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 13.Henderson DM, Hanson S, Allen T, Wilson K, Coulter-Karis DE, Greenberg ML, Hershfield MS, Ullman B. Cloning of the gene encoding Leishmania donovani S-adenosylhomocysteine hydrolase, a potential target for antiparasitic chemotherapy. Mol Biochem Parasitol. 1992;53(1–2):169–183. doi: 10.1016/0166-6851(92)90019-g. [DOI] [PubMed] [Google Scholar]
- 14.Shuman S. The mRNA capping apparatus as drug target and guide to eukaryotic phylogeny. Cold Spring Harb Symp Quant Biol. 2001;66:301–12. doi: 10.1101/sqb.2001.66.301. [DOI] [PubMed] [Google Scholar]
- 15.Teixeira RA, Nitz N, Guimaro MC, Gomes C, Santos-Buch CA. Chagas disease. Postgrad Med J. 2006;82(974):788–798. doi: 10.1136/pgmj.2006.047357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang X, Hu Y, Yin DH, Turner MA, Wang M, Borchardt RT, Howell PL, Kuczera K, Schowen RL. Catalytic strategy of S-adenosyl-L-homocysteine hydrolase: transition-state stabilization and the avoidance of abortive reactions. Biochemistry. 2003;42(7):1900–9. doi: 10.1021/bi0262350. [DOI] [PubMed] [Google Scholar]
- 17.Wang M, Borchardt RT, Schowen RL, Kuczera K. Domain motions and the open-to-closed conformational transition of an enzyme: a normal mode analysis of S-adenosyl-L-homocysteine hydrolase. Biochemistry. 2005;44(19):7228–39. doi: 10.1021/bi047524m. [DOI] [PubMed] [Google Scholar]
- 18.Wang M, Unruh JR, Johnson CK, Kuczera K, Schowen RL, Borchardt RT. Effects of ligand binding and oxidation on hinge-bending motions in S-adenosyl-L-homocysteine hydrolase. Biochemistry. 2006;45(25):7778–86. doi: 10.1021/bi0523106. [DOI] [PubMed] [Google Scholar]
- 19.Hu Y, Komoto J, Huang Y, Gomi T, Ogawa H, Takata Y, Fujioka M, Takusagawa F. Crystal structure of S-adenosylhomocysteine hydrolase from rat liver. Biochemistry. 1999;38(26):8323–33. doi: 10.1021/bi990332k. [DOI] [PubMed] [Google Scholar]
- 20.Takata Y, Yamada T, Huang Y, Komoto J, Gomi T, Ogawa H, Fujioka M, Takusagawa F. Catalytic mechanism of S-adenosylhomocysteine hydrolase. Site-directed mutagenesis of Asp-130, Lys-185, Asp-189, and Asn-190. J Biol Chem. 2002;277(25):22670–6. doi: 10.1074/jbc.M201116200. [DOI] [PubMed] [Google Scholar]
- 21.Yin D, Yang X, Hu Y, Kuczera K, Schowen RL, Borchardt RT, Squier TC. Substrate binding stabilizes S-adenosylhomocysteine hydrolase in a closed conformation. Biochemistry. 2000;39(32):9811–8. doi: 10.1021/bi000595a. [DOI] [PubMed] [Google Scholar]
- 22.Hu C, Fang J, Borchardt RT, Schowen RL, Kuczera K. Molecular dynamics simulations of domain motions of substrate-free S-adenosyl- L-homocysteine hydrolase in solution. Proteins. 2008;71(1):131–43. doi: 10.1002/prot.21664. [DOI] [PubMed] [Google Scholar]
- 23.Palmer JL, Abeles RH. Mechanism for enzymatic thioether formation. Mechanism of action of S-adenosylhomocysteinase. J Biol Chem. 1976;251(18):5817–9. [PubMed] [Google Scholar]
- 24.Palmer JL, Abeles RH. The mechanism of action of S-adenosylhomocysteinase. J Biol Chem. 1979;254(4):1217–26. [PubMed] [Google Scholar]
- 25.Hu Y, Yang X, Yin DH, Mahadevan J, Kuczera K, Schowen RL, Borchardt RT. Computational characterization of substrate binding and catalysis in S-adenosylhomocysteine hydrolase. Biochemistry. 2001;40(50):15143–52. doi: 10.1021/bi015690d. [DOI] [PubMed] [Google Scholar]
- 26.Porter DJ, Boyd FL. Mechanism of bovine liver S-adenosylhomocysteine hydrolase. Steady-state and pre-steady-state kinetic analysis. J Biol Chem. 1991;266(32):21616–25. [PubMed] [Google Scholar]
- 27.Porter DJ, Boyd FL. Reduced S-adenosylhomocysteine hydrolase. Kinetics and thermodynamics for binding of 3'-ketoadenosine, adenosine, and adenine. J Biol Chem. 1992;267(5):3205–13. [PubMed] [Google Scholar]
- 28.Porter DJ. S-adenosylhomocysteine hydrolase. Stereochemistry and kinetics of hydrogen transfer. J Biol Chem. 1993;268(1):66–73. [PubMed] [Google Scholar]
- 29.Yuan C-S, Liu S, Wnuk S, Robins MJ, Borchardt RT. Design and synthesis of S-adenosylhomocysteine hydrolase as broad-spectrum antiviral agents. In: De Clercq E, editor. Advances in Antiviral Drug Design. Vol. 2. JAI press, Inc; Greenwicl, CT: 1996. pp. 41–88. [Google Scholar]
- 30.Wolfe MS, Borchardt RT. S-adenosyl-L-homocysteine hydrolase as a target for antiviral chemotherapy. J Med Chem. 1991;34:1521–30. doi: 10.1021/jm00109a001. [DOI] [PubMed] [Google Scholar]
- 31.Borchardt RT, Keller BT, Patel-Thombre U. Neplanocin A. A potent inhibitor of S-adenosylhomocysteine hydrolase and of vaccinia virus multiplication in mouse L929 cells. J Biol Chem. 1984;259(7):4353–8. [PubMed] [Google Scholar]
- 32.Yuan CS, Wnuk SF, Liu S, Robins MJ, Borchardt RT. (E)-5',6'-didehydro-6'-deoxy-6'-fluorohomoadenosine: a substrate that measures the hydrolytic activity of S-adenosylhomocysteine hydrolase. Biochemistry. 1994;33(40):12305–11. doi: 10.1021/bi00206a038. [DOI] [PubMed] [Google Scholar]
- 33.Li Q-S, Cai S, Borchardt RT, Fang J, Kuczera K, Middaugh CR, Schowen RL. Comparative kinetics of cofactor association and dissociation for the human and trypanosomal s-adenosylhomocysteine hydrolases. 1. Basic features of the association and dissociation processes. Biochemistry. 2007;46(19):5798–809. doi: 10.1021/bi700170m. [DOI] [PubMed] [Google Scholar]
- 34.Li QS, Cai S, Fang J, Borchardt RT, Kuczera Krzysztof, Middaugh CR, Schowen RL. Comparative Kinetics of Cofactor Association and Dissociation for the Human and Trypanosomal S-Adenosylhomocysteine Hydrolases. 2. The Role of Helix 18 Stability. Biochemistry. 2008;47(17):4983–4991. doi: 10.1021/bi800175g. [DOI] [PubMed] [Google Scholar]
- 35.Rossmann MG, Liljas A, Brandén CI, Banaszak LJ. Evolutionary and structural relationships among dehydrogenases. In: Boyer PD, editor. The Enzymes. 3rd edn XI. Academic Press; New York: 1975. pp. 61–102. [Google Scholar]
- 36.Wierenga RK, De Maeyer MCH, Hol WGJ. Interaction of pyrophosphate moieties with helixes in dinucleotide binding proteins. Biochemistry. 1985;24(6):1346–1357. [Google Scholar]
- 37.Elrod P, Zhang J, Yang X, Yin D, Hu Y, Borchardt RT, Schowen RL. Contributions of active site residues to the partial and overall catalytic activities of human S-adenosylhomocysteine hydrolase. Biochemistry. 2002;41(25):8134–42. doi: 10.1021/bi025771p. [DOI] [PubMed] [Google Scholar]
- 38.Cai S, Li Q-S, Borchardt RT, Kuczera K, Schowen RL. The antiviral drug ribavirin is a selective inhibitor of S-adenosyl-L-homocysteine hydrolase from Trypanosoma cruzi. Bioorganic and Medicinal Chemistry. 2007;15:7281–7287. doi: 10.1016/j.bmc.2007.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]