

Abstract
We utilized mouse models to elucidate the immunologic mechanisms of functional graft loss during mixed antibody mediated rejection of renal allografts (mixed AMR), in which humoral and cellular responses to the graft occur concomitantly. Although the majority of T cells in the graft at the time of rejection were CD8 T cells with only a minor population of CD4 T cells, depletion of CD4 but not CD8 cells prevented acute graft loss during mixed AMR. CD4 depletion eliminated anti-donor alloantibodies and conferred protection from destruction of renal allografts. ELISPOT revealed that CD4 T effectors responded to donor alloantigens by both the direct and indirect pathways of allorecognition. In transfer studies, CD4 T effectors primed to donor alloantigens were highly effective at promoting acute graft dysfunction, and exhibited the attributes of effector T cells. Laser capture microdissection and confirmatory immunostaining studies revealed that CD4 T cells infiltrating the graft produced effector molecules with graft destructive potential. Bioluminescent imaging confirmed that CD4 T effectors traffic to the graft site in immune replete hosts. These data document that host CD4 T cells can promote acute dysfunction of renal allografts by directly mediating graft injury in addition to facilitating anti-donor alloantibody responses.
Keywords: antibody mediated rejection, T cell mediated rejection, graft infiltrating lymphocytes, adoptive transfer, ELISPOT
Introduction
Despite the now routine nature of clinical renal transplantation, the adaptive immune response to transplanted tissues remains poorly defined. Clearly, both the cellular and humoral arms of the immune response have the potential to contribute to the immunologic destruction of renal allografts, but the relative contributions of the individual pathways remain unclear. There is compelling evidence that antibodies to donor alloantigens are causally related to destruction of clinical renal transplants (1). For example, deposition of complement split products such as C4d on the graft peritubular capillaries (PTC) correlates closely with the presence of circulating donor-reactive antibodies and eventual development of graft dysfunction (2–5). Moreover, antibodies reactive with the graft endothelium promote subclinical alterations in graft endothelial cells (6, 7). However, the vast majority of antibody mediated rejection (AMR) is accompanied by concomitant T-cell infiltration (mixed AMR) (8), raising the possibility that T cells contribute to development of graft dysfunction. Consistent with this possibility, treatment with anti-T cell reagents reverse mixed AMR rejection episodes (9). However, the salient mechanisms of graft injury in this common transplant scenario remain largely a matter of speculation.
We have previously defined the mechanisms of AMR of human renal allografts (10). We herein used mouse models to elucidate the role of host T cells in promoting acute loss of renal allografts during mixed AMR episodes. We provide evidence that CD4 T cells not only play a dominant role in promoting acute graft dysfunction in this rejection scenario by facilitating anti-donor antibody responses but also serve as T effectors that directly mediate graft injury. Surprisingly, these data indicate that CD8 T cells play little if any role in promoting graft dysfunction during mixed AMR. These data provide mechanistic insight into an important clinical problem, and have implications for effective management of clinical renal allograft recipients.
Materials and Methods
Mice
C57Bl/6 (B6, H-2b), BALB/c and DBA/2 (H-2d), FVB/N (H-2q), CD8 KO (B6.129S2-Cd8atm1Mak/J), and RAG−KO (B6;129S7-Rag1tm1Mom/J)mice were purchased from Jackson Laboratories (Bar Harbor, MA). Mice transgenic for firefly luciferase on the B6 background (L2G85.B6) were a kind gift from Dr. Robert Negrin (Stanford, CA). All mice were housed and treated in accordance with Animal Care Guidelines established by the National Institute of Health and The Ohio State University. All experiments described in this manuscript were approved by the OSU IACUC.
ELISPOT assays
Splenic lymphocytes (SC) were isolated from skin primed renal allograft rejectors or controls and CD4 T cells were purified using reagents and columns from Miltenyi Biotec (San Diego, CA). The resulting cells were >95% CD4+CD3+ cells. Unseparated or purified CD4+ T cells were cultured with irradiated DBA/2 splenocytes (SC) for 24 hours and assayed for IFNG or IL-17 production using kits from R&D Systems (Minneapolis, MN). The resulting spots were counted with an ImmunoSpot Series I analyzer (Cellular Technology, Cleveland, OH).
T cell depletion
CD8 T cells were depleted by treatment of mice with 100 mg mixture of monoclonal antibodies to CD8 (TIB 105 and YTS 169) on days −3, −2, −1, +5, and +10 relative to renal allograft transplantation. CD4 T cells were depleted by treatment with 100 mg of mAb GK1.5 on days −3, −2, −1 relative to renal allograft transplantation. Treatment strategies were confirmed to be effective in eliminating the corresponding cells from the graft site.
Histologic examination
3 μm sections of paraffin embedded grafts were stained with hematoxylin and eosin (H&E), trichrome, or immunostained for CD4d deposition using an antibody provided by Dr. Wink Baldwin of the Cleveland Clinic. For immunohistochemistry (IHC) studies, 4u frozen sections of rejecting renal allografts were stained singly with mAb to mouse CD8 (BD Biosciences, San Diego, CA) or doubly stained with anti-CD4 mAb (BD Biosciences), in combination with biotinylated rabbit anti-rat IgG, mouse adsorbed (Vector Laboratories, Burlingame, CA) visualized with the chromagen vina green (Biocare) followed byprimary rabbit polyclonal antibodies to FasL (Abcam ab15285, Cambridge, MA), Granzyne B (Abcam ab4059), perforin (Santa Cruz Biotechnology sc-9105, Dallas Tx), or IFNG (Life technologies AMC4034, Grand Island, NY) followed by anti-rabbit IgG-alkaline phosphatase (Biocare) visualized with warp red chromagen (Biocare), then counterstained with Richard Allan Hematoxylin (Fisher Scientific, Pittsburgh, PA). All studies were performed in the Pathology Core Laboratory at the Ohio State University Wexner Medical Center.
Isolation of graft infiltrating lymphocytes
After removal of the renal capsule, the graft cortex was cut into approximately 1 mm3 pieces, rinsed to remove contaminating peripheral blood, then incubated for 30 min in DMEM:F12 (50:50) containing 0.1% collagenase (type IV; Worthington Biochemical), 0.1% soybean trypsin inhibitor (Sigma-Aldrich), and 0.01% DNase I (Boehringer Mannheim). Following vigorous agitation, the resulting cell suspension was centrifuged on Lympholyte-M (Cedarlane Laboratories) to isolate lymphocytes.
Flow Cytometry
Flow cytometry was performed using a FACSCalibur (BD Biosciences). Data were analyzed using FlowJo (Tree Star Inc, Ashland, OR). and WinMDI software (Scripps Institute, Lo Jolla, CA). The percentage of positive cells for a given marker was based on cutoff points chosen to exclude >99% of the negative control population. Fluorochrome-conjugated mAbs to mouse CD3e (145-2C11), CD4 (GK1.5), CD8a (53-6.7), CD8b (H35-17.2), CD44 (IM7), and CD103 (M290) and the respective species- and isotype matched negative control mAbs were purchased from BD Biosciences.
The presence of donor-reactive antibodies was determined by the ability of sera to bind to DBA/2 splenocytes detected with goat anti-mouse IgG (γ chain specific) antibodies (Southern Biotechnology, Birmingham, AL) measured as mean fluorescence intensity (MFI) of DBA/2 SC targets. Background fluorescence was determined for each subtype by taking the MFI plus three standard deviations of sera from five naïve C57BL/6 mice. Negative controls included SC plus each detection antibody; MFI values ranged from 25 to 35.
Bioluminescent imaging (BLI)
Purified CD4 T cells were isolated from DBA/2 primed L2G85.B6 mice and then adoptively transferred into syngeneic recipients previously primed to donor (DBA/2) alloantigens and transplanted with either renal isografts or DBA/2 renal allografts. Imaging was performed 3–5 days after transplantation to localize CD4 T cells by injecting mice i.v. with 4 mg D-luciferin. An In Vivo Imaging System 100 (PerkinElmer, Waltham, MA) was used for data acquisition. Living Image software (PerkinElmer) was used for data analysis. All BLI studies were performed at the Small Animal Research Imaging Core at the OSU College of Veterinary Medicine.
Lasar capture microdissection (LCM)
RAG KO mice received DBA/2 renal allografts and after one week were injected intravenously with 15 million purified CD4 T cells from CD8 KO donors on the C57Bl/6 background that had previously rejected DBA/2 skin allografts. For LCM, CD4 T cells in the graft were identified using anti-CD4 mAb (Abcam) followed by captures. RNA was then isolated from CD4+ cells in the graft and analyzed by qRT-PCR to determine mRNA levels. Data were normalized to values obtained for kidneys from naive DBA/2 mice.
Statistical analysis
Analyses were performed using Prism 5 program (GraphPad Software). Graft survival curves were compared using the Log-rank test. Other analyses performed included Students t-test and one-way ANOVA. Differences between means of experimental groups were considered significant at p< 0.05.
RESULTS
Key role for host CD4 T cells in promoting acute loss of renal allografts during mixed AMR
We have previously described a mouse model of mixed AMR in which B6 hosts are primed to DBA/2 alloantigens with a DBA/2 skin allograft 14 to 21 days prior to receiving a DBA/2 renal allograft (11). As shown in Figure S1, identical results were obtained with recipients primed to donor alloantigens at times ranging from 11–91 days prior to kidney transplantation, indicating that the time of renal transplantation relative to the priming stage was not critical. As shown in Fig. 1A, FACS analyses of DBA/2 renal allografts undergoing acute rejection revealed a predominance of CD8 T cells in the kidney allografts accompanied by a minor population of CD4 T cells at the time of rejection. To assess the functional contributions of CD4 and CD8 T cells in this model, we depleted CD4 or CD8 T cells in sensitized mice at the time of kidney transplantation. Surprisingly, CD4 depletion was highly effective at preventing graft loss whereas CD8 depletion was totally ineffectual (Figure 1B). Moreover, sensitized CD8 KO mice rejected grafts acutely (at days 9–10), documenting that CD8 T cells are not required for acute graft dysfunction in this model. It should be noted that mortality of hosts depleted of CD4 T cells was nearly 40% (Figure 1B). The vast majority of these deaths occurred immediately after the final injection suggesting the involvement of a toxic agent or an immune reaction rather than rejection per se.
Figure 1.
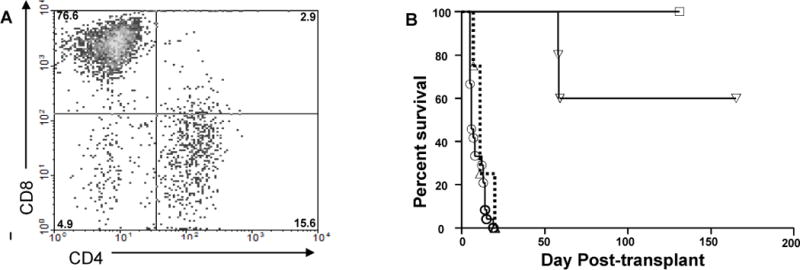
Host CD4 T cells represent a minority of T cells infiltrating renal allografts but play a dominant role in promoting acute graft dysfunction. C57Bl/6 mice were primed with DBA/2 skin allografts then received DBA/2 renal allografts as described in Materials and Methods. A. Cells from grafts undergoing rejection (serum creatinine > 70 mmol/L) were isolated and subjected to multicolor FACS analysis using mAbs to CD3, CD4, and CD8. Numbers in each quadrant represent percentages of total CD3+ T lymphocytes. Data are representative of four independent experiments. B. Primed C57BL/6 recipients received DBA/2 renal allografts and were either untreated (●, n=24) or depleted of CD8 T cells (▲, n=9) or CD4 T cells (▼, n=16) at the time of transplantation.
A disadvantage of the above model is that DBA/2 renal allografts are spontaneously accepted in B6 hosts, a scenario quite unlike that seen clinically. However, as shown in Figure 2A, depletion of CD4, but not CD8, T cells also prevented graft loss when BALB/c renal allografts were transplanted into naïve B6 hosts, a model in which grafts undergo slow spontaneous rejection in naive hosts, similar to the clinical situation. Note that rejection in this model also exhibits classic features of mixed AMR with deposition of complement split products on the peritubular capillaries in the context of massive graft inflammation (Figures 2B, C and D). These data are consistent with the possibility that CD8-independent rejection extends to mixed AMR occurring in non-sensitized recipients.
Figure 2.
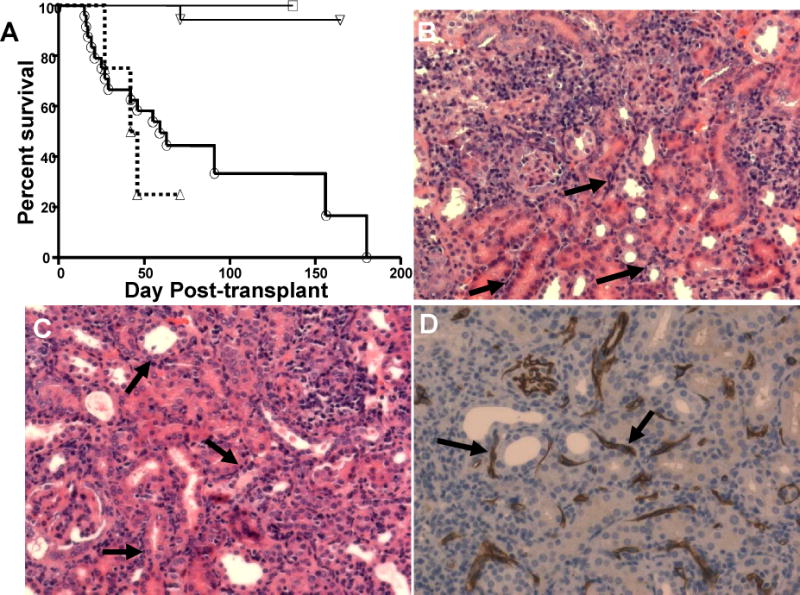
Mixed AMR in non-primed renal allograft recipients. (A.) Survival of BALB/c renal allografts in naïve C57BL/6 (B6) recipients. Experimental group indicators as for Figure 1B. Rejected renal allografts (BALB/c->B6) were harvested at the time of rejection. (B and C) H&E sections demonstrate focal inflammatory cell infiltrates, peri-tubular capillary (PTC) margination (arrows), H&E, 200×. (D) C4d in PTC (arrows) as detected by immunoperoxidase staining. Data are representative of four or more grafts.
Figure 3A shows that sensitization to donor alloantigens followed by renal transplantation was accompanied by an ~4-fold increase in the frequency of CD4 T cells capable of responding to donor alloantigens via production of IFNG. The response was specific in that the same cells failed to mount responses in lieu of stimulation (responders aloneAs shown in Fig 3B, mice primed to skin allografts alone exhibited an increase in frequencies of CD4 T cells producing IFNG in response to DBA/2 alloantigens approximately one half of that of hosts receiving both skin and renal allografts, suggesting that the two allografts types make comparable contributions to the increased frequency. The response was highly specific to DBA/2 alloantigens as the frequency of splenic CD4 cells responding to syngeneic or third party stimulators did not significantly increase.). Note that a true third party stimulator is not shown in Fig. 3A as the BALB/c and DBA/2 strains share the H-2d haplotype.
Figure 3.
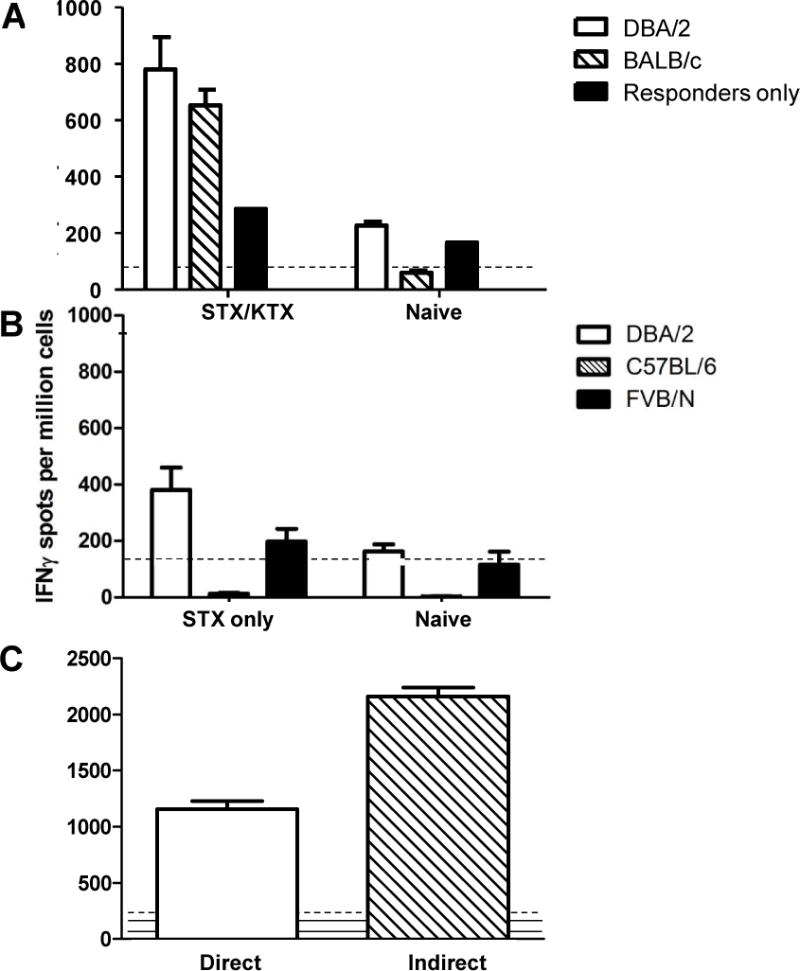
Sensitization to donor alloantigens followed by renal transplantation increases the frequency of donor-reactive CD4 T cells. A. Purified splenic CD4 T cells from B6 hosts that had rejected both renal and skin allografts from DBA/2 donors (STX/RTX) or from naïve recipients (Naïve) were tested in an IFNG ELISPOT assay against the indicated targets. B. Purified splenic CD4 T cells from B6 hosts that had rejected DBA/2 skin allografts only (STX only) or from normal B6 hosts (Naïve) were tested in an IFNG ELISPOT assay for reactivity to B6 (syngeneic), DBA/2 (allogeneic), or FVB/N (third party) SC targets. C. Unseparated SC from primed B6 hosts at the time of rejection of DBA/2 renal allografts were tested in an IFNG ELISPOT assay against either intact (direct) or subcellular (indirect) irradiated stimulator cells. Data shown are the mean (+ SEM) IFNG spots per million cells. Dashed lines show reactivity of stimulators only; solid lines show reactivity of responders only.
Note that splenic CD4 T cells mounted comparable responses to MHC-matched BALB/c (H-2d) (MHC matched with the donor strain but disparate for multiple mHA) stimulator cells (Fig. 3) indicating specificity for MHC-encoded alloantigens via the direct pathway of allorecognition. As shown in Figure S2, the frequency of T cells producing IL-17 in response to alloantigen mirrored that of CD4 T cells producing IFNG, suggesting that all potential mediators of graft injury are increased following combined skin/renal allotransplantation.
Highly purified splenic CD4 T cells were used as responders in the above assays thereby precluding the detection of indirect alloantigen presentation due to the absence of host APC. However, as shown in Figure 3B, unseparated splenocytes (SC) mounted vigorous responses to subcellular alloantigen. Thus, these data indicate that sensitization to donor alloantigens followed by renal transplantation increases the frequency of T cells capable of responding to renal allografts by both the direct and indirect pathways of allorecognition.
Antibody responses in CD4 depleted recipients
To assess the impact of CD4 depletion on the development of alloantibodies in this model, serum from CD4 depleted recipients was collected over time and analyzed for donor-reactive antibodies by FACS analyses. Consistent with the findings of Steele et al. (12), CD4 depletion completely abrogated donor-reactive IgG antibody levels elicited in response to skin allografts (Fig. 4A). Staining for complement split products (C4d) in the graft peritubular capillaries (Fig. 4B) failed to reveal C4d deposition in the vast majority (14/16) of grafts (Figure 4B, left). In contrast, grafts in CD4 replete (untreated) mice showed strong positivity (Figure 4B, right). These data indicate that the loss of antibodies detected in the circulation in this model does not reflect absorption by the graft.
Figure 4.
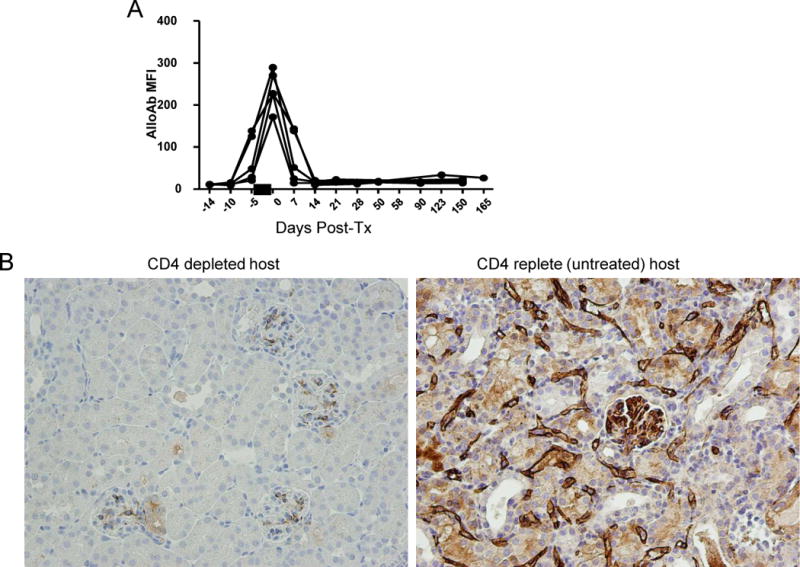
Antibody responses in CD4 depleted recipients. A. Sera were collected from skin primed recipients (n=5) at the indicated times post-transplantation and tested for reactivity to DBA/2 splenocytes. IgG binding was detected by the mean fluorescence intensity (MFI) of fluorochrome conjugated anti-mouse IgG for each sample. Each line represents a single recipient mouse. The black bar indicates the time of CD4 depletion (days −3, −2, −1). B. Immunostaining for C3d deposition in peritubular capillaries (PTC) (arrows).
CD4 T effectors but not alloantibodies promote acute injury to renal allografts
To further assess the contribution of anti-donor alloantibodies to acute destruction of renal allografts, we adoptively transferred alloantibodies into Rag KO mice (B6;129S7-Rag1tm1Mom/J) bearing DBA/2 renal allografts. Rag KO mice lack T and B cells (13) and therefore do not reject transplanted tissues without transferred antibodies or T cells. Antibodies examined in these studies included sera from sensitized WT mice that had rejected DBA/2 renal allografts, and a mAb (HB159) directed to donor MHC class I (H-2Kd) alloantigens. As shown in Fig. 5A, sera from mice that had rejected renal allografts did not cause graft dysfunction nor did anti-donor MHC I mAb even when administered at high doses. Long term followup confirmed the absence of detectable injury out to day 30, and identical (negative) results were obtained when antibodies were transferred prior to renal transplantation (data not shown). Graft survival was not due to an absence of antibodies because these were detectable in recipient sera throughout the course of the experiment (Fig. 5B) and led to antibody deposition (Figure S3A) and fixation of complement (Fig. S3D) in the graft. Notably, comparable doses of HB159 mAb elicit vascular injury to cardiac allografts (14), arguing that the failure to promote injury to renal allografts does not reflect an inadequate dose or specificity of the mAb. Rejector sera did not cause detectable immunoglobulin or complement deposition in RAG KO mice (Figures S3B and S3E), probably because of the low concentration of anti-MHC antibodies that could be achieved through the transfer of sera.
Figure 5.
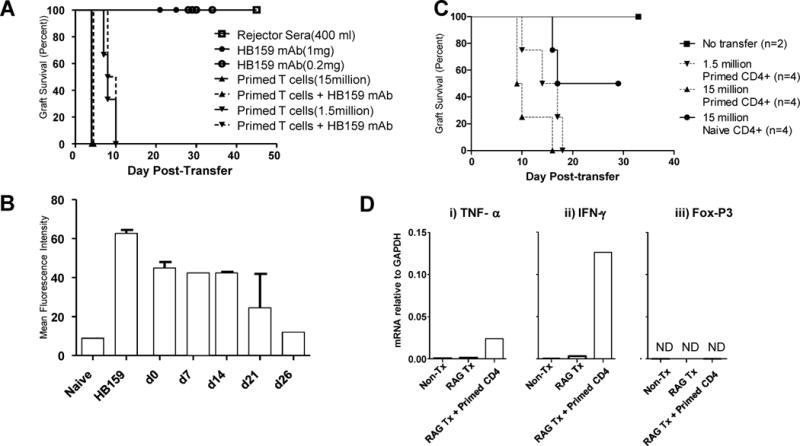
Primed CD4 T cells promote acute destruction of renal allografts. A. Rag−/− mice received DBA/2 renal allografts and after one week were injected intravenously with sera from WT mice that had rejected DBA/2 renal allografts (squares), HB159 monoclonal antibody (circles), or donor-primed T cells (triangles). B. Sera from RAG−/− recipients of DBA/2 renal allografts and HB159 mAb were collected at the indicated times post transplantation, diluted 1:16 and then tested for reactivity to DBA/2 splenocytes. C. Rag−/− mice received DBA/2 renal allografts and after one week were injected intravenously with 15 million naïve CD4+ T cells (solid line, circle) or with primed CD4+ T cells at a dose of 1.5 million (inverted triangle, dashed line)or 15 million (triangle, dashed line). D. Renal allografts were retrieved at the time of rejection in RAG−/− recipients receiving no (RAG TX) or 15 million primed CD4+ T cells (RAG TX + Primed CD4) and RNA isolated from total graft homogenates was analyzed by qRT-PCR to determine the indicated mRNA levels. Negative controls included RNA from normal DBA/2 kidneys (NON-TX); positive controls included RNA from grafts transplanted into primed wildtype recipients (WT TX) undergoing mixed AMR. Data shown in D are mRNA levels relative to GAPDH; Not detected (ND).
In contrast, transferred primed CD4 T cells were highly effective at destroying renal allografts in RAG KO mice as evidenced by rapid graft loss (Figure 5A,C) and histologic injury to the graft (not shown). Note that the combination of HB159 mAb and CD4 T effectors did not change renal allograft survival relative to that of CD4 T effectors alone at either high or low doses (Fig 5A). In experiments not shown, purified CD4 T cells from sensitized CD8 knockout mice yielded identical results, documenting that the destructive capacity observed was not due to contamination with CD8 T cells. As shown in Fig. 5D, CD4 T cells mediating rejection of renal allografts transplanted into RAG KO recipients elicited production of the effector cytokines TNFα, and IFNG in the absence FoxP3 expression consistent with the possibility that CD4 T cells entering the graft were T effectors rather than regulatory T cells. These data suggest that CD4 T effectors are fully capable of promoting renal allograft dysfunction without the involvement of host B cells.
Graft infiltrating CD4 T cells express effector molecules
To gain insight into the mechanisms by which CD4 T cells infiltrating renal allografts cause graft dysfunction, we used laser capture microdissection (LCM). In these experiments, purified CD4 T cells from sensitized CD8 KO mice were adoptively transferred into RAG KO mice. As shown in Fig. 6A, graft infiltrating CD4 T cells produced high levels of FasL (CD95L), granzyme B, perforin and IFNG following entry into the graft. Note that the donor cells in these experiments are homozygous for targeted disruption of the CD8a locus, so do not contain CD8a-expressing cells including CD8 T cells and NK cells, but that the RAG KO recipients potentially do as do DBA/2 renal allografts. However, most if not all leukocytes (CD45-postive cells) in the graft co-expressed CD4 (data not shown) suggesting that non-CD4 T cells do not make a significant contribution to gene expression in these experiments. As shown in Fig. 7, IHC studies confirmed that subsets of CD4 cells expressing the above effector molecules infiltrate the graft site concomitant with acute graft dysfunction, strongly suggesting that these cells are key mediators of the rejection process.
Figure 6.
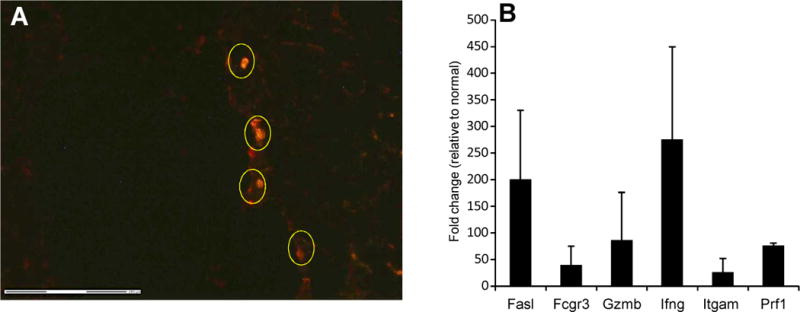
Graft infiltrating CD4 T cells produce mRNA for effector cytokines. RAG KO mice received DBA/2 renal allografts and after one week were injected intravenously with 15 million donor primed purified CD4+ T cells from CD8 KO donors on the C57Bl/6 background that had previously rejected DBA/2 skin allografts. For laser capture microdissection (LCM), CD4+ cells were identified using anti-CD4 antibody followed by captures. RNA was then isolated from CD4+ cells in the graft (A) and analyzed by qRT-PCR to determine mRNA levels (B). Data shown in B are normalized to values obtained for kidneys from non-transplanted DBA/2 mice. C) Cytotoxicity of purified CD4 or CD8 T cells from normal and skin grafted C57BL6 mice to Con A blast targets of DBA/2 or C57BL/6 origin.
Figure 7.
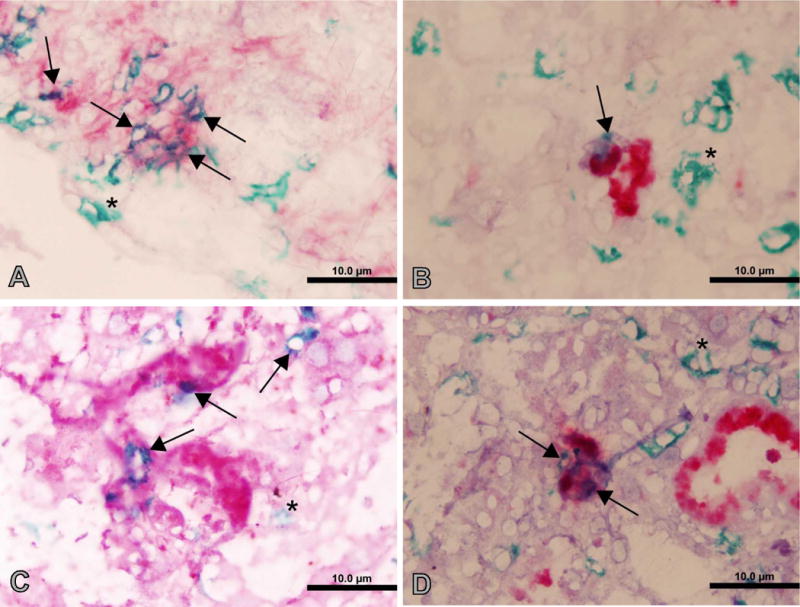
Immunohistochemistry of graft infiltrating CD4 cells. A rejecting renal allograft was stained with anti-CD4 (green) in combination with antibodies to Fasl (A), granzyme B (B), IFNγ (C), or perforin (D) (red). Singly stained CD4 cells are indicated by arrows; doubly stained cells are indicated by astericks.
Primed CD4 T cells migrate to the site of renal allografts
An important limitation of the transfer models described above is that naïve T cells can artifactually acquire effector function following transfer into immunodeficient hosts (15, 16). This was confirmed in our model by transfer of purified CD4 T cells from naïve mice (Fig. 5C). To circumvent this limitation, bioluminescent imaging was used to track migration of donor-primed CD4 T cells transferred into immune replete WT recipients. As shown in Figure 8A, by day 5 post-transplantation, the majority of primed CD4 T cells following renal transplantation resided in the allograft, with the remainder residing in the host spleen. Such accumulation was specific in that transferred CD4 cells failed to accumulate within renal isografts (Figure 8B). These data indicate that host CD4 T cells responding to donor alloantigens in immune replete hosts specifically migrate to the site of the renal allograft at the time of graft injury, consistent with a key role for these cells in direct attack of the graft.
Figure 8.
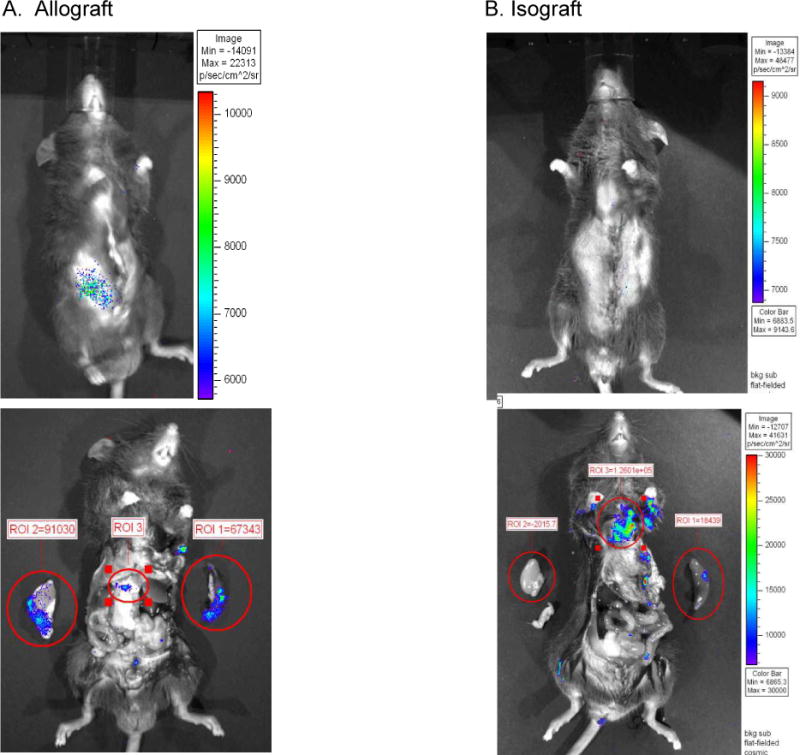
Bioluminescent imaging of CD4+ T cells. Splenic CD4 T cells were isolated from skin primed L2G85.B6 donors and injected intravenously into previously skin primed congenic B6 recipients. Within 24 hours after injection recipients received a DBA/2 renal allograft (A) or a B6 renal isograft (B). Five days after transplant, whole body imaging revealed bioluminescent signal detectable from the renal allograft but not the isograft. Data shown are a whole body image (upper) and an image of exposed organs (lower). The scale to the right of the images indicates the photon count (and hence the relative numbers of CD4 T cells) designated by the different colors. Boxes show relative light emission values (proportional to cell abundance) obtained for the circled regions of interest (ROI) which included the graft, spleen, and other secondary lymphoid organs.
Discussion
The salient finding of the present study is that CD4 T effector cells promote acute dysfunction of renal allografts during mixed AMR, serving both as T helpers for antibody responses and as CD4 T effectors that directly attack the graft. However, we find no evidence that antibodies contribute to acute graft dysfunction in this model. That CD4 T effectors directly promote injury to renal allografts is supported by the results of adoptive transfer studies indicating that host CD4 T effectors are highly efficient at eliciting functional destruction of renal allografts in both immune deficient (Figure 5A and 5C) and immune sufficient hosts (Figures 1B and 2A). We further show that host CD4 T cells migrate to the site of renal allografts but not isografts in immune replete hosts (Figure 8), and produce cytotoxic effector molecules therein (Figures 6 and 7), consistent with a role in direct attack of the graft.
Our data provide novel insight into mechanisms of renal allograft rejection. Surprisingly, our data indicate CD8 T cells are not necessary in promoting acute dysfunction of renal allografts in either naïve (Fig. 2) or sensitized (Fig. 1) recipients. This finding is in accord with results in mouse cardiac allograft models in which CD8 depletion has a barely discernible impact on rejection events, simply altering the type of CD4 T cell response (TH1 vs. TH2) leading to graft destruction (17). These data also are in line with prior studies showing that CD4 T cells alone are adequate to reject mouse skin and cardiac allografts (18), and that CD4 T effectors can be more efficient in killing activity than CD8 T cells even across MHC I disparities (19). These data argue that classical CD8 T effector populations play little if any role in promoting graft injury during mixed AMR. Our LCM and IHC data showing that a subset of graft infiltrating CD4 T cells express CD95L, perforin, granzyme B, and IFNG following entry into the graft (Figures 6 and 7) provide insight into the mechanisms by which these cells promote acute graft dysfunction. Notably, IFNG production identifies these cells as Th1 cells (as opposed to Th17 or Th2 cells), known to play critical roles in regulating immunopathology (20, 21). Our ELISPOT data (Fig. 3) indicate that host CD4 T cells comprise a mixed population responding to donor alloantigens by both the direct and indirect pathways of allorecognition. The presence of an IgG response, which is known to depend on CD4 T cells recognizing donor alloantigen by the indirect pathway of allorecognition (13), further supports the involvement of the latter cells in our model. Brennan et al. (24) documented that indirect pathway cells are initially a minor population but undergo rapid expansion following transplantation, and there is evidence that that such cells contribute to chronic injury to renal allografts (25). We postulate that direct pathway CD4 T cells directly attack the graft, whereas indirect pathway CD4 T cells provide help for B cell responses, but our data do not discriminate between these possibilities.
The failure of antibodies to elicit acute allograft dysfunction (Figure 5A) was an unexpected finding. This was not due to inadequate doses of antibody because the present study used mAb levels comparable or higher than those used in cardiac allograft studies which document the capacity of this mAb to induce graft injury (26). Moreover, we documented in our studies that excess antibody binding activity (Figure 5B) was present in circulation for >30 days, with clear evidence of antibody and complement deposition at the graft site (Figure S3). Thus, while there is evidence that the historic failure of passively transferred anti-donor antibodies to elicit acute graft loss reflects inadequate antibody titers (27, 28) or an inability of anti-donor mAbs to fix complement (1), this is not a viable explanation in our experiments. We speculate that this difference reflects the fact that unlike the renal allografts examined in this study, cardiac allografts are not life supporting – i.e., these are heterotopic grafts that do not pump blood – and thus are susceptible to immune mechanisms distinct from those that mediate rejection of renal allografts.
Wasowska et al. (29) showed that transfer of donor-specific mAb into Ig KO mice results in acute rejection of mouse cardiac allografts. Given that the IgKO hosts used in this study were undergoing cellular rejection at the time of transfer, these data suggest that an ongoing cellular immune response is critical for development of acute renal allograft dysfunction. Our data indicate that transfer of antibody plus T cells into Rag−/− hosts does not increase the pace of graft loss (Fig. 5A). Based on these data, we postulate that interaction between antibody and non-T cells – i.e., macrophages and/or neutrophils as described by Bickerstaff et al. (28) – is necessary for reconstitution of acute antibody mediated rejection of renal allografts. Our studies indicate that non-T cells are not significantly present during CD4-dependent rejection of renal allografts in RAG KO recipients.
Our data indicate that the peak of the donor-reactive antibody response occurs in this model at the time of the skin allograft rejection then gradually disappears in about 2 weeks following CD4 depletion (Figure 4A). Given that we observe no evidence of C4d deposition in the graft PTC in the vast majority of grafts (14/16) the simplest explanation for these data is that the gradual disappearance of circulating antibodies reflects a lack of cd4 help for anti-donor B cell responses rather than absorption by the graft. Moreover, it should be noted, however, that the interpretation of these data is complicated by the fact that the time of CD4 depletion (day 10 post-priming) in our model coincides with the peak of germinal center formation potentially abrogating the process of affinity maturation and the generation of donor-reactive alloantibodies.
The clinical relevance of our data is uncertain given that the time intervals for priming used in our mouse model are likely much shorter than those that occur in clinical renal allograft recipients. Thus, there is likely inadequate time for development of true memory B cell responses, potentially resulting in incomplete elimination of alloantibodies following CD4 depletion in the clinical situation. While our data (Supplemental Figure 1) indicate that priming at late time points results in survival times similar to those primed earlier, it is not clear that the mechanisms of injury are the same in the two situations given the likely differences in populations of memory cells. This limitation to data interpretation is compounded by the fact that the mechanistic studies reported in this manuscript were performed using samples from mice primed shortly (14 days or less) before renal transplantation.
Our data provide an alternative explanation for the observation that anti-T cell reagents successfully reverse antibody associated graft injury in clinical renal allograft recipients (9, 30). In this scenario, the capacity of anti-B cell therapies such as rituximab and IVIg to reverse rejection episodes may be explained by an impact on anti-donor CD4 T cell responses. In addition to their well documented role in Ab production, B cells play important roles as APC for alloreactive CD4 T cell responses (31, 32), and IVIg preparations contain specificities that impact T cell activation events (33). Based on these data, we postulate that immunosuppressive strategies in current clinical usage ameliorate mixed AMR rejection episodes because they inhibit CD4 T cell responses. Our data support ours and others’ experience that in acute AMR, unless vascular thrombosis develops, subsequent/concomitant cellular rejection may be the most important direct pathogenic factor causing graft injury. However, the role of anti-donor antibodies should not be neglected; there is mounting evidence implicating antibodies in the development of chronic graft injury in human renal allografts, a condition now called chronic AMR (5). It will be important to develop good animal models for chronic AMR to define mechanisms of late loss of renal allografts.
Supplementary Material
Figure S1. Skin primed renal allograft rejection model. C57Bl/6 recipients were primed with DBA/2 skin allografts at day 0 then transplanted with DBA/2 renal allografts at the indicated times. Data shown are survival times of the renal allografts. The brackets show the level of statistical significance for differences between the groups.
Figure S2. Sensitization increases the frequency of immune cells capable of producing IL-17 in response to donor alloantigens. A. Purified splenic CD4+ T cells from primed B6 hosts that had rejected DBA/2 renal allografts at the time of renal allograft rejection (STX/RTX), normal B6 hosts (Naive), or media alone (Media) were tested in an IL-17 ELISPOT assay against intact DBA/2 irradiated stimulators. B. Purified splenic CD4+ T cells from normal B6 hosts (Naive) B6 hosts that had rejected DBA/2 skin allografts only (STX only), or media alone (Media) were tested in an IL-17 ELISPOT assay for reactivity to DBA/2 SC stimulators. Data shown are the mean (+ SEM) IL-17 spots per million cells.
Figure S3. Antibody and C4d deposition in renal allografts following adoptive transfer of alloantibodies. Renal allografts were harvested 30 days after transplantation and Ig (A, B, C) and C4d (D, E, F) were detected by immunohistochemistry. Data are representative of four or more grafts.
Acknowledgments
We gratefully acknowledge the outstanding contributions of Cherri Bott, Viy McGaughy, and Kristin Kovach in providing invaluable expertise and guidance in analyses of graft histology and immunohistochemistry. We also would like to thank Dr. Wink Baldwin for his kind gift of polyclonal antibodies with specificity for CD4d.
These studies were supported by a grant from the National Institutes of Health to G.A.H (AI036532).
Footnotes
Disclosures
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
Supporting materials include supplemental figures S1 – S2 with associated legends. Additional supporting information may be found in the online version of this article.
References
- 1.Baldwin WM, 3rd, Valujskikh A, Fairchild RL. Antibody-mediated rejection: emergence of animal models to answer clinical questions. Am J Transplant. 2010 May;10(5):1135–42. doi: 10.1111/j.1600-6143.2010.03065.x. Epub 2010/03/30. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collins AB, Schneeberger EE, Pascual MA, Saidman SL, Williams WW, Tolkoff-Rubin N, et al. Complement activation in acute humoral renal allograft rejection: diagnostic significance of C4d deposits in peritubular capillaries. J Am Soc Nephrol. 1999 Oct;10(10):2208–14. doi: 10.1681/ASN.V10102208. Epub 1999/10/03. eng. [DOI] [PubMed] [Google Scholar]
- 3.Bohmig GA, Exner M, Habicht A, Schillinger M, Lang U, Kletzmayr J, et al. Capillary C4d deposition in kidney allografts: a specific marker of alloantibody-dependent graft injury. J Am Soc Nephrol. 2002 Apr;13(4):1091–9. doi: 10.1681/ASN.V1341091. Epub 2002/03/26. eng. [DOI] [PubMed] [Google Scholar]
- 4.Nakashima S, Qian Z, Rahimi S, Wasowska BA, Baldwin WM., 3rd Membrane attack complex contributes to destruction of vascular integrity in acute lung allograft rejection. J Immunol. 2002 Oct 15;169(8):4620–7. doi: 10.4049/jimmunol.169.8.4620. Epub 2002/10/09. eng. [DOI] [PubMed] [Google Scholar]
- 5.Colvin RB. Antibody-mediated renal allograft rejection: diagnosis and pathogenesis. J Am Soc Nephrol. 2007 Apr;18(4):1046–56. doi: 10.1681/ASN.2007010073. Epub 2007/03/16. eng. [DOI] [PubMed] [Google Scholar]
- 6.Smith JD, Lawson C, Yacoub MH, Rose ML. Activation of NF-kappa B in human endothelial cells induced by monoclonal and allospecific HLA antibodies. Int Immunol. 2000 Apr;12(4):563–71. doi: 10.1093/intimm/12.4.563. Epub 2000/04/01. eng. [DOI] [PubMed] [Google Scholar]
- 7.Jin YP, Singh RP, Du ZY, Rajasekaran AK, Rozengurt E, Reed EF. Ligation of HLA class I molecules on endothelial cells induces phosphorylation of Src, paxillin, and focal adhesion kinase in an actin-dependent manner. J Immunol. 2002 Jun 1;168(11):5415–23. doi: 10.4049/jimmunol.168.11.5415. Epub 2002/05/23. eng. [DOI] [PubMed] [Google Scholar]
- 8.Nickeleit V, Andreoni K. The classification and treatment of antibody-mediated renal allograft injury: where do we stand? Kidney Int. 2007 Jan;71(1):7–11. doi: 10.1038/sj.ki.5002003. Epub 2006/12/15. eng. [DOI] [PubMed] [Google Scholar]
- 9.Sun Q, Liu ZH, Cheng Z, Chen J, Ji S, Zeng C, et al. Treatment of early mixed cellular and humoral renal allograft rejection with tacrolimus and mycophenolate mofetil. Kidney Int. 2007 Jan;71(1):24–30. doi: 10.1038/sj.ki.5001870. Epub 2006/09/14. eng. [DOI] [PubMed] [Google Scholar]
- 10.Singh N, Sun Q, Nadasdy T, Adams P, Dipaola NR, Pesavento T, et al. The pathogenesis of acute allograft dysfunction in desensitized renal transplant recipients. Clin Transplant. 2012 Jul-Aug;26(4):E402–11. doi: 10.1111/j.1399-0012.2012.01684.x. [DOI] [PubMed] [Google Scholar]
- 11.Bickerstaff A, Pelletier R, Wang JJ, Nadasdy G, DiPaola N, Orosz C, et al. An experimental model of acute humoral rejection of renal allografts associated with concomitant cellular rejection. The American journal of pathology. 2008 Aug;173(2):347–57. doi: 10.2353/ajpath.2008.070391. Epub 2008/06/28. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steele DJ, Laufer TM, Smiley ST, Ando Y, Grusby MJ, Glimcher LH, et al. Two levels of help for B cell alloantibody production. J Exp Med. 1996 Feb 1;183(2):699–703. doi: 10.1084/jem.183.2.699. Epub 1996/02/01. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992 Mar 6;68(5):869–77. doi: 10.1016/0092-8674(92)90030-g. Epub 1992/03/06. eng. [DOI] [PubMed] [Google Scholar]
- 14.Jindra PT, Hsueh A, Hong L, Gjertson D, Shen XD, Gao F, et al. Anti-MHC class I antibody activation of proliferation and survival signaling in murine cardiac allografts. J Immunol. 2008 Feb 15;180(4):2214–24. doi: 10.4049/jimmunol.180.4.2214. Epub 2008/02/06. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldrath AW, Bogatzki LY, Bevan MJ. Naive T cells transiently acquire a memory-like phenotype during homeostasis-driven proliferation. J Exp Med. 2000 Aug 21;192(4):557–64. doi: 10.1084/jem.192.4.557. Epub 2000/08/22. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho BK, Rao VP, Ge Q, Eisen HN, Chen J. Homeostasis-stimulated proliferation drives naive T cells to differentiate directly into memory T cells. J Exp Med. 2000 Aug 21;192(4):549–56. doi: 10.1084/jem.192.4.549. Epub 2000/08/22. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan SY, DeBruyne LA, Goodman RE, Eichwald EJ, Bishop DK. In vivo depletion of CD8+ T cells results in Th2 cytokine production and alternate mechanisms of allograft rejection. Transplantation. 1995 Apr 27;59(8):1155–61. Epub 1995/04/27. eng. [PubMed] [Google Scholar]
- 18.Krieger NR, Yin DP, Fathman CG. CD4+ but not CD8+ cells are essential for allorejection. J Exp Med. 1996 Nov 1;184(5):2013–8. doi: 10.1084/jem.184.5.2013. Epub 1996/11/01. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perez-Diez A, Joncker NT, Choi K, Chan WF, Anderson CC, Lantz O, et al. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood. 2007 Jun 15;109(12):5346–54. doi: 10.1182/blood-2006-10-051318. Epub 2007/03/01. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y, Janeway CA., Jr Interferon gamma plays a critical role in induced cell death of effector T cell: a possible third mechanism of self-tolerance. J Exp Med. 1990 Dec 1;172(6):1735–9. doi: 10.1084/jem.172.6.1735. Epub 1990/12/01. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gattoni A, Parlato A, Vangieri B, Bresciani M, Derna R. Interferon-gamma: biologic functions and HCV therapy (type I/II) (1 of 2 parts) Clin Ter. 2006 Jul-Aug;157(4):377–86. Epub 2006/10/21. eng. [PubMed] [Google Scholar]
- 22.Xu Q, Lee J, Jankowska-Gan E, Schultz J, Roenneburg DA, Haynes LD, et al. Human CD4+CD25low adaptive T regulatory cells suppress delayed-type hypersensitivity during transplant tolerance. J Immunol. 2007 Mar 15;178(6):3983–95. doi: 10.4049/jimmunol.178.6.3983. Epub 2007/03/07. eng. [DOI] [PubMed] [Google Scholar]
- 23.Salama AD, Najafian N, Clarkson MR, Harmon WE, Sayegh MH. Regulatory CD25+ T cells in human kidney transplant recipients. J Am Soc Nephrol. 2003 Jun;14(6):1643–51. doi: 10.1097/01.asn.0000057540.98231.c1. Epub 2003/05/23. eng. [DOI] [PubMed] [Google Scholar]
- 24.Brennan TV, Jaigirdar A, Hoang V, Hayden T, Liu FC, Zaid H, et al. Preferential priming of alloreactive T cells with indirect reactivity. Am J Transplant. 2009 Apr;9(4):709–18. doi: 10.1111/j.1600-6143.2009.02578.x. Epub 2009/04/07. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poggio ED, Clemente M, Riley J, Roddy M, Greenspan NS, Dejelo C, et al. Alloreactivity in renal transplant recipients with and without chronic allograft nephropathy. J Am Soc Nephrol. 2004 Jul;15(7):1952–60. doi: 10.1097/01.asn.0000129980.83334.79. Epub 2004/06/24. eng. [DOI] [PubMed] [Google Scholar]
- 26.Jindra PT, Hsueh A, Hong L, Gjertson D, Shen XD, Gao F, et al. Anti-MHC class I antibody activation of proliferation and survival signaling in murine cardiac allografts. J Immunol. 2008 Feb 15;180(4):2214–24. doi: 10.4049/jimmunol.180.4.2214. Epub 2008/02/06. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nozaki T, Amano H, Bickerstaff A, Orosz CG, Novick AC, Tanabe K, et al. Antibody-mediated rejection of cardiac allografts in CCR5-deficient recipients. J Immunol. 2007 Oct 15;179(8):5238–45. doi: 10.4049/jimmunol.179.8.5238. Epub 2007/10/04. eng. [DOI] [PubMed] [Google Scholar]
- 28.Bickerstaff A, Nozaki T, Wang JJ, Pelletier R, Hadley G, Nadasdy G, et al. Acute humoral rejection of renal allografts in CCR5(−/−) recipients. Am J Transplant. 2008 Mar;8(3):557–66. doi: 10.1111/j.1600-6143.2007.02125.x. Epub 2008/02/26. eng. [DOI] [PubMed] [Google Scholar]
- 29.Wasowska BA, Qian Z, Cangello DL, Behrens E, Van Tran K, Layton J, et al. Passive transfer of alloantibodies restores acute cardiac rejection in IgKO mice. Transplantation. 2001 Mar 27;71(6):727–36. doi: 10.1097/00007890-200103270-00007. Epub 2001/05/02. eng. [DOI] [PubMed] [Google Scholar]
- 30.Nickeleit V, Zeiler M, Gudat F, Thiel G, Mihatsch MJ. Detection of the complement degradation product C4d in renal allografts: diagnostic and therapeutic implications. J Am Soc Nephrol. 2002 Jan;13(1):242–51. doi: 10.1681/ASN.V131242. Epub 2001/12/26. eng. [DOI] [PubMed] [Google Scholar]
- 31.Crawford A, Macleod M, Schumacher T, Corlett L, Gray D. Primary T cell expansion and differentiation in vivo requires antigen presentation by B cells. J Immunol. 2006 Mar 15;176(6):3498–506. doi: 10.4049/jimmunol.176.6.3498. Epub 2006/03/07. eng. [DOI] [PubMed] [Google Scholar]
- 32.Noorchashm H, Reed AJ, Rostami SY, Mozaffari R, Zekavat G, Koeberlein B, et al. B cell-mediated antigen presentation is required for the pathogenesis of acute cardiac allograft rejection. J Immunol. 2006 Dec 1;177(11):7715–22. doi: 10.4049/jimmunol.177.11.7715. Epub 2006/11/23. eng. [DOI] [PubMed] [Google Scholar]
- 33.Seite JF, Shoenfeld Y, Youinou P, Hillion S. What is the contents of the magic draft IVIg? Autoimmun Rev. 2008 Jun;7(6):435–9. doi: 10.1016/j.autrev.2008.04.012. Epub 2008/06/19. eng. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Skin primed renal allograft rejection model. C57Bl/6 recipients were primed with DBA/2 skin allografts at day 0 then transplanted with DBA/2 renal allografts at the indicated times. Data shown are survival times of the renal allografts. The brackets show the level of statistical significance for differences between the groups.
Figure S2. Sensitization increases the frequency of immune cells capable of producing IL-17 in response to donor alloantigens. A. Purified splenic CD4+ T cells from primed B6 hosts that had rejected DBA/2 renal allografts at the time of renal allograft rejection (STX/RTX), normal B6 hosts (Naive), or media alone (Media) were tested in an IL-17 ELISPOT assay against intact DBA/2 irradiated stimulators. B. Purified splenic CD4+ T cells from normal B6 hosts (Naive) B6 hosts that had rejected DBA/2 skin allografts only (STX only), or media alone (Media) were tested in an IL-17 ELISPOT assay for reactivity to DBA/2 SC stimulators. Data shown are the mean (+ SEM) IL-17 spots per million cells.
Figure S3. Antibody and C4d deposition in renal allografts following adoptive transfer of alloantibodies. Renal allografts were harvested 30 days after transplantation and Ig (A, B, C) and C4d (D, E, F) were detected by immunohistochemistry. Data are representative of four or more grafts.