

Abstract
BACKGROUND AND PURPOSE
Inhibitors of DNA methyltransferases (DNMTs), such as azacytidine, decitabine and zebularine, are used for the epigenetic treatment of cancer. Their action may depend upon their translocation across the plasma membrane. The aim of this study was to identify transporter proteins contributing to DNMT inhibitor action.
EXPERIMENTAL APPROACH
Drug interactions with selected hCNT and hENT proteins were studied in transiently transfected HeLa and MDCK cells. Interaction with human organic cation transporters (hOCTs) was assessed in transiently transfected HeLa cells and Xenopus laevis oocytes.
KEY RESULTS
Zebularine uptake was mediated by hCNT1, hCNT3 and hENT2. Decitabine interacted with but was not translocated by any nucleoside transporter (NT) type. hCNT expression at the apical domain of MDCK cells promoted net vectorial flux of zebularine. Neither hOCT1 nor hOCT2 transported decitabine, but both were involved in the efflux of zebularine, suggesting these proteins act as efflux transporters. hOCT1 polymorphic variants, known to alter function, decreased zebularine efflux.
CONCLUSIONS AND IMPLICATIONS
This study highlights the influence of human NTs and hOCTs on the pharmacokinetics and pharmacodynamics of selected DNMT inhibitors. As hOCTs may also behave as efflux transporters, they could contribute either to chemoresistance or to chemosensitivity, depending upon the nature of the drug or combination of drugs being used in cancer therapy.
Introduction
Among the nucleoside derivatives currently used in cancer treatment, some cytidine analogues represent a class of drugs which target epigenetic changes caused by gene hypermethylation, some of them relevant to cancer stem cell reprogramming and tumour growth (Heyn and Esteller, 2012; Munoz et al., 2012; Rius and Lyko, 2012). These nucleoside derivatives exert their action mostly by inhibiting DNA methyltransferases (DNMTs), but as for other nucleoside-derived drugs, they require metabolic activation to be incorporated into either RNA or DNA (Rius and Lyko, 2012). FDA-approved epigenetic nucleoside-based drugs are 5-azacytidine (Vidaza) and decitabine (2′-deoxy-5-azacitidine, Dacogen), generally known as azanucleosides. Both appear to be suitable for the treatment of myelodysplastic syndromes and acute myeloid leukaemia, although they might prove useful for other types of cancer, including solid tumours (Quintas-Cardama et al., 2010; Garcia-Manero, 2012; Rius and Lyko, 2012; Ghai et al., 2013). Moreover, other cytidine analogues, such as zebularine (1-β-D-ribofuranosyl-2-(1H)-pyrimidinone), are currently at preclinical stages (Robak, 2011). Zebularine also inhibits DNMT despite not being an azanucleoside (Zhou et al., 2002). These three DNMT inhibitors share metabolic activation routes and can eventually incorporate into nucleic acids, but major differences in their mechanisms of action have also been reported (Hollenbach et al., 2010; Robak, 2011; Schmiedel et al., 2011; Rius and Lyko, 2012).
DNMT inhibitors require membrane transporters in order to be internalized into target cells and their transportability profiles are likely to affect their anti-tumour efficacy. Nucleoside analogues can use a broad set of plasma membrane transporters, which is not restricted to SLC28 and SLC29 family members [encoding human concentrative nucleoside transporters (hCNTs) and human equilibrative nucleoside transporters (hENTs) respectively] (Errasti-Murugarren and Pastor-Anglada, 2010; Minuesa et al., 2011; nomenclature follows Alexander et al., 2013). Several members of the SLC22 gene family, including human organic cation transporters 1, 2 and 3 (hOCT1, hOCT2, hOCT3) and human organic anion transporters 1, 2 and 3 (hOAT1, hOAT2, hOAT3), have been reported to interact with a great variety of nucleoside-derived drugs (Errasti-Murugarren and Pastor-Anglada, 2010; Minuesa et al., 2011). Interaction of selected nucleoside analogues with hOCT proteins might be mechanistically complex. For instance, whereas hOCT1, 2 and 3 can all bind and translocate lamivudine (3TC), the three isoforms show high-affinity binding sites but no translocation for abacavir (ABC), zidovudine (AZT), emtricitabine (FTC) and tenofovir disoproxil fumarate (Minuesa et al., 2009). The high-affinity binding results in inhibition of 3TC uptake, thereby suggesting the occurrence of drug-drug interactions in AIDS therapy (Minuesa et al., 2009; 2011).
The transportability profile of cytidine-derived DNMT inhibitors is not well known, its analysis being restricted so far to azacytidine. In particular, it has been shown that this drug is a high-affinity substrate for hCNT1 and hCNT3, with apparent Km values of 63 and 147 μM respectively (Rius et al., 2009; 2010). When apically expressed in epithelia, both transporters determine the vectorial flux of azacytidine, a process that can be further activated by the basal expression of the export pump MRP4 (ABCC4) (Rius et al., 2009; 2010).
Here, we have addressed the question of how these three DNMT inhibitors (azacytidine, decitabine and zebularine) interact with selected SLC28, SLC29 and SLC22 encoded transporter proteins, all of them expressed, albeit to different extent, in immune cells and epithelial barriers and determining drug pharmacokinetics. Interestingly, a novel role for hOCT1 in nucleoside-derived drug action is proposed based upon the evidence that this protein may also behave as an export transporter.
Methods
Transporter cDNA cloning for heterologous expression
The hCNT1 cDNA (GenBank™ Accession No. U62966) was cloned from human fetal liver (Mata et al., 2001). The hCNT3 cDNA (GenBank Accession No. AF305210) was cloned from human kidney (Errasti-Murugarren et al., 2007). hOCT1 (GenBank Accession No. X98322) and hOCT2 cDNAs (GenBank Accession No. X98333) were cloned from human kidney (Gorboulev et al., 1997). The C88R, M408V, M420del and G465R substitutions were respectively introduced into hOCT1 using compatible reverse and forward primers: 5′GGGCGAGGCCTTCCTTGGCCAGCGCAGGCGCTATGAAGTGGACTGG-3′; 5′-GGGCCGCATCTACCCCATGGCCGTGTCAAATTTGTTGGCGGGGGCAG 3′; 5′-GGCGGGGGCAGCCTGCCTCGTCATTTTTATCTCACCTGACCTGC-3′; 5′-CCCCACATTCGTCAGGAACCTCCGAGTGATGGTGTGTTCCTCCCTG-3′. All DNA constructs were verified by DNA sequencing (BigDye Terminator v3.1, Applied Biosystems, Foster City, CA, USA) in both directions.
For the generation of the stable-expressing cell lines, hOCT1, hOCT1M420del and hOCT1G465R encoding cDNAs were recloned into the pcDNA5/FRT/TO vector (Life Technologies, Paisley, UK). Restriction sites were added using the following primers (restriction sites underlined): 5′-CGGGGTACCATCATGCCCACCGTGGATGACA-3′ and 5′- CGCGGATCCCTCTCAGGTGCCCGAGGGTTC-3′; and the amplification product was subcloned into KpnI and BamHI sites of pcDNA5/FRT/TO vector.
Commercially available vector pOG44 (Life Technologies) codifying for a Flp recombinase was co-transfected with pcDNA5/FRT/TO so the gene was inserted in a previously added restriction site using pFRT/lacZeo (Life Technologies) in order to generate HEK293 cells stably expressing hOCT1.
For expression in Xenopus laevis oocytes, the cDNAs encoding hOCT1 wild type (wt) and the genetic variants were subcloned into the pOG1 vector (a gift of Dr. Michael Kavanaugh, Montana, MO, USA).
Cell culture
Human cervix carcinoma cells (HeLa) and Madin–Darby canine kidney (MDCK) cells were maintained at 37°C/5% CO2 in DMEM (Lonza Verviers SPRL, Verviers, Belgium) supplemented with 10% FBS (vol/vol) (Life Technologies), 2 mM glutamine and a mixture of antibiotics (100 U penicillin, 0.1 mg·mL−1 streptomycin and 0.25 mg·mL−1 fungizone).
The HEK293-FlpIn cell line was cultured in DMEM supplemented with 10% heat-inactivated FBS (vol/vol), 50 U·mL−1 penicillin, 50 μg·mL−1 streptomycin, 2 mM L-glutamine and 200 μg·mL−1 zeocin (Life Technologies).
HEK293 cells stably expressing hOCT proteins were cultured in the same medium supplemented with 100 μg·mL−1 hygromycin B (Life Technologies) instead of zeocin.
Cell transfection and generation of a HEK293-hOCT1 stable cell line
Nucleoside transporters (hCNT1 and hCNT3) were transiently expressed both in HeLa and MDCK cells, whereas hOCT1 was stably expressed, as detailed below, in a HEK293 cell background. HeLa cells were transiently transfected using Lipofectamine 2000 (Life Technologies) as described by the manufacturer. Nucleoside uptake experiments were carried out 24 h after transfection, as explained below. MDCK cells were plated on 12 mm diameter, 0.3 μm pore Transwell plates (Corning Incorporated, Corning, NY, USA) and transfected as described previously (Errasti-Murugarren et al., 2007). Proper expression and insertion of transporter proteins at the plasma membrane has been verified using YFP-tagged transporters, as previously reported (Errasti-Murugarren et al., 2007; 2010a,b), and transport assays, as detailed below, using model substrates of hCNT- and hOCT-type proteins (uridine and cytidine for hCNT1 and hCNT3, and MPP+ for hOCT1). Uptake rates were determined as a control of transporter expression levels at the plasma membrane.
The plasmid constructions described above, as well as an empty vector (pcDNA5/FRT/TO) used as a control in uptake experiments, were co-transfected using calcium phosphate with a vector containing the Flp recombinase (pOG44) into the Flp-In-HEK293 cell line. This is the host cell line with a Flp recombination target (FRT) site, previously generated following the manufacture's protocol (Life Technologies). Positive clones were selected with 100 μg·mL−1 hygromycin B and tested for transport activity.
Uptake experiments
Nucleoside and nucleoside-derived drug uptake rates were measured as previously described (del Santo et al., 1998). In brief, for nucleoside transporters (NTs), replicate cultures were exposed at room temperature to [3H]uridine (1 μM, 1 μCi·mL−1) and to the antineoplastic drugs [3H]decitabine and [3H]zebularine either in a sodium-repleted (137 mM NaCl) or a sodium-free (137 mM choline chloride) transport buffer containing 5.4 mM KCl, 1.8 mM CaCl2, 1.2 mM MgSO4 and 10 mM HEPES, pH 7.4. Initial rates of transport were determined using an incubation period of 1 min and uptake measurements were terminated by washing the cells off with an excess volume of chilled stop buffer (137 mM NaCl, 10 mM HEPES, pH 7.4). To assess whether a selected nucleoside or nucleoside derivative interacts with the transporter protein and to determine Ki values, we monitored sodium-dependent uridine uptake by concentrative transporters in the presence of increasing concentrations of the non-labelled putative inhibitor. For equilibrative transport measurements, the same assay was performed using ENT inhibitors, 1 μM NBTI for hENT1 and 10 μM dipyridamole for both hENT1 and hENT2. Data were fitted to non-linear regression analysis (variable slope) using GraphPad Prism 4.0 software (GraphPad Software, San Diego, CA, USA) to calculate Ki values.
[3H]MPP+ uptake rates mediated by hOCTs were measured in a N-methyl-D-glucamine (137 mM) medium containing 5.4 mM KCl, 1.8 mM CaCl2, 1.2 mM MgSO4 and 10 mM HEPES, pH 7.4, as described above. The hOCT-related activity was calculated by subtracting basal [3H]MPP+ uptake rates present in mock-transfected cells from those determined in hOCT1-expressing cells.
At the end of the transport assays, cells were solubilized with 0.5% Triton-X100/100 mM NaOH and the accumulated intracellular radioactivity was measured by liquid scintillation counting.
Determination of nucleoside-derived drug transport and vectorial flux on MDCK cell epithelial barriers
MDCK cells were grown on Transwell filters. To ensure that cells had polarized and formed tight junctions, transport experiments were conducted when the transepithelial electrical resistance values (measured by millicell-ERS; Millipore, Bedford, MA, USA) reached 300–500 Ω·cm2 in representative wells (routinely 2 days after seeding the cells at high density). Transport measurements were done as previously described (Harris et al., 2004; Errasti-Murugarren et al., 2007). In brief, filter inserts were washed three times with sodium-replete or sodium-free buffer, and then 1 μM, 2 μCi·mL−1 of [3H]nucleoside-derived antineoplastic drug (zebularine, decitabine) or natural nucleoside (cytidine) was added either to the apical or to the basolateral side. Transport experiments were conducted using buffers containing either sodium or choline chloride on both sides of the Transwell filters (0.5 mL in both apical and basal compartments). At various time points (up to 20 min), 50 μL of medium was collected from the opposite compartment to which radiolabelled substrates were added. Transport experiments were terminated by aspirating the buffer and washing filters with chilled buffer. The entire filter was wiped with tissue to remove excess buffer and removed from the plastic support. Cells on the filters were lysed with 0.1% SDS/100 mM NaOH and extracts counted on a scintillation counter.
Cytotoxicity assays
Cytotoxicity assays were performed by seeding HeLa cells (40 000 cell/cm−2) on 96-well culture plates. Twenty-four hours after plating, cells were transfected using Lipofectamine 2000 with either a empty pcDNA3.1 vector or the same vector containing either hCNT1 or hCNT3 cDNAs. The transfection buffer was kept for 5 h and later replaced by fresh media. Twenty-four hours after transfection, cells were exposed to increasing concentrations (from 100 nM to 1 mM) of azacytidine, decitabine or zebularine. Viability was assessed 48 h after the addition of the drugs using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. MTT reagent (7.5 mg·mL−1) was added to each well after removal of medium and incubated for 45 min at 37°C. Subsequently, the MTT reagent was discarded and the formazan crystals were solubilized in dimethyl sulfoxide. The absorbance of formazan was measured photometrically at 595 nm. Data were fitted to a dose–response curve using GraphPad Prism 4.0 software to calculate EC50 values.
Functional expression of hOCT1 in X. laevis oocytes
To allow the expression of hOCT1 in X. laevis oocytes, the pOG1 vectors containing its corresponding cDNA and its mutants as well were linearized with NotI. cRNAs were transcribed as previously described (Arndt et al., 2001). X. laevis oocytes were prepared and stored in Ori buffer (5 mM MOPS, 100 mM NaCl, 3 mM KCl, 2 mM CaCl2 and 1 mM MgCl2, adjusted to pH 7.4, using NaOH) supplemented with 50 mg·L−1 gentamycin as described (Arndt et al., 2001). Per oocyte, 50 nL of H2O containing 10 ng of cRNA encoding the wt hOCT1, hOCT2 and the selected hOCT1 genetic variants was injected into oocytes. Oocytes were then stored for 3–4 days in Ori buffer at 16°C before transport measurements were performed.
Oocytes expressing either hOCT1 or hOCT2 were incubated for 30 min at room temperature in Ori buffer containing either [3H]MPP+ (10 nM, 1 μCi) or [3H]zebularine (100 nM, 1 μCi). Non-injected oocytes from the same batch were used as control. At the end of the incubation period, oocytes were washed three times with ice-cold Ori buffer and solubilized in a 5% SDS solution. Then intracellular radioactivity was analysed by liquid scintillation counting.
For efflux experiments, control oocytes and oocytes expressing hOCT1, hOCT2 or its polymorphic variants were injected with either [3H]MPP+ (0.025 μCi, 6 nM) or [3H]zebularine (0.013 μCi, 26.8 nM), and washed with ice-cold Ori buffer. The efflux was initiated by adding one oocyte to 100 μL of Ori buffer with or without quinine 100 μM, and the efflux rate was measured at different time points, taking samples and quantifying the radioactivity released in the buffer.
Efflux experiments in HEK293 cells
Efflux measurements were performed in HEK293 cells either stably expressing the hOCT1 protein or not (transfected with an empty pcDNA5 vector) which were either transiently transfected with the concentrative nucleoside transporter hCNT3 or with its corresponding empty vector pcDNA3. Transfection was done after seeding the cells in 24-well plates using the calcium phosphate method. Cells were washed twice 40 h post-transfection with sodium-repleted transport buffer (see composition above) and incubated for 5 min with transport buffer containing [3H]zebularine (1 μM, 1 μCi·mL−1). This allowed the cells to accumulate the radiolabelled drug to further monitor its efflux out to the medium. For that purpose, cells were washed twice with ice-cold transport buffer, either containing or not 100 μM quinine. Then 200 μL of transport medium, maintained at room temperature, either with or without 100 μM quinine, was placed in each well. After 1 min of incubation, 50 μL of supernatant was collected for radioactivity quantification. Cells were then lysed and total well protein was quantified.
Data analysis
Data are expressed as the mean ± SEM of at least three independent experiments. The unpaired Student's t-test was used for the statistical comparison of experimental data. These analyses were carried out using GraphPad Prism 4.0 software.
Materials
Uridine, cytidine, azacytidine, decitabine (2′deoxy-5-azacytidine), zebularine, 1-methyl-4-phenylpyridinium iodide (MPP+), quinine, 4-nitrobenzyl-6-thioinosine (NBTI) and dipyridamole were obtained from Sigma-Aldrich (St. Louis, MO, USA). [5,6-3H]uridine was purchased from Perkin Elmer (Waltham, MA, USA). [5-3H(N)]cytidine, [6-3H]5-aza-2′-deoxycytidine and [5′-3H]zebularine were purchased from Hartmann Analytic GmbH (Braunschweig, Germany). [Methyl-3H]-N-methyl-4-phenylpyridinium iodide ([3H]MPP+) was obtained from American Radiolabeled Chemicals, Inc. (St. Louis). The structures of the three cytidine analogues being studied in this contribution are shown in Figure 1.
Figure 1.
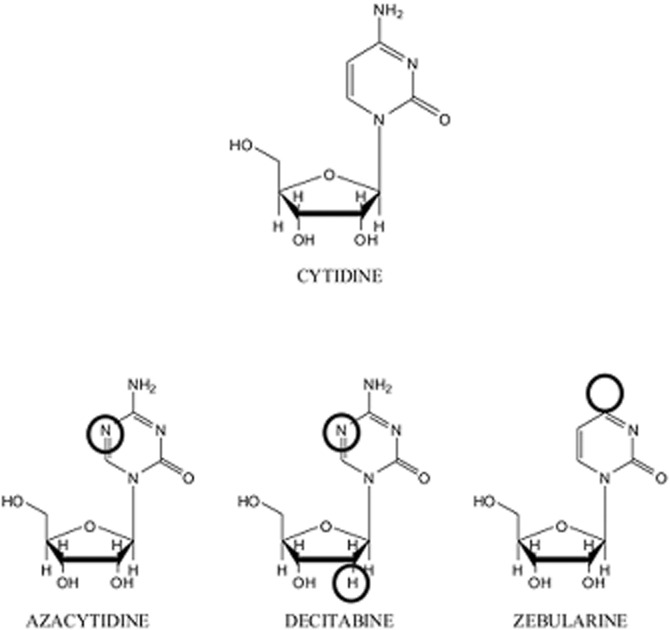
Structure of cytidine and cytidine-analogue inhibitors of DNA methyltransferases. The differences between the cytidine analogues and the natural nucleoside are highlighted.
Results
Interaction of DNMT inhibitors with NTs
To study the ability of nucleoside-derived DNMT inhibitors to interact with NTs, we assayed the effect of increasing concentrations of these drugs on the uptake of 1 μM [3H]uridine in either hCNT1- or hCNT3-transfected HeLa cells. The same cis-inhibition approach was used for equilibrative nucleoside transporters. hENT1- and hENT2-related activities can be pharmacologically discriminated by using inhibitors (1 μM NBTI for hENT1 and 10 μM dypiridamole for both), thereby allowing the analysis of hENT-type contribution to the uptake of DNMT inhibitors by taking advantage of the endogenous human transporter isoforms present in HeLa cells. Although dypiridamole is not a specific inhibitor of hENT-type transporters (Koepsell et al., 2007), the inhibition of uridine uptake can be directly correlated to hENT activity. As all nucleoside analogues tested in this study were pyrimidine analogues (Figure 1), hCNT2 was not considered a candidate for mediating drug uptake because of its well-known purine selectivity (Pastor-Anglada et al., 2008; Errasti-Murugarren and Pastor-Anglada, 2010). The Ki values calculated from the drug concentration-dependent inhibition of uridine uptake are listed in Table 1. Ki values for the interaction of DNMT inhibitors with hCNTs and hENT2 were in the low-medium micromolar range, whereas interaction with hENT1 was comparatively poor, Ki values being in the mM range, particularly for decitabine and zebularine.
Table 1.
Inhibitor constants of DNMT inhibitors for nucleoside transporters
| Ki (μM) | hENT1 | hENT2 | hCNT1 | hCNT3 |
|---|---|---|---|---|
| Azacytidine | 379.5 ± 3.5 | 25.0 ± 0.9 | 11.4 ± 1.6 | 3.5 ± 1.2 |
| Decitabine | 1000–5000 | 5.6 ± 0.5 | 21.6 ± 3.0 | 14.4 ± 4.6 |
| Zebularine | ≍1000 | 22.6 ± 6.1 | 31.3 ± 5.0 | 4.6 ± 0.5 |
Ki values for inhibition of [3H]uridine uptake by DNA-methyltransferase inhibitors in HeLa cells transiently transfected with either hCNT1 or hCNT3 encoding cDNAs. Drug interactions with hENT1 and hENT2 were determined based upon the endogenous activities present in HeLa cells. Ki values were obtained by fitting data by non-linear regression in GraphPad Prism 4.0 software. Data are shown as Ki values ± SEM from two to three independent experiments.
Role of NTs in the uptake of DNMT inhibitors
To check whether the nucleoside-derived DNMT inhibitors were substrates of the hCNT-type transporters, direct uptake measurements using radiolabelled drugs were performed. Because azacytidine is not commercially available in any radiolabelled form, the study was restricted to decitabine and zebularine. Nevertheless, as pointed out above, azacytidine is the only azanucleoside for which transport properties by hCNT1 and hCNT3 had been determined earlier using a radiolabelled form that had been synthesized by the authors (Rius et al., 2009; 2010). [3H]Decitabine and [3H]zebularine uptake was measured in transiently transfected HeLa cells. The results, shown in Figure 2A, confirmed that zebularine was a suitable substrate for both hCNTs, which is consistent with its low Ki value obtained from the cis-inhibition experiments (Table 1). Zebularine uptake rates via hCNT1 and hCNT3 were significantly higher than those of the natural hCNT substrate uridine when used at the same concentrations (Figure 2A). A sodium-independent component of transport was also observed for zebularine which corresponds to the sum of the hENT1- and hENT2-related activities (Figure 2A). Interestingly, decitabine was not internalized by either hCNT1 or hCNT3 although it showed high-affinity interaction with hCNT1, hCNT3 and hENT2 (Figure 2A and Table 1). A minor, almost negligible uptake of decitabine via hENTs was observed (Figure 2A).
Figure 2.
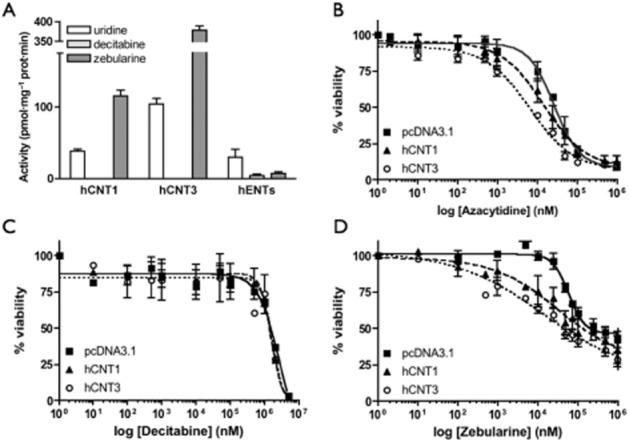
Role of nucleoside transporters in the uptake of DNMT inhibitors and in drug-induced chemosensitivity. (A) Uptake of [3H]-labelled nucleoside-derived drugs and uridine (1 μM, 1 min) by hCNT1 or hCNT3 was measured in transport medium containing 137 mM NaCl or 137 mM choline chloride. Sodium-dependent transport was calculated as uptake in NaCl medium minus uptake in choline chloride medium in HeLa transiently transfected cells. For equilibrative transport, the endogenous activity was measured in HeLa cells using sodium-depleted medium (n = 3). (B–D) HeLa cell viability was measured after 48 h drug exposure to hCNT1, hCNT3 or mock-transfected HeLa cells. Values were fitted to a non-linear regression curve (n = 3).
To determine the relevance of hCNT1 and hCNT3 in assessing intracellular drug bioavailability and action, cell proliferation studies in the presence of these drugs were performed. The experiments were initially done by incubating the cells in the presence of a particular drug for 24, 48 and 72 h. The 48 h time point was chosen because 24 h treatments did not result in maximum cytotoxicity, whereas the 72 h time point yielded similar results to those obtained after incubating the cells for 48 h. Results are shown in Figure 2B–D and the EC50 values derived from these curves listed in Table 2. Independent of expression of hCNT1 and hCNT3, decitabine showed very low toxicity. The EC50 values could not be precisely calculated but were in the high micromolar/low mM range. Azacytidine and zebularine, which are internalized by hCNT1 and hCNT3 proteins, induced cytotoxicity at lower concentrations in those cells expressing the transporters compared with pcDNA3.1-transfected cells (Figure 2B,D). The evidence that the highest drug concentrations used did not apparently reach maximal toxicity (100%) can be explained by the fact that these DNMT inhibitors are likely to induce cell cycle arrest rather than apoptosis. Transporter-mediated sensitization to both analogues resulted in significant decreases in the EC50 values (Table 2). For zebularine, these effects were more pronounced. Expression of hCNT3 seemed to induce greater sensitivity to both azacytidine and zebularine compared with hCNT1. Overall, the effect of transporter expression on cell sensitivity of the DNMT inhibitors was consistent with the interaction and transport-kinetic values reported above.
Table 2.
EC50 values for DNA-methyltransferase inhibitors
| EC50 (μM) 48 h | pcDNA3.1 | hCNT1 | hCNT3 |
|---|---|---|---|
| Azacytidine | 29.5 ± 2 | 13.6 ± 0.7** | 6.8 ± 0.7*** |
| Decitabine | >1000 | >1000 | >1000 |
| Zebularine | 62.0 ± 5.9 | 18.5 ± 6.6** | 8.9 ± 2.0*** |
EC50 values were calculated by fitting data by non-linear regression in GraphPad Prism 4.0 software. Data are expressed as the mean ± SEM of at least three independent experiments performed on different days using different passages of cells. Unpaired Student's t-test was performed comparing the EC50 value for mock-transfected cells and hCNT1, hCNT3. (**P < 0.01; ***P < 0.001).
Role of NTs in determining the vectorial flux of DNMT inhibitors across epithelial barriers
The impact of the apical insertion of hCNT-type proteins on the transepithelial fluxes of DNMT inhibitors was assessed on polarized MDCK cells grown on transwell inserts, as previously reported (Errasti-Murugarren et al., 2007; Pastor-Anglada et al., 2008). Apical-to-basolateral and basolateral-to-apical fluxes of the DNMT inhibitors were measured. For decitabine, no sodium-dependent flux was observed in either direction (Figure 3A). This is consistent with its lack of transport by hCNT1 and hCNT3 (Figure 2A). Additionally, sodium-independent decitabine fluxes in both directions, mediated by endogenous equilibrative nucleoside transporters, were smaller than those observed for cytidine (data not shown). For zebularine, a net apical-to-basal sodium-dependent flux was detected after expression of either hCNT1 or hCNT3 (Figure 3B). Nevertheless, the magnitude of the apical-to-basolateral zebularine flux in the presence of hCNT3 was nearly fourfold greater than that observed when hCNT1 was expressed. Because hCNT-type function is totally dependent upon the occurrence of a Na+ transmembrane gradient, fluxes in its absence are low and similar to those measured in non-transfected MDCK cells (not shown). Basal-to-apical flux of zebularine after transfection of either hCNT1 or hCNT3 in the presence of sodium was much lower than the apical-to-basal flux and likely to be mediated by the drug concentration gradient across the epithelial barrier, which can be supported by hENT-type proteins (Figure 3B). This finding demonstrates that apical insertion of hCNT proteins does contribute to drug vectorial flux across this epithelial barrier. As previously reported for other nucleoside analogues (Errasti-Murugarren et al., 2007), the basal-to-apical flux of zebularine, although much lower than that found in the apical-to-basal direction, was even lower in the presence of sodium than when fluxes were measured in the choline chloride medium. This can be explained by the fact that hCNT3 proteins at the apical side of the barrier are highly efficient in removing the drug from the apical compartment, thereby reducing the net basal-to-apical flux. In fact, this had been previously demonstrated by blocking the apical hCNT3 function with phloridzin (Errasti-Murugarren et al., 2007).
Figure 3.
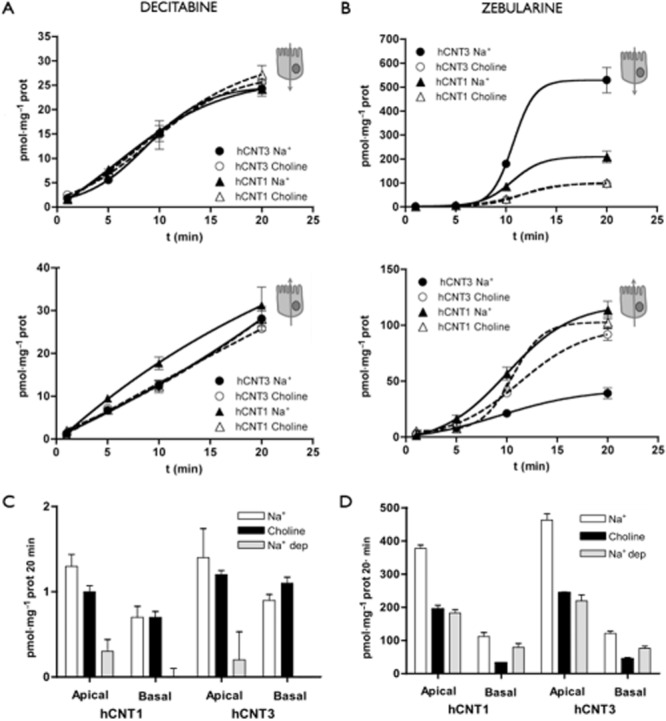
Transepithelial flux and cellular accumulation of DNMT inhibitors in hCNT1- or hCNT3-transfected MDCK cells grown as monolayers. Transepithelial fluxes of [3H]decitabine (A) and [3H]zebularine (B) (1 μM, 2 μCi·mL−1) in sodium and choline chloride medium from apical to basolateral compartments (upper panel) and from basolateral to apical compartments (lower panel) are shown. The MDCK cells transfected with hCNT1 or hCNT3 were grown on filters. Accumulation of decitabine (C) and zebularine (D) after 20 min of incubation with radioactive DNMT inhibitors added to the apical or basal compartments. The measurements were performed in sodium and choline media and the differences between sodium-containing and sodium-free corresponding to the hCNT activities are indicated as sodium-dependent (Na+-dep) accumulation. Data are expressed as the mean ± SEM of uptake values obtained in three wells. Data are representative of three experiments carried out on different days using different batches of cells.
Drug cellular accumulations were measured 20 min after addition of the DNMT inhibitors under the same conditions used for the analysis of vectorial fluxes (Figure 3C,D). As expected, accumulation of decitabine was negligible (around 1 pmol·mg−1 protein/20 min) and totally independent of the presence of sodium in either compartment (Figure 3C). Zebularine accumulated within hCNT1- and hCNT3-MDCK expressing cells more than decitabine (Figure 3D). The highest intracellular accumulation of zebularine was observed, as expected, in the presence of sodium when the drug was added to the apical compartment, where hCNT-type proteins are expressed.
Role of OCTs in the uptake of DNMT inhibitors
As reviewed above, hOCTs have also been shown to mediate the uptake of certain nucleoside analogues (Errasti-Murugarren and Pastor-Anglada, 2010; Minuesa et al., 2011). Therefore, we investigated whether hOCT1 and hOCT2 transported decitabine and/or zebularine. HeLa cells transiently transfected with either hOCT1 or hOCT2 showed a sixfold higher accumulation of the hOCT model substrate [3H]MPP+ than control cells (transfected with the empty vector) (47.3 ± 1.1 and 45.5 ± 5 vs. 7.5 ± 1.1). This resulted in similar net hOCT1 and hOCT2 MPP+-mediated influx uptake rates (Figure 4A), which is consistent with both transporter proteins being similarly expressed and properly inserted at the plasma membrane of HeLa cells. hOCT-mediated [3H]decitabine transport was low (hOCT2) or even negligible (hOCT1). Surprisingly, the heterologous expression of hOCT1 and hOCT2 resulted in reduced accumulation of [3H]zebularine compared with control cells (Figure 4A). This observation could be interpreted on the basis that the transporter rather than mediating the uptake of zebularine facilitates its release from cells. When these experiments were done in the presence of 10 μM dipyridamol, an inhibitor of hENT-type transporters, no differences were observed between hOCT-expressing and non-expressing cells and no zebularine accumulated at all (data not shown). In fact, HeLa cells express significant basal hENT-related transport activities (i.e. 43 ± 2.5 and 11 ± 2.5 pmol uridine/min·mg protein via hENT1 and hENT2, respectively, n = 4, mean ± SEM), thereby suggesting that zebularine is preferentially taken into hOCT-expressing cells via hENT-type transporters, although once inside cells it is rapidly released via hOCTs.
Figure 4.
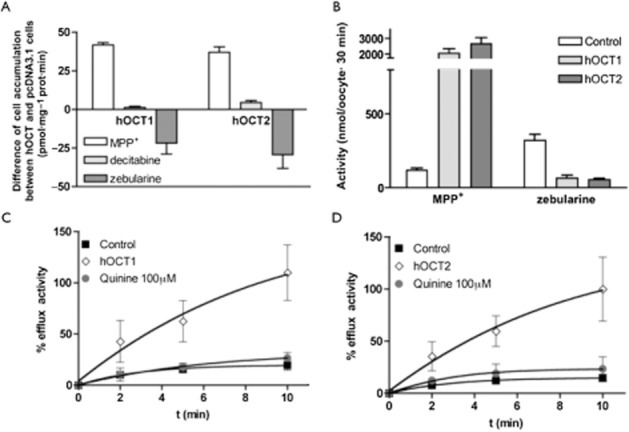
hOCT's implication in zebularine efflux. (A) Difference between the accumulation in hOCT-expressing cells and the accumulation in mock-transfected cells after 1 min of uptake measurement (n = 3). (B) [3H]MPP+ (10 nM, 1 μCi·mL−1) and [3H]zebularine (100 nM, 1 μCi·mL−1) accumulation in oocytes after 30 min of drug exposure. Data shown as mean ± SEM of 20 single oocytes from two different batches. (C,D) Efflux of [3H]zebularine from previously injected (0.013 μCi·mL−1, 26.8 nM) control oocytes in ORI buffer or oocytes expressing hOCT1 (C) or hOCT2 (D) in ORI buffer or in ORI buffer containing quinine 100 μM. All values have been calculated considering the maximum efflux 100%. Data are representative of four experiments carried out on different days using different batches of oocytes.
We decided to corroborate the role of hOCTs in zebularine efflux using X. laevis oocytes, a suitable model that allows substrate injection to load the cells and monitor efflux phenomena. While hOCT1 and hOCT2, as expected, promoted a significant uptake of radiolabelled MPP+ into oocytes, zebularine accumulation into hOCT1- and hOCT2-expressing cells was significantly reduced when compared with non-injected oocytes (Figure 4B). This mimics the results using a human cell background (HeLa cells) (Figure 4A). To definitely validate that both hOCT1 and hOCT2 were responsible for zebularine efflux, the radiolabelled drug was injected into oocytes expressing either hOCT1 or hOCT2, and control oocytes as well, and the release of [3H]zebularine to the extracellular milieu was measured at different time points in Ori buffer either containing or not 100 μM quinine, a well-known hOCT inhibitor (Koepsell et al., 2007). A large efflux of zebularine was observed only in hOCT1/2-injected oocytes, whereas efflux from control oocytes was minimal. Of note, hOCT1- and hOCT2-mediated zebularine efflux was completely blocked by quinine (Figure 4C,D).
Role of hOCT1 in mediating the efflux of zebularine in HEK293 cells
To further reassess the role hOCTs can play as efflux transporters in human cells, a similar type of study as the one described above using oocytes was performed using HEK293 cells. This cell line was engineered to make it express either hOCT1 (stable expression) or hCNT3 (transient expression) or both, by transiently expressing hCNT3 in a hOCT1-HEK293 background (see Methods). Results using these cell models are shown in Figure 5. In the absence of either transporter, HEK293 cells showed a low basal efflux rate, which was insensitive to the hOCT1 inhibitor quinine. Expression of hCNT3, prior to zebularine loading, resulted in a significant increase in drug efflux, which was equally insensitive to quinine. HEK293 cells stably expressing hOCT1 showed higher zebularine efflux rates than control cells, and this potentiation of drug efflux was inhibited by quinine. Cells expressing both hCNT3 and hOCT1 proteins showed the highest zebularine efflux rate. In the presence of quinine, this efflux activity was reduced to the same levels as those found in hCNT3-expressing HEK293 cells, lacking hOCT1 expression. All these data, taken together, strongly support the view that hOCT1 is a zebularine efflux transporter also in human cells. These observations are also consistent with hCNT3 being a highly concentrative nucleoside transporter protein, thereby facilitating zebularine accumulation above extracellular levels. The accumulated drug will be extruded from cells at a faster rate if hOCT1 is also expressed.
Figure 5.
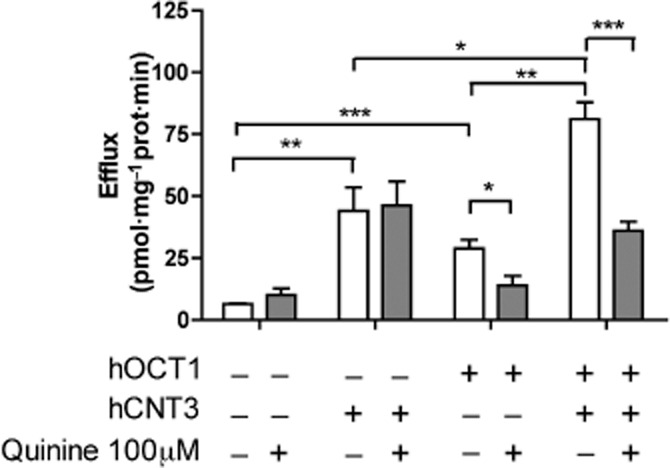
hOCT1-mediated efflux of zebularine in HEK293 cells. [3H]zebularine released by HEK293 cells expressing or not hCNT3 and hOCT1 in the extracellular milieu during 1 min after preloading the cells for 5 min with [3H]zebularine (1 μM; 1 μCi·mL−1) in the presence or absence of quinine 100 μM. Data are expressed as the mean ± SEM of three to six individual experiments. (*P < 0.05; **P < 0.01; ***P < 0.001).
Effect of hOCT1 polymorphic variants on zebularine efflux
The gene encoding the hOCT1 protein, SLC22A1, is highly polymorphic, and several of its genetic variants also show relatively high allelic frequency in humans (Shu et al., 2003; Errasti-Murugarren and Pastor-Anglada, 2010). The functional consequences on zebularine efflux of some of the most frequent polymorphic variants found in Caucasian population were assessed. Site-directed mutagenesis of the wt transporter allowed the generation of the following variants: hOCT1C88R, hOCT1M408V, hOCT1M420del and hOCT1G465R. Once demonstrated that hOCT1 is also a zebularine efflux transporter in HEK293 cells (Figure 5), X. laevis oocytes were used again for the functional analysis of hOCT1 polymorphic variants, because efflux experiments were more readily feasible in this background than in HEK293 cells. These cDNAs were transcribed in vitro and cRNAs injected into oocytes to allow transporter protein synthesis and efflux measurements. Efflux was determined by quantifying the accumulation of the radiolabelled substrate in the Ori buffer 10 min after injection of the hOCT substrates inside the oocytes. The efflux profile for MPP+ was very similar to that of its uptake (Figure 6A and Shu et al., 2003). In the case of zebularine, all tested polymorphic variants promoted a significantly lower efflux than the wt transporter (Figure 6B).
Figure 6.
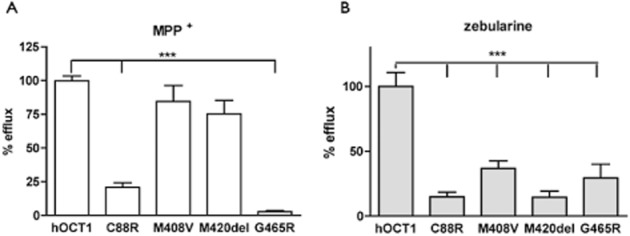
Effects of hOCT1 polymorphisms on zebularine efflux. (A) [3H]MPP+ (0.025 μCi·mL−1, 6 nM) and (B) [3H]zebularine (0.013 μCi·mL−1, 26.8 nM) effluxes measured in 15 single oocytes from three different experiments performed at different days with different batches of oocytes injected with the cRNA of hOCT1, hOCT1C88R, hOCT1M408V, hOCT1M420del, hOCT1G465R. All hOCT1 values were expressed as percentage of maximal efflux, which was set at a value of 100% (***P < 0.001).
Discussion
This study demonstrates that three cytidine-derived DNMT inhibitors, two of them azanucleosides currently used in the clinics, all bearing minor changes in their molecular structures (Figure 1), show significant differences in their transportability profiles. Interaction analysis of these analogues with nucleoside transporter hCNT1 and hCNT3 proteins yielded Ki values for the three assayed nucleoside analogues (azacytidine, decitabine and zebularine) within the low micromolar concentration range. Analysis of the endogenous hENT2-type protein also revealed a high-affinity interaction with these drugs. The three molecules retain the 3′ hydroxyl group, which is known to be essential for high-affinity interaction with NTs (Chang et al., 2004; Cano-Soldado et al., 2011; Cano-Soldado and Pastor-Anglada, 2012), but not necessarily for substrate translocation. In fact, decitabine did interact with all these NT proteins with high affinity but could not be efficiently transported, which is in agreement with a recent report showing very poor decitabine uptake via NT-type proteins, when compared with other nucleosides and analogues (i.e. uridine, adenosine, gemcitabine and azacitidine) (Damaraju et al., 2012). This suggests that the 2′ hydroxyl plays a key role in determining substrate transportability in these cytidine analogues.
The interaction of the three DNMT inhibitors with hENT1 was of lower affinity than that found for the other transporter proteins, but within the range of what has been reported for its natural substrate cytidine (Molina-Arcas et al., 2009). This observation is particularly interesting because hENT1 expression is normally retained or even high in tumours (Farre et al., 2004; Bhutia et al., 2011; Bock et al., 2011). Moreover, as recently shown, hENT1 expression in leukaemia cell lines appears to be a key determinant of azacytidine-triggered cytotoxicity (Hummel-Eisenbeiss et al., 2013). On the contrary, expression of hCNT proteins in tumours can be more variable (Farre et al., 2004; Bhutia et al., 2011; Bock et al., 2011), thus anticipating that response to DNMT inhibitors, such as azacytidine and zebularine, would be also dependent upon hCNT-type expression levels in target cells.
In fact, heterologous expression of hCNT1 and hCNT3 in HeLa cells resulted in a significant sensitization of cells to azacytidine and zebularine, whereas whether these transporters were present or not, cells remained resistant to decitabine, due to its poor or even lack of permeability via NT-type transporter proteins. The highest sensitization of cells to azacytidine and zebularine associated with hCNT3 expression is consistent with the observed high-affinity interaction of both drugs with this transporter protein, and with the fact that hCNT3 is the only member within the SLC28 gene family able to translocate 2 Na+ per nucleoside (Errasti-Murugarren and Pastor-Anglada, 2010; Minuesa et al., 2011; Cano-Soldado and Pastor-Anglada, 2012). This would explain a probable higher hCNT3-driven capacity to concentrate these two cytidine analogues within the cells than that associated with hCNT1 expression. Moreover, the finding that decitabine cannot be translocated via these transporter proteins, but can inhibit them with high affinity, suggests that, if combined therapies implicating hCNT-related substrates would have to be used, undesired drug-drug interactions may occur. This possibility might be also relevant to the pharmacokinetics of these DNMT inhibitors. In fact, hCNT proteins are highly expressed in small intestine (Govindarajan et al., 2007) and in the proximal convoluted tubule of the nephron (Rodriguez-Mulero et al., 2005), where most of the (re)absorption of natural nucleosides and nucleoside-derived drugs occurs. Moreover, it is known that hCNT and hENT expression in (re)absorptive epithelia is asymmetric, thereby facilitating vectorial flux of substrates across these barriers (Mangravite et al., 2003; Govindarajan et al., 2007; 2008). As for other nucleoside analogues used in anticancer and antiviral therapies (Errasti-Murugarren et al., 2007), the apical expression of hCNT-type transporters in polarized epithelia (in this case polarized MDCK cells grown on transwell inserts) determined the apical-to-basal vectorial flux of zebularine.
hCNT expression has been found not only in epithelia and epithelial-derived tumours (Farre et al., 2004; Bhutia et al., 2011; Bock et al., 2011) but also in cells from patients with lymphoproliferative diseases. Chronic lymphocytic leukaemia (CLL) cells express hCNT2 and hCNT3 (Molina-Arcas et al., 2003). hCNT3 function in CLL is regulated in a complex manner and might be highly variable among patients thereby contributing to different therapeutic responses to the nucleoside-derived drug, fludarabine (Fernandez-Calotti and Pastor-Anglada, 2010; Fernandez-Calotti et al., 2012). SLC28 and SLC29 genes and the protein transporters encoded by these genes have also been comprehensively screened in human immune cells. Although hENT1 and hENT2 appear to be highly expressed in all analysed cell types, hCNT3, which according to this study is a suitable transporter for azacytidine and zebularine, is highly expressed in monocytes, monocyte-derived macrophages (MDMs) and monocyte-derived dendritic cells (MDDCs). However, hCNT3 expression in peripheral blood mononucleated cells (PBMCs) and CD4+ T-cells is negligible although significantly induced in phytohemagglutinin (PHA)-stimulated cells (Minuesa et al., 2008). Overall, this anticipates that all immune cell types but especially myeloid populations and CLL cells can be targeted by selected DNMT inhibitors based upon their transporter expression patterns.
Chemical modification of nucleosides for therapeutic purposes might determine a relative shift in their transportability profiles (Cano-Soldado and Pastor-Anglada, 2012). In fact, selected SLC22 gene family members, in particular hOCT1, hOCT2 and hOCT3, might play a role in the uptake of some nucleoside analogues, such as 3TC (Minuesa et al., 2009). Interestingly, as for hCNT proteins, hOCT expression has also been found in immune cells. hOCT1 and hOCT3 have been shown to be expressed at significant levels in monocytes, MDMs and MDDCs (Minuesa et al., 2008). Both transporter proteins are also expressed in PBMCs and CD4+ T-cells, being their expression up-regulated after PHA stimulation (Minuesa et al., 2008). Interestingly, hOCT1 has also been found in primary CLL cells where it appears to contribute to irinotecan- and paclitaxel-induced cytotoxicities (Gupta et al., 2012). In this context, the analysis of the putative interaction of DNMT inhibitors with hOCT proteins seemed to be of interest, particularly considering hOCT1 shows high genetic variability in humans and its polymorphic variants might determine drug (i.e. metformin) pharmacokinetics and pharmacodynamics (Shu et al., 2007; 2008; Tzvetkov et al., 2009; Gong et al., 2012).
In this study, it was found that transient expression of hOCT1 and hOCT2 resulted in a low ability to concentrate zebularine into HeLa cells. This was prevented when hENT1 and hENT2 (zebularine influx transporters) were blocked using uridine as a competitive inhibitor. This suggests that hOCTs are contributing to zebularine efflux from cells. Thus, some asymmetry in the translocation properties of hOCTs can be anticipated. This has been already reported for the interaction of corticosterone with hOCT1, being its binding properties different depending upon whether the molecule binds to the inward or to the outward pocket of the protein (Volk et al., 2009). The ability of hOCT1 to release intracellular zebularine to the extracellular milieu was unequivocally demonstrated when measuring efflux rates in hOCT1-expressing X. laevis oocytes, but also in HEK293 cells stably expressing the hOCT1 protein. Interestingly, and consistently with the discussed role of hCNT3 as a high concentrative NT, the heterologous expression of this protein in HEK293 cells induced a high capacity to accumulate zebularine inside cells, thereby promoting an increased efflux of the drug, which was further stimulated when hOCT1 was present.
If hOCT1 plays a role in zebularine efflux instead of facilitating its uptake into target cells, hOCT1 expression would not result in increased sensitivity to the drug but, instead, in chemoresistance. More interestingly, selected genetic hOCT1 variants known to determine poor response to therapy (Shu et al., 2007; Tzvetkov et al., 2009) would confer more sensitivity to zebularine than the wt. In this particular case, these variants would contribute to retain zebularine inside tumour cells once previously internalized via hENT-type proteins. Caution should be taken when facing combined therapies, because hOCT1 can interact with DNMT inhibitors but also translocate a broad variety of tyrosine kinase inhibitors targeting multiple intracellular pathways (Minematsu and Giacomini, 2011). hOCT1 mRNA levels are fourfold higher in primary CLL cells than in normal lymphocytes (Gupta et al., 2012), but its expression in hepatocarcinoma (Schaeffeler et al., 2011; Martinez-Becerra et al., 2012), hepatoblastoma and cholangiocarcinoma (Martinez-Becerra et al., 2012) is significantly down-regulated, also at the mRNA level. Interestingly, this effect, at least in hepatocarcinoma, seems to be related to increased DNA methylation of the hOCT1-encoding gene SLC22A1 when compared with normal adjacent tissue (Schaeffeler et al., 2011). In this regard, it was postulated that targeting SLC22A1 methylation by demethylating agents (i.e. DNMT inhibitors) may offer a novel strategy for treatment (Schaeffeler et al., 2011). Nevertheless, this possibility, although theoretically interesting, should take into consideration not only the characteristics of the interaction between DNMT inhibitors with hOCT1, but also the expression levels of other transporters of the hCNT family. In fact, hCNT1 expression, another high-affinity transporter for these inhibitors, is also significantly down-regulated in hepatic cancers (Martinez-Becerra et al., 2012). Overall, it could be anticipated that those individuals with high hNT-related activity expressing hOCT1 polymorphic variants conferring increased retention of epigenetic drugs in cancer cells would have an optimal chemotherapeutic response.
In summary, this study outlines the scenario for the transport phenomena determining the intracellular levels of DNMT inhibitors in cells. Immune system cells do express the combination of transporter proteins (hENT1, hENT2, hCNT3 and hOCT1) here reported to interact with these inhibitors in a differential manner (see Figure 7). Unexpectedly, interaction with hOCT1 was shown to be variable depending on the substrate used but, more importantly, asymmetry in the interaction could confer this transporter protein a novel role in target cells as contributor to chemoresistance, thereby changing the classical view of how selected polymorphic variants of hOCT1 can determine drug action. DNMT inhibitors are used with variable results in highly heterogeneous myelodysplastic syndromes, for which patterns of drug transporter expression are not yet known. This study strongly supports the need for the analysis of drug transporter profiles in target cells, and eventually genotyping for hOCT1 polymorphisms, when dealing with DNMT inhibitor-based therapies.
Figure 7.
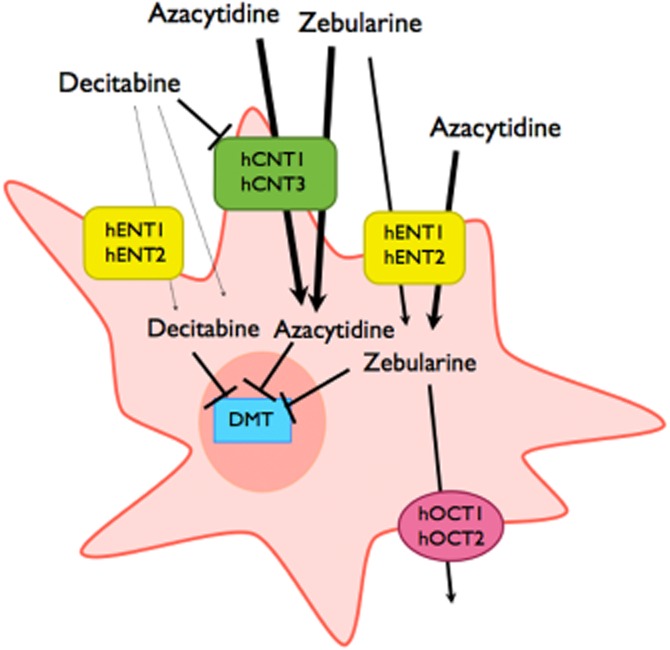
Model showing the role of nucleoside transporters and organic cation transporters in the internalization and efflux of DNMT inhibitors. The thickness of the arrows represents the different magnitude of transport processes.
Acknowledgments
The UB laboratory is member of the Oncology Program of the National Biomedical Research Institute of Liver and Gastrointestinal Diseases (CIBERehd). CIBERehd is an initiative of Instituto de Salud Carlos III (Spain). This work has been funded by projects SAF2011-23660 (MINECO, Government of Spain) and 2009SGR624 (Generalitat de Catalunya) to M. P-A. and by the Deutsche Forschungsgemeinschaft Grant KO 872/6-1 to H. K. Work at J. M-P. laboratory is supported by the Spanish Ministry of Science and Innovation (Grant SAF2010-21224), the Spanish AIDS Network (RD06/0006) and by the European Union's Seventh Framework Programme (FP7/2007-2013) under the ‘Collaborative HIV and Anti-HIV Drug Resistance Network (CHAIN)’ Project Grant Agreement 223131. C. A-N. is a Formación de Personal Investigador (FPI) Fellow (MINECO, Government of Spain). E. E-M. was funded by CIBERehd. We thank Ingrid Iglesias for helpful and continuous technical support.
Glossary
- DNMT
DNA methyltransferase
- hCNT
human concentrative nucleoside transporter
- hENT
human equilibrative nucleoside transporter
- hOCT
human organic cation transporter
- MDDC
monocyte-derived dendritic cell
- MDM
monocyte-derived macrophage
- MPP+
1-methyl-4-phenylpyridinium iodide
- NBTI
nitrobenzylthioinosine
- NT
nucleoside transporter
Author contributions
C. A-N., E. E-M., H. K. and M. P-A. participated in research design. C. A-N., E. E-M., G. M. and V. G. conducted the experiments. C. A-N., E. E-M. and M. P-A. performed the data analysis. C. A-N., E. E-M., G. M., J. M-P., H. K. and M. P-A. wrote or contributed to writing the manuscript.
Conflict of interest
None.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Transporters. Br J Pharmacol. 2013;170:1706–1796. doi: 10.1111/bph.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arndt P, Volk C, Gorboulev V, Budiman T, Popp C, Ulzheimer-Teuber I, et al. Interaction of cations, anions, and weak base quinine with rat renal cation transporter rOCT2 compared with rOCT1. Am J Physiol Renal Physiol. 2001;281:F454–F468. doi: 10.1152/ajprenal.2001.281.3.F454. [DOI] [PubMed] [Google Scholar]
- Bhutia YD, Hung SW, Patel B, Lovin D, Govindarajan R. CNT1 expression influences proliferation and chemosensitivity in drug-resistant pancreatic cancer cells. Cancer Res. 2011;71:1825–1835. doi: 10.1158/0008-5472.CAN-10-2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock AJ, Dong HP, Trope CG, Staff AC, Risberg B, Davidson B. Nucleoside transporters are widely expressed in ovarian carcinoma effusions. Cancer Chemother Pharmacol. 2011;69:467–475. doi: 10.1007/s00280-011-1716-7. [DOI] [PubMed] [Google Scholar]
- Cano-Soldado P, Pastor-Anglada M. Transporters that translocate nucleosides and structural similar drugs: structural requirements for substrate recognition. Med Res Rev. 2012;32:428–457. doi: 10.1002/med.20221. [DOI] [PubMed] [Google Scholar]
- Cano-Soldado P, Gorraitz E, Errasti-Murugarren E, Casado FJ, Lostao MP, Pastor-Anglada M. Functional analysis of the human concentrative nucleoside transporter-1 variant hCNT1S546P provides insight into the sodium-binding pocket. Am J Physiol Cell Physiol. 2011;302:C257–C266. doi: 10.1152/ajpcell.00198.2011. [DOI] [PubMed] [Google Scholar]
- Chang C, Swaan PW, Ngo LY, Lum PY, Patil SD, Unadkat JD. Molecular requirements of the human nucleoside transporters hCNT1, hCNT2, and hENT1. Mol Pharmacol. 2004;65:558–570. doi: 10.1124/mol.65.3.558. [DOI] [PubMed] [Google Scholar]
- Damaraju VL, Mowles D, Yao S, Ng A, Young JD, Cass CE, et al. Role of human nucleoside transporters in the uptake and cytotoxicity of azacitidine and decitabine. Nucleosides Nucleotides Nucleic Acids. 2012;31:236–255. doi: 10.1080/15257770.2011.652330. [DOI] [PubMed] [Google Scholar]
- Errasti-Murugarren E, Pastor-Anglada M. Drug transporter pharmacogenetics in nucleoside-based therapies. Pharmacogenomics. 2010;11:809–841. doi: 10.2217/pgs.10.70. [DOI] [PubMed] [Google Scholar]
- Errasti-Murugarren E, Pastor-Anglada M, Casado FJ. Role of CNT3 in the transepithelial flux of nucleosides and nucleoside-derived drugs. J Physiol. 2007;582(Pt 3):1249–1260. doi: 10.1113/jphysiol.2007.130138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errasti-Murugarren E, Casado FJ, Pastor-Anglada M. Different N-terminal motifs determine plasma membrane targeting of the human concentrative nucleoside transporter 3 in polarized and nonpolarized cells. Mol Pharmacol. 2010a;78:795–803. doi: 10.1124/mol.110.065920. [DOI] [PubMed] [Google Scholar]
- Errasti-Murugarren E, Molina-Arcas M, Casado FJ, Pastor-Anglada M. The human concentrative nucleoside transporter-3 C602R variant shows impaired sorting to lipid rafts and altered specificity for nucleoside-derived drugs. Mol Pharmacol. 2010b;78:157–165. doi: 10.1124/mol.110.063552. [DOI] [PubMed] [Google Scholar]
- Farre X, Guillen-Gomez E, Sanchez L, Hardisson D, Plaza Y, Lloberas J, et al. Expression of the nucleoside-derived drug transporters hCNT1, hENT1 and hENT2 in gynecologic tumors. Int J Cancer. 2004;112:959–966. doi: 10.1002/ijc.20524. [DOI] [PubMed] [Google Scholar]
- Fernandez-Calotti P, Pastor-Anglada M. All-trans-retinoic acid promotes trafficking of human concentrative nucleoside transporter-3 (hCNT3) to the plasma membrane by a TGF-beta1-mediated mechanism. J Biol Chem. 2010;285:13589–13598. doi: 10.1074/jbc.M109.055673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Calotti PX, Lopez-Guerra M, Colomer D, Pastor-Anglada M. Enhancement of fludarabine sensitivity by all-trans-retinoic acid in chronic lymphocytic leukemia cells. Haematologica. 2012;97:943–951. doi: 10.3324/haematol.2011.051557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Manero G. Myelodysplastic syndromes: 2012 update on diagnosis, risk-stratification, and management. Am J Hematol. 2012;87:692–701. doi: 10.1002/ajh.23264. [DOI] [PubMed] [Google Scholar]
- Ghai V, Sharma K, Abbi KK, Shimko S, Epner EM. Current approaches to epigenetic therapy for the treatment of mantle cell lymphoma. Adv Exp Med Biol. 2013;779:257–266. doi: 10.1007/978-1-4614-6176-0_11. [DOI] [PubMed] [Google Scholar]
- Gong L, Goswami S, Giacomini KM, Altman RB, Klein TE. Metformin pathways: pharmacokinetics and pharmacodynamics. Pharmacogenet Genomics. 2012;22:820–827. doi: 10.1097/FPC.0b013e3283559b22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorboulev V, Ulzheimer JC, Akhoundova A, Ulzheimer-Teuber I, Karbach U, Quester S, et al. Cloning and characterization of two human polyspecific organic cation transporters. DNA Cell Biol. 1997;16:871–881. doi: 10.1089/dna.1997.16.871. [DOI] [PubMed] [Google Scholar]
- Govindarajan R, Bakken AH, Hudkins KL, Lai Y, Casado FJ, Pastor-Anglada M, et al. In situ hybridization and immunolocalization of concentrative and equilibrative nucleoside transporters in the human intestine, liver, kidneys, and placenta. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1809–R1822. doi: 10.1152/ajpregu.00293.2007. [DOI] [PubMed] [Google Scholar]
- Govindarajan R, Endres CJ, Whittington D, LeCluyse E, Pastor-Anglada M, Tse CM, et al. Expression and hepatobiliary transport characteristics of the concentrative and equilibrative nucleoside transporters in sandwich-cultured human hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2008;295:G570–G580. doi: 10.1152/ajpgi.00542.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Wulf G, Henjakovic M, Koepsell H, Burckhardt G, Hagos Y. Human organic cation transporter 1 is expressed in lymphoma cells and increases susceptibility to irinotecan and paclitaxel. J Pharmacol Exp Ther. 2012;341:16–23. doi: 10.1124/jpet.111.190561. [DOI] [PubMed] [Google Scholar]
- Harris MJ, Kagawa T, Dawson PA, Arias IM. Taurocholate transport by hepatic and intestinal bile acid transporters is independent of FIC1 overexpression in Madin-Darby canine kidney cells. J Gastroenterol Hepatol. 2004;19:819–825. doi: 10.1111/j.1440-1746.2004.03347.x. [DOI] [PubMed] [Google Scholar]
- Heyn H, Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet. 2012;13:679–692. doi: 10.1038/nrg3270. [DOI] [PubMed] [Google Scholar]
- Hollenbach PW, Nguyen AN, Brady H, Williams M, Ning Y, Richard N, et al. A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS ONE. 2010;5:e9001. doi: 10.1371/journal.pone.0009001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel-Eisenbeiss J, Hascher A, Hals PA, Sandvold ML, Muller-Tidow C, Lyko F, et al. The role of human equilibrative nucleoside transporter 1 on the cellular transport of the DNA methyltransferase inhibitors 5-azacytidine and CP-4200 in human leukemia cells. Mol Pharmacol. 2013;84:438–450. doi: 10.1124/mol.113.086801. [DOI] [PubMed] [Google Scholar]
- Koepsell H, Lips K, Volk C. Polyspecific organic cation transporters: structure, function, physiological roles, and biopharmaceutical implications. Pharm Res. 2007;24:1227–1251. doi: 10.1007/s11095-007-9254-z. [DOI] [PubMed] [Google Scholar]
- Mangravite LM, Badagnani I, Giacomini KM. Nucleoside transporters in the disposition and targeting of nucleoside analogs in the kidney. Eur J Pharmacol. 2003;479:269–281. doi: 10.1016/j.ejphar.2003.08.076. [DOI] [PubMed] [Google Scholar]
- Martinez-Becerra P, Vaquero J, Romero MR, Lozano E, Anadon C, Macias RI, et al. No correlation between the expression of FXR and genes involved in multidrug resistance phenotype of primary liver tumors. Mol Pharm. 2012;9:1693–1704. doi: 10.1021/mp300028a. [DOI] [PubMed] [Google Scholar]
- Mata JF, Garcia-Manteiga JM, Lostao MP, Fernandez-Veledo S, Guillen-Gomez E, Larrayoz IM, et al. Role of the human concentrative nucleoside transporter (hCNT1) in the cytotoxic action of 5[Prime]-deoxy-5-fluorouridine, an active intermediate metabolite of capecitabine, a novel oral anticancer drug. Mol Pharmacol. 2001;59:1542–1548. doi: 10.1124/mol.59.6.1542. [DOI] [PubMed] [Google Scholar]
- Minematsu T, Giacomini KM. Interactions of tyrosine kinase inhibitors with organic cation transporters and multidrug and toxic compound extrusion proteins. Mol Cancer Ther. 2011;10:531–539. doi: 10.1158/1535-7163.MCT-10-0731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minuesa G, Purcet S, Erkizia I, Molina-Arcas M, Bofill M, Izquierdo-Useros N, et al. Expression and functionality of anti-human immunodeficiency virus and anticancer drug uptake transporters in immune cells. J Pharmacol Exp Ther. 2008;324:558–567. doi: 10.1124/jpet.107.131482. [DOI] [PubMed] [Google Scholar]
- Minuesa G, Volk C, Molina-Arcas M, Gorboulev V, Erkizia I, Arndt P, et al. Transport of lamivudine [(-)-beta-L-2′,3′-dideoxy-3′-thiacytidine] and high-affinity interaction of nucleoside reverse transcriptase inhibitors with human organic cation transporters 1, 2, and 3. J Pharmacol Exp Ther. 2009;329:252–261. doi: 10.1124/jpet.108.146225. [DOI] [PubMed] [Google Scholar]
- Minuesa G, Huber-Ruano I, Pastor-Anglada M, Koepsell H, Clotet B, Martinez-Picado J. Drug uptake transporters in antiretroviral therapy. Pharmacol Ther. 2011;132:268–279. doi: 10.1016/j.pharmthera.2011.06.007. [DOI] [PubMed] [Google Scholar]
- Molina-Arcas M, Bellosillo B, Casado FJ, Montserrat E, Gil J, Colomer D, et al. Fludarabine uptake mechanisms in B-cell chronic lymphocytic leukemia. Blood. 2003;101:2328–2334. doi: 10.1182/blood-2002-07-2236. [DOI] [PubMed] [Google Scholar]
- Molina-Arcas M, Casado FJ, Pastor-Anglada M. Nucleoside transporter proteins. Curr Vasc Pharmacol. 2009;7:426–434. doi: 10.2174/157016109789043892. [DOI] [PubMed] [Google Scholar]
- Munoz P, Iliou MS, Esteller M. Epigenetic alterations involved in cancer stem cell reprogramming. Mol Oncol. 2012;6:620–636. doi: 10.1016/j.molonc.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor-Anglada M, Cano-Soldado P, Errasti-Murugarren E, Casado FJ. SLC28 genes and concentrative nucleoside transporter (CNT) proteins. Xenobiotica. 2008;38:972–994. doi: 10.1080/00498250802069096. [DOI] [PubMed] [Google Scholar]
- Quintas-Cardama A, Santos FP, Garcia-Manero G. Therapy with azanucleosides for myelodysplastic syndromes. Nat Rev Clin Oncol. 2010;7:433–444. doi: 10.1038/nrclinonc.2010.87. [DOI] [PubMed] [Google Scholar]
- Rius M, Lyko F. Epigenetic cancer therapy: rationales, targets and drugs. Oncogene. 2012;31:4257–4265. doi: 10.1038/onc.2011.601. [DOI] [PubMed] [Google Scholar]
- Rius M, Stresemann C, Keller D, Brom M, Schirrmacher E, Keppler D, et al. Human concentrative nucleoside transporter 1-mediated uptake of 5-azacytidine enhances DNA demethylation. Mol Cancer Ther. 2009;8:225–231. doi: 10.1158/1535-7163.MCT-08-0743. [DOI] [PubMed] [Google Scholar]
- Rius M, Keller D, Brom M, Hummel-Eisenbeiss J, Lyko F, Keppler D. Vectorial transport of nucleoside analogs from the apical to the basolateral membrane in double-transfected cells expressing the human concentrative nucleoside transporter hCNT3 and the export pump ABCC4. Drug Metab Dispos. 2010;38:1054–1063. doi: 10.1124/dmd.110.032664. [DOI] [PubMed] [Google Scholar]
- Robak T. New nucleoside analogs for patients with hematological malignancies. Expert Opin Investig Drugs. 2011;20:343–359. doi: 10.1517/13543784.2011.554822. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Mulero S, Errasti-Murugarren E, Ballarin J, Felipe A, Doucet A, Casado FJ, et al. Expression of concentrative nucleoside transporters SLC28 (CNT1, CNT2, and CNT3) along the rat nephron: effect of diabetes. Kidney Int. 2005;68:665–672. doi: 10.1111/j.1523-1755.2005.00444.x. [DOI] [PubMed] [Google Scholar]
- del Santo B, Valdes R, Mata J, Felipe A, Casado FJ, Pastor-Anglada M. Differential expression and regulation of nucleoside transport systems in rat liver parenchymal and hepatoma cells. Hepatology. 1998;28:1504–1511. doi: 10.1002/hep.510280609. [DOI] [PubMed] [Google Scholar]
- Schaeffeler E, Hellerbrand C, Nies AT, Winter S, Kruck S, Hofmann U, et al. DNA methylation is associated with downregulation of the organic cation transporter OCT1 (SLC22A1) in human hepatocellular carcinoma. Genome Med. 2011;3:82. doi: 10.1186/gm298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmiedel BJ, Arelin V, Gruenebach F, Krusch M, Schmidt SM, Salih HR. Azacytidine impairs NK cell reactivity while decitabine augments NK cell responsiveness toward stimulation. Int J Cancer. 2011;128:2911–2922. doi: 10.1002/ijc.25635. [DOI] [PubMed] [Google Scholar]
- Shu Y, Leabman MK, Feng B, Mangravite LM, Huang CC, Stryke D, et al. Evolutionary conservation predicts function of variants of the human organic cation transporter, OCT1. Proc Natl Acad Sci U S A. 2003;100:5902–5907. doi: 10.1073/pnas.0730858100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y, Sheardown SA, Brown C, Owen RP, Zhang S, Castro RA, et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Invest. 2007;117:1422–1431. doi: 10.1172/JCI30558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y, Brown C, Castro RA, Shi RJ, Lin ET, Owen RP, et al. Effect of genetic variation in the organic cation transporter 1, OCT1, on metformin pharmacokinetics. Clin Pharmacol Ther. 2008;83:273–280. doi: 10.1038/sj.clpt.6100275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzvetkov MV, Vormfelde SV, Balen D, Meineke I, Schmidt T, Sehrt D, et al. The effects of genetic polymorphisms in the organic cation transporters OCT1, OCT2, and OCT3 on the renal clearance of metformin. Clin Pharmacol Ther. 2009;86:299–306. doi: 10.1038/clpt.2009.92. [DOI] [PubMed] [Google Scholar]
- Volk C, Gorboulev V, Kotzsch A, Muller TD, Koepsell H. Five amino acids in the innermost cavity of the substrate binding cleft of organic cation transporter 1 interact with extracellular and intracellular corticosterone. Mol Pharmacol. 2009;76:275–289. doi: 10.1124/mol.109.054783. [DOI] [PubMed] [Google Scholar]
- Zhou L, Cheng X, Connolly BA, Dickman MJ, Hurd PJ, Hornby DP. Zebularine: a novel DNA methylation inhibitor that forms a covalent complex with DNA methyltransferases. J Mol Biol. 2002;321:591–599. doi: 10.1016/S0022-2836(02)00676-9. [DOI] [PMC free article] [PubMed] [Google Scholar]