

Abstract
BACKGROUND AND PURPOSE
GPR18 is a candidate cannabinoid receptor, but its classification as such is controversial. The rationale of the study presented herein was to consider the effects of N-arachidonoyl glycine (NAGly) and cannabinoids via differential G-protein coupled pathways, in addition to β-arrestin signalling. Cellular localization of GPR18 receptors was also examined.
EXPERIMENTAL APPROACH
Calcium mobilization and ERK1/2 phosphorylation were quantified in a cell line stably expressing GPR18 (HEK293/GPR18 cells). In addition, using the DiscoveRx PathHunter® CHO-K1 GPR18 β-arrestin cell line, recruitment of β-arrestin was quantified.
KEY RESULTS
Concentration-dependent increases in intracellular calcium and ERK1/2 phosphorylation were observed in the presence of NAGly, abnormal cannabidiol (AbnCBD), O-1602, O-1918 and Δ9-tetrahydrocannabinol (Δ9-THC) in HEK293/GPR18 cells. The initial rise in intracellular calcium in the presence of NAGly, O1918 and THC was blocked by either Gαq or Gαi/o inhibition. The ERK1/2 phosphorylation was inhibited by Pertussis toxin and N-arachidonoyl-L-serine (NARAS). Recruitment of β-arrestin in the PathHunter CHO-K1 GPR18 cell line revealed a differential pattern of GPR18 activation; of all the ligands tested, only Δ9-THC produced a concentration-dependent response. The localization of GPR18 receptors within the HEK293/GPR18 cells is both intracellular, and on the plasma membrane.
CONCLUSIONS AND IMPLICATIONS
These findings suggest that GPR18 activation involves several signal transduction pathways indicative of biased agonism, thereby providing a plausible explanation for the apparent discrepancies in GPR18 activation found in the literature. Additionally, the results presented herein provide further evidence for GPR18 as a candidate cannabinoid receptor.
Introduction
The endogenous cannabinoid system, including cannabinoid receptors and endocannabinoids, has been implicated in many physiological processes; including neurotransmission, inflammation/migration, intraocular pressure, neuropathic and inflammatory pain, obesity, osteoporosis, drug addiction, cardiovascular disorders, and liver disease (Pacher et al., 2006; Qin et al., 2011; Battista et al., 2012; McHugh, 2012; Caldwell et al., 2013; Oliere et al., 2013). Presently there are two bonafide cannabinoid receptors, CB1 and CB2, several endocannabinoids, of which anandamide (AEA) and 2-arachidonoyl glycerol are the most extensively studied, and their synthetic and degradative enzymes (Pertwee et al., 2010; Alexander et al., 2013; Console-Bram et al., 2012). Cannabinoid receptors were so named as they were found to bind, and elicit the effects of, the main component of the Cannabis sativa plant, Δ9-tetrahydrocannabinol (Δ9-THC). However, it is widely accepted that the actions of cannabinoid compounds are not limited to these two receptors. Consequently, it is imperative to elucidate the existence of additional cannabinoid receptors, the compounds with which they interact, and the mechanism(s) of signal transduction. The term cannabinoid compound encompasses three categories of compounds, those derived from the C. sativa plant (phytocannabinoids), synthetically derived compounds (synthetic cannbinoids), as well as endogenous compounds that were shown to bind to CB1 and CB2 receptors (endocannabinoids).
Current opinion describes GPR18 as a candidate cannabinoid receptor (for receptor nomenclature see Alexander et al., 2013). Transfection of GPR18 into CHO, K562 and L929 cells resulted in the mobilization of calcium in the presence of N-arachidonyl glycine (NAGly), a metabolite of the endocannabinoid AEA (Kohno et al., 2006; Bradshaw et al., 2009). Consequently, identification of NAGly as the endogenous ligand of GPR18 was postulated (Kohno et al., 2006). Further support for GPR18 as a candidate cannabinoid receptor has recently emerged as Δ9-THC was found to activate GPR18 in migration assays (McHugh et al., 2012) and in the PathHunter β-arrestin assay (Rempel et al., 2013). In addition, as a consequence of the overlap in the pharmacology of NAGly and abnormal cannabidiol (AbnCBD), and the inhibition of these ligand-induced effects by O-1918, McHugh et al. (2010) proposed that the AbnCBD receptor and GPR18 are one in the same.
Anatomical localization of GPR18 was initially reported to be highest in spinal cord and small intestine, with lower levels in testis and cerebellum (Gantz et al., 1997). In a more recent study, localization of GPR18 mRNA in mouse tissue revealed highest levels in spleen and bone marrow, followed by thymus, lung and cerebellar tissue (Regard et al., 2008). This receptor is also expressed in endometrial cells (McHugh et al., 2012), murine microglial cells (McHugh et al., 2010), and is one of the most highly expressed GPCRs in metastatic melanoma (Qin et al., 2011). As a consequence of its localization, it is quite possible that GPR18 may be involved in a myriad of cellular functions. However, very little is known about the physiological effects of GPR18. Thus far, cellular migration, apoptosis, immunostimulant activity in catfish, increased levels of intracellular calcium and augmented MAPK activity are ascribed to this GPCR (Kohno et al., 2006; McHugh et al., 2010; Takenouchi et al., 2012; Pridgeon and Klesius, 2013). The signal transduction mechanism involved in GPR18 activation by NAGly has been reported to be the consequence of Gαi/o coupling, as Pertussis toxin (PTX) was found to attenuate GPR18-mediated inhibition of cAMP production (Kohno et al., 2006). In studies with BV-2 murine microglia, PTX was found to attenuate NAGly-induced migration (McHugh et al., 2010).
Progress towards acceptance of NAGly as the endogenous ligand of GPR18, and activation of GPR18 by AEA, AbnCBD and O-1602 has been brought into question, as a recent report indicated that none of these ligands activate GPR18 expressed in sympathetic cervical ganglia (Lu et al., 2013). This study examined calcium mobilization via N-type voltage-gated calcium channels of rat superior ganglion cells heterologously expressing GPR18. Low voltage-gated channels of the superior ganglion are described as being inhibited by Gαi/o proteins via an interaction between Gβγ dimers and β subunits of this type of calcium channel (Ikeda, 1996; Herlitze et al., 1999). Consequently, in the presence of Gαi/o agonists, the expectation was that a decrease in calcium would be observed. However, Lu et al. (2013) observed an increase in calcium mobilization and thereby concluded neither NAGly, AbnCBD, anandamide nor O-1602 are agonists at GPR18.
As a consequence of the apparent discrepancies in the literature, the rationale of the present study was to examine the effects of phytocannabinoids (Δ9-THC), synthetic cannabinoids (AbnCBD, O-1602 and O-1918), and endocannabinoids (NAGly) on calcium mobilization in HEK293 cells heterologously expressing GPR18. HEK293 cells were chosen as they are known to be devoid of N-type voltage-gated calcium channels (Berjukow et al., 1996) and ryanodine receptors (Aoyama et al., 2004). Consequently, any change in calcium mobilization would be a consequence of GPR18 activation, not channel activity. In addition, we investigated MAPK activity following exposure to O-1918, NAGly, O-1602, AbnCBD, Δ9-THC and another endocannabinoid, N-arachidonyl serine (NARAS), to extend the findings of McHugh et al. (2012). In an attempt to establish mechanism(s) involved in these GPR18-mediated biological events, the following effector pathway inhibitors were employed: wortmannin (PI3K inhibitor), bisindolymalemide (BI, PKC inhibitor); PTX (Gαi/o inhibitor), and the Gαq inhibitor, [D-Trp7,9,10]-substance P (Mukai et al., 1992). The GPR18-mediated effects of AbnCBD, NAGly, NARAS, O-1602, O-1918 and Δ9-THC were also investigated in the β-arrestin signal transduction pathway. In addition, the activity of AEA and CBD in this signalling mechanism was also explored.
Methods
Cell culture
HEK293/GPR18 cells (a generous gift from Drs. D. McHugh and H. Bradshaw, Indiana University) were maintained in high-glucose DMEM (Gibco, Carlsbad, CA, USA) supplemented with 5% FBS and G418 (350 μg·mL−1) at 37°C, 5% CO2. Cells were passaged every 2–3 days for a maximum of 30 passages. The GPR18 cDNA contains a triple sequence of haemagglutinin (HA) at the N-terminus. The PathHunter β-arrestin CHO-K1/GPR18 cells (DiscoveRx) were maintained in PathHunter select cell culture media at 37°C, 5% CO2 (according to the manufacturer specifications, and passaged every 2–3 days for a maximum of 10 passages).
Poly-D-lysine coating
All cover slips and 60 mm plates were coated with a solution of poly-D-lysine (5 μg·mL−1) for 24 h (4°C), rinsed three times with sterile water, and allowed to air dry at 37°C.
Calcium imaging
HEK293/GPR18 cells were grown on poly-D-lysine-coated coverslips (25 mm in diameter) in 5% FBS/DMEM overnight (20–24 h). Media was then removed and cells were incubated overnight in serum-free DMEM. For Gαq or Gαi studies, cells were exposed to inhibitors overnight in serum-free DMEM. Intracellular Ca2+ measurements were performed as described previously (Brailoiu et al., 2011). Briefly, HEK293/GPR18 cells were incubated with 5 μM fura-2/AM in HBSS at room temperature (RT, 19–23°C) for 45 min in the dark, washed with dye-free buffer, and incubated for another 45 min to allow for complete de-esterification of the dye (in calcium and calcium-free buffer). Coverslips were subsequently mounted in an open bath chamber (RP-40LP, Warner Instruments, Hamden, CT, USA) on the stage of an inverted microscope Nikon Eclipse TiE (Optical Apparatus Co., Ardmore, PA, USA), and cells were exposed to agonists. The microscope was equipped with a Perfect Focus System and a Photometrics CoolSnap HQ2 CCD camera (Roper Scientific, Optical Apparatus Co.). During the experiments the Perfect Focus System was activated.
Fura-2/AM fluorescence (emission = 510 nm) following alternate excitation at 340 and 380 nm was acquired at a frequency of 0.25 Hz. Images were acquired and analysed using Nikon NIS-Elements AR 3.1 software (Optical Apparatus Co.). The ratio of the fluorescence signals (340/380 nm) was converted to Ca2+ concentrations (Grynkiewicz et al., 1985).
MAPK assay
HEK293/GPR18 cells were plated on poly-D-lysine-coated 60 mm plates in 5% FBS/DMEM until 90% confluent. Following 20–24 h in a serum-free medium, cells were rinsed once with HBSS and incubated in fresh HBSS for 30 min at RT. Cells were then incubated with agonists for 5 min at RT or pre-incubated with PTX (100 ng·mL−1) overnight, in serum-free DMEM, before exposure to agonists. NARAS, wortmannin and BI were pre-incubated with cells for 20 min at RT before addition of agonists. Cells were lysed with the addition of: HEPES (50 mM), NaCl (150 mM), EDTA (1 mM), EGTA (1 mM), glycerol (10%, w v−1), Triton X-100 (1%, w v−1), MgCl2 (10 μM), 20 mm p-nitrophenyl phosphate, 1 mm Na3VO4, 25 mm NaF, and a EDTA-free protease inhibitor mixture (1:50, pH 7.5). Cell lysates were incubated on ice for 30 min followed by centrifugation at 16 100× g (4°C), 30 min. Cytosolic fractions (20 μg) were separated on a 10% acrylamide gel by SDS-PAGE followed by standard Western protocols (phospho ERK1/2, 1:5000; total ERK1/2, 1:1000; conjugated secondary antibodies, IRdye 800CW, 1:8000; IRDye 680, 1:10 000). Quantification of MAPK activity was determined using the Odyssey Infrared Imaging System and software (Li-Cor Biosciences, St. Lincoln, NE, USA).
β-Arrestin translocation assay
Quantification of β-arrestin recruitment was determined using DiscoveRx PathHunter CHO-K1 cells stably expressing GPR18 fused with a β-galactosidase enzyme fragment, and β-arrestin fused to an N-terminal deletion mutant of β-galactosidase. Activation of GPR18 induces β-arrestin recruitment, forcing complementation of the two β-galactosidase enzyme fragments. Levels of this active enzyme are a direct result of GPR18 activation and are quantitated using chemiluminescent PathHunter detection reagents containing the β-galactoside substrate. The assay was performed as outlined by the manufacturer. Briefly, cells were plated in 384-well plates at 5000 cells per well in the manufacturer's cell plating two reagent overnight. Cells were then incubated with ligand (AbnCBD, AEA, CBD, NAGly, NARAS, O-1602, O-1918, or Δ9-THC) for 90 min at 37°C. Ligands were dissolved in ethanol and serial dilutions were made in cell plating two reagent. Following a 1 h incubation (RT) in detection reagent, the chemiluminesence signal was measured using a Perkin Elmer Envision plate reader for 1 s (Perkin Elmer, Waltham, MA, USA). Ligand readings were subtracted by their corresponding vehicle reading and analysed in GraphPad Prism (GraphPad Software, La Jolla, CA, USA).
Immunocytochemistry
Cells were plated on poly-D-lysine-coated coverslips and incubated overnight at 37°C in 24-well plates before immunocytochemical protocols. Localization of GPR18 receptors in the HEK293/GPR18 cell line was visualized through the use of two different antibodies. The HA epitope located at the N-terminal end of the receptor was identified using a mouse antibody to HA and an epitope within the cytoplasmic loop was recognized by a rabbit antibody to GPR18. Detection of GPR18 was visualized using Alexafluor 568. Antibody validation was attained by subjecting untransfected HEK293 cells to the same immunocytochemical protocols. Coverslips were mounted onto slides using DAPI Fluromount-G for nuclear identification. Corresponding images from both channels were overlayed in Adobe Photoshop® CS5 (Adobe Systems Inc., San Jose, CA, USA).
Reagents
AbnCBD (Cayman Chemical, Ann Arbor, MI, USA); Alexa Fluor 568 goat anti-mouse, and Alexa Fluor555 goat anti-rabbit (Invitrogen, Carlsbad, CA, USA); anti-GPR18 (Prestige Antibodies, Sigma, St. Louis, MO, USA); anti-HA mouse monoclonal antibody (Covance, Princeton, NJ, USA); BI (LC Laboratories, St. Woburn, MA, USA); [D-Trp7,9,10]-substance P (Tocris Biosciences, R&D Systems Inc., Minneapolis, MN, USA); DAPI Fluoromount-G (Southern Biotech, Birmingham, AL, USA); FBS (Hyclone/Fisher Scientific, Pittsburgh, PA, USA); G418, (A.G. Scientific, San Diego, CA, USA); HBSS and high-glucose DMEM (Cellgro, MediaTech Inc., Manassas, VA, USA); IRDye 800-conjugated goat anti-mouse IgG, IRDye 680-conjugated goat anti-rabbit IgG, and blocking buffer (Li-Cor Biosciences); monoclonal anti-phosphorylated ERK 1/2, Poly-D-Lysine and wortmannin (Sigma); NAGly and O-1602 (Tocris Bioscience); NARAS (Cayman Chemical); O-1918 (Cayman Chemical); PTX salt-free (EMD Sciences); rabbit anti-total ERK1/2 (Cell Signaling); Δ9-THC (Research Triangle Institute, NC); Triton X-100 (Fisher Scientific); zeocin (Invitrogen). All other chemicals were obtained from standard laboratory chemical suppliers. All drug/molecular target nomenclature conforms to the British Journal of Pharamcology's Concise Guide to Pharmacology (Alexander et al., 2013).
Statistical analysis
Results were graphed using GraphPad Prism 5 (GraphPad Software). Data points are presented as a mean ± SEM and results were analysed using a one-way anova followed by Bonferroni correction.
Results
Calcium mobilization in HEK293/GPR18 cells
Extracellular application of Δ9-THC, NAGly, O-1602, AbnCBD or O-1918 resulted in significant transient calcium mobilization within a second of exposure (Figure 1). Real-time photographs of calcium mobilization in basal and treated states are depicted in Figure 1A. The specificity of the GPR18-mediated calcium response was evaluated through the measurement of intracellular calcium in HEK293 cells devoid of GPR18 expression in the presence of ligand (Figure 1B, upper right tracing). Concentration–response curves for the ligands are depicted in Figure 1C. Note that the concentration–response curve for AbnCBD is furthest to the right. At equimolar concentrations, this ligand was the least efficacious, whereas O-1918 was the most efficacious (O1918 > NAGly > Δ9-THC > O-1602 > AbnCBD). Levels of significance (P < 0.05) for AbnCBD-mediated increases in intracellular calcium were attained at 10 and 3 μM, whereas NAGly, O-1602, and Δ9-THC yielded significance at 10, 3 and 1 μM. O-1918-mediated increases in calcium were demonstrated over the largest range of concentrations; 0.3 through 10 μM. To identify the G-proteins involved in agonist-mediated calcium mobilization, HEK293/GPR18 cells were pre-incubated overnight with either the Gαi/o inhibitor PTX (100 ng·mL−1), or the Gαq inhibitor [D-Trp7,9,10]-substance P (10 μM). Calcium mobilization was completely inhibited by both G-protein inhibitors (Figure 1C). Furthermore, in the absence of extracellular calcium, significant agonist-induced GPR18-mediated calcium mobilization occurred (P < 0.05), albeit at lower levels (10 μM NAGly, 299 ± 14.51; 10 μM O-1918, 346 ± 10.92; 10 μM Δ9-THC, 273 ± 5.1 nM). An example of trace recordings with an associated graph of intracellular calcium levels in cells incubated with agonists in the absence of extracellular calcium is shown with Δ9-THC in Figure 2. Note that the time of peak intracellular calcium levels in the absence of extracellular calcium is similar to that observed in the presence of extracellular calcium.
Figure 1.
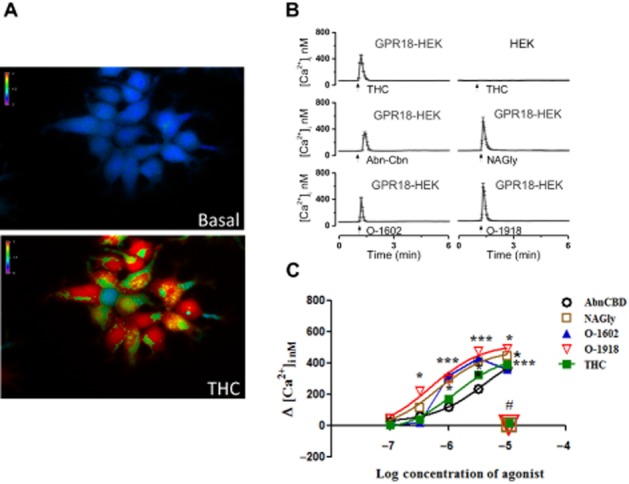
Calcium mobilization of HEK293/GPR18 cells in the presence of NAGly and cannabinoid ligands. (A) Representative micrograph of the change of intracellular calcium [Ca2+]i in HEK293/GPR18 cells before (basal, upper) and after exposure to cannabinoid ligands (Δ9-THC, lower). (B) Real-time traces of the change in [Ca2+]i prior to (basal), and following exposure to cannabinoid ligands. (C) Concentration–response curves of [Ca2+]i in HEK293/GPR18 cells in the presence of cannabinoid ligands (THC, n = 43–67; NAGly, n = 64–86; AbnCBD, n = 48–74; O-1602, n = 46–72; O-1918, n = 71–91; one-way anova, P < 0.05*, P < 0.01**, P < 0.001*** with Bonferroni correction). The lack of change in [Ca2+]i following blockade of Gαi/o and Gαq subunits in the presence of cannabinoid ligands is depicted in this graph by the placement of the number symbol (#) above representative symbols for NAGly; O-1918; Δ9-THC; n = 59–62.
Figure 2.
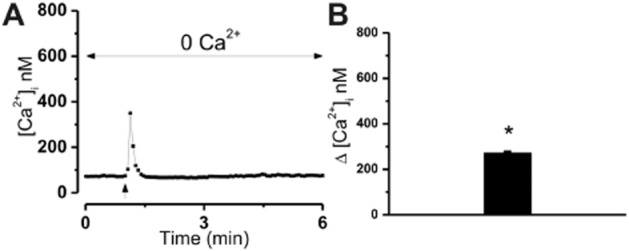
Calcium mobilization of HEK293/GPR18 in the absence of extracellular calcium. (A) Representative tracing illustrating an increase in intracellular calcium [Ca2+]i following exposure of HEK293/GPR18 cells to ligands (Δ9-THC) in the absence of extracellular calcium. (B) Bar graph of the mean increase in [Ca2+]i following exposure to 10 μM THC in the absence of extracellular calcium (one-way anova, *P < 0.05, n = 31–37 with Bonferroni correction).
MAPK activity in HEK293/GPR18 cells
Initial time course studies revealed that levels of ERK1/2 phosphorylation remain elevated for 2 h post exposure to NAGly whereas levels returned to basal levels within 40 min following exposure to AbnCBD and O-1602 (Figure 3). Peak increases were attained 5 min post ligand exposure; consequently, levels of MAPK activity were quantitated at this time point. Significant concentration-dependent increases in ERK1/2 phosphorylation were observed in HEK293/GPR18 cells following exposure to NAGly, O-1602, AbnCBD and Δ9-THC (Figure 4A, C, D and E), similar to previous reports (McHugh et al., 2012). In addition, we demonstrate significant concentration-dependent increases in ERK phosphorylation in the presence of O-1918 (Figure 4B). As shown in Figure 4, pre-incubation with 100 μg·mL−1 of PTX or 1 μM NARAS inhibited the ligand-induced increases in ERK1/2 phosphorylation. No effect was observed with incubation of either NARAS or PTX in the absence of cannabinoid ligands as illustrated in Figure 4C.
Figure 3.

Time course of MAPK activity in HEK293/GPR18 cells. Quantification of ERK1/2 phosphorylation 5–120 min post exposure to 3 μM of NAGly, AbnCBD or O-1602. Values are normalized to basal levels, and are expressed as % of control. The dotted line is indicative of 100% basal activation.
Figure 4.
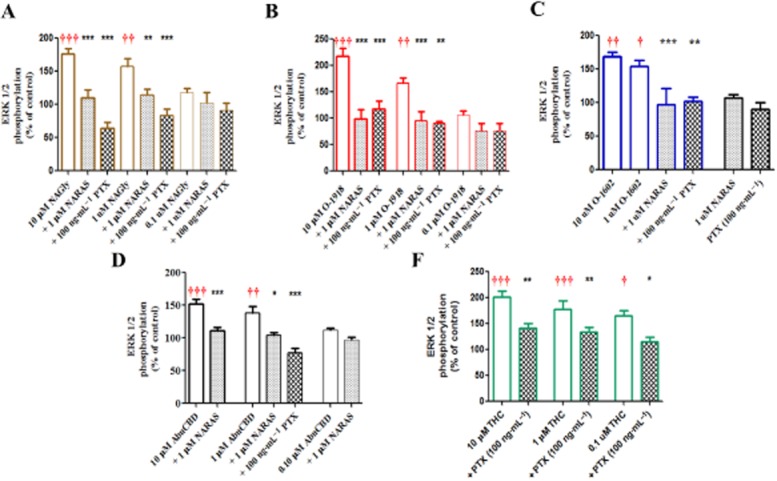
Concentration-dependent changes in ERK1/2 phosphorylation of HEK293/GPR18 cells in the presence of NAGly (A), O-1918 (B), O-1602 (C), AbnCBD (D), and Δ9-THC (E). Significance (†P < 0.05, ††0.01, †††0.001) was determined by one-way anova with Bonferroni correction in comparison with control. * Indicates significant inhibition of the ligand response in the presence of PTX or NARA-S; one-way anova (*P < 0.05, **0.01, ***0.001) with Bonferroni correction.
Further characterization of the MAPK response was performed using the most efficacious ligand, O-1918, with the PI3K inhibitor wortmannin (100 nM), or the PKC inhibitor BI (10 nM). As illustrated in Figure 5, application of either inhibitor attenuated O-1918-dependent ERK1/2 phosphorylation. Because activation of Gαq leads to production of diacylglycerol and inositol-3-phosphate via PLC cleavage and concomitant PKC activation, the results of these experiments supports the involvement of Gαq in GPR18 activation.
Figure 5.
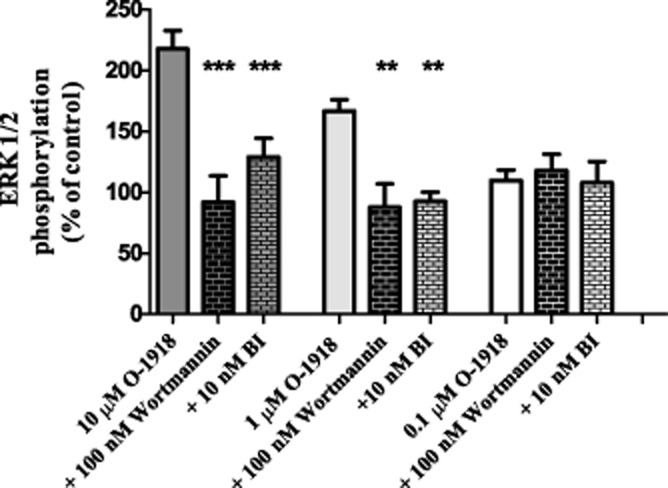
Inhibition of O-1918 induced increases of ERK1/2 phosphorylation in HEK293/GPR18 cells in the presence of the PKC inhibitor, Wortmannin (100 nm) and the PI3K inhibitor, BI (10 nM). Significance (**P < 0.01, ***0.001) was determined by one-way anova with Bonferroni correction.
β-Arrestin activation
Of all the agonists investigated in the MAPK assay only Δ9-THC, had a concentration-dependent effect on β-arrestin translocation (Figure 6). Increases in β-arrestin activity were significantly different at 10 and 30 μM Δ9-THC in comparison with the other cannabinoid ligands (AbnCBD, AEA, NAGly, NARAS, O-1602 and O-1918). It is interesting to note that slight significance in β-arrestin activity was observed at 30 μM CBD, yet the CBD analogues O-1602 and O-1918 had no effect on β-arrestin translocation. These findings further demonstrate multiple signalling avenues for GPR18 and suggest biased agonism at this GPCR.
Figure 6.
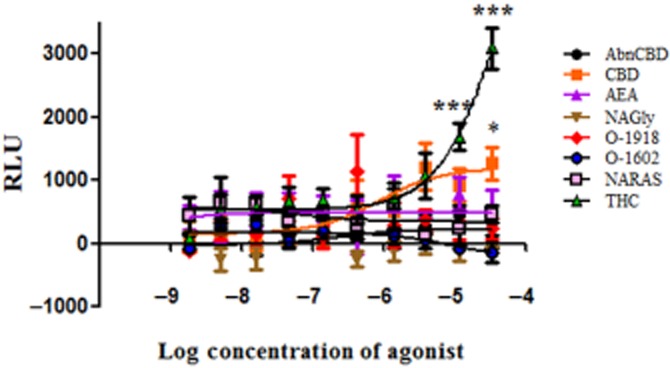
β-Arrestin activity in DiscoveRx CHO-K1 GPR18 cells exposed to NAGly and cannabinoid ligands. Levels of control β-arrestin were subtracted from ligand values and reported as relative luminescence units (RLU). Significant differences between Δ9-THC and the other ligands; AbnCBD, AEA, NAGly, O-1918, O-1602, N-ARAS were determined by one-way anova (***P < 0.001) with Bonferroni correction. CBD showed significance (*P < 0.05) at the 30 μM concentration.
Cellular localization of GPR18
Distribution of GPR18 in HEK293 and HEK293/GPR18 cells using immunocytochemistry is illustrated in Figure 7. Figure 7A (live, nonpermeabilized) and 7B (permeabilized) depict staining of cells with an anti-HA antibody illustrating both plasma membrane and intracellular localization of GPR18. A similar pattern was observed using an anti-GPR18 antibody in permeabilized cells (Figure 7C). Figure 7D illustrates the absence of detectable GPR18 in untransfected HEK293 cells using both antibodies thereby demonstrating specificity of the antibodies.
Figure 7.
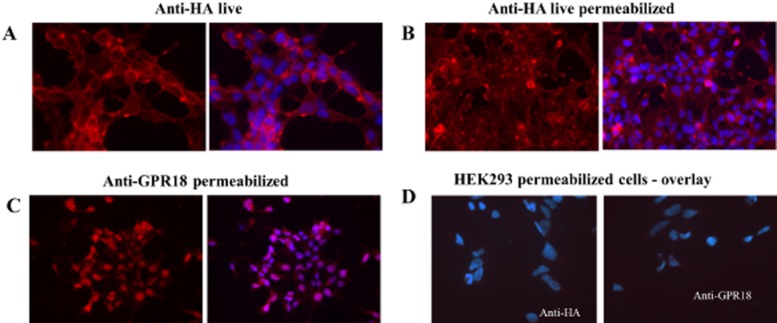
Immunocytochemistry of HEK293/GPR18 cells using anti-HA, and anti-GPR18 antibodies. Antibody staining is depicted in the left panel and nuclear staining using DAPI overlaid with the antibody staining image is depicted in the right panel. (A) Localization of receptor on the plasma membrane of live, non-permeabilized HEK293/GPR18 cells using anti-HA antibodies. (B) Intracellular localization of GPR18 receptors using anti-HA antibodies in live permeabilized cells. (C) Fixed, permeabilized cells depicting intracellular GPR18 receptors using anti-GPR18 antibodies. (D) Specificity of anti-HA and anti-GPR18 antibodies is demonstrated by a lack of staining in fixed, permeabilized wild-type HEK293 cells.
Discussion
Findings from the study presented here indicate that GPR18 signalling is rather complex. Results from our study suggest the occurrence of biased agonism at GPR18 as activation of this receptor by a particular ligand was dependent upon the biological endpoint. Increases in both intracellular calcium and MAPK, two cellular signalling pathways, were observed in HEK/GPR18 cells in the presence of five cannabinoid ligands; Δ9-THC, AbnCBD, NAGly, O-1602 and O-1918. However, the only agonist that exhibited concentration-dependent GPR18-mediated β-arrestin activity was Δ9-THC.
Our research is the first to suggest multiple G-protein involvement in GPR18 activation. The Gαi/o pathway is implicated in view of the findings that ligand-induced increases in MAPK and calcium mobilization were attenuated by PTX treatment. Given that Δ9-THC-induced increases in MAPK were not completely blocked by PTX, and that the GPR18-mediated rises of intracellular calcium induced by NAGly, Δ9-THC and O-1918 were also inhibited by the Gαq inhibitor, [D-Trp7,9,10]-substance P (Mukai et al., 1992), the Gαq pathway is probably involved as well. Additional support for involvement of both G-proteins comes from the observation that the Gαq inhibitor completely blocked the initial and secondary rises in intracellular calcium in the presence of NAGly, Δ9-THC and O-1918; whereas, only the initial rise in intracellular calcium was inhibited by PTX (the secondary rise, observed 10 min following agonist exposure, was not; data not shown). Furthermore, the demonstrated inhibition of ERK1/2 phosphorylation by inhibition of PKC activity, known to be activated via Gαq, reinforces the involvement of Gαq in GPR18 activation. Participation of both Gαi and Gαq transduction pathways suggests effector redundancy; perhaps for indispensable cellular function(s) and/or maintenance of homeostasis. Several GPCRs have been shown to exhibit promiscuity in coupling to G-proteins; for example, rhodopsin and β2-adrenoceptor (Cerione et al., 1985), GPR55 (Lauckner et al., 2008; Obara et al., 2011), melanin-concentrating hormone receptor (Hawes et al., 2000) and CB1 receptor (Ahn et al., 2013). Furthermore, crosstalk among G-protein subunits has been observed for Gαs- and Gαq-coupled adenosine receptors (Benovic et al., 1985). Hence, the involvement of multiple G-proteins in signal transduction via GPR18 is quite plausible.
The results presented here corroborate the agonist ligand profiles of AbnCBD, NAGly, O-1602 and Δ9-THC as reported by McHugh et al. (2012). Our study is the first report of NARAS inhibition of MAPK activity induced by AbnCBD, NAGly, O-1602 and O-1918. This finding complements the reported inhibition of NAGly–GPR18-mediated cellular migration by NARAS (McHugh et al., 2010). However, our finding that O-1918 mediates increases in MAPK activity via GPR18 activation is in contrast with previously reported GPR18 antagonist activity of O-1918 in migration studies using BV-2 and HEK/GPR18 cells (McHugh et al., 2010). Additionally, attenuation of AbnCBD-induced MAPK activity by high concentrations of O-1918 has been reported in HUVEC cells (Offertaler et al., 2003).
Further characterization of O-1918-mediated increases in MAPK activity was performed as a consequence of enhanced calcium mobilization with this ligand. Increases in intracellular calcium, such as triggered by the activation of GPR18, are known to activate several isoforms of PKC. Once activated, either via calcium and/or directly by Gαq activation, PKC may lead to activation of the MAPK and PI3K pathways (Breitkreutz et al., 2007; Rozengurt, 2007). In addition, βγ dimer formation is known to activate PI3K (Naor et al., 2000). We therefore examined whether inhibition of PKC and PI3K pathways would affect O-1918-mediated ERK1/2 phosphorylation. Both the PKC inhibitor (BI), and the PI3K inhibitor (wortmannin), attenuated increases in ERK1/2 phosphorylation. Thus, our findings demonstrate the multiplicity of cellular pathways involved in GPR18-mediated increases in MAPK activity.
As indicated earlier, increases in MAPK induced by AbnCBD, NAGly, O-1602, O-1918 and Δ9-THC were sensitive to PTX. In both BV-2 and human endometrial cells, PTX also attenuates migration induced by NAGly (McHugh et al., 2010; 2012); however, sensitivity to PTX by the other ligands was not demonstrated. In addition, there are discrepancies in the literature with respect to PTX sensitivity of endothelial-dependent AbnCBD-induced vasorelaxation of mesenteric arteries. For example, AbnCBD-induced vasorelaxation of mesenteric arteries has been reported to be both sensitive (Offertaler et al., 2003) and insensitive to PTX treatment (Ho and Hiley, 2003). Whereas aortic relaxation induced by AbnCBD has been demonstrated to be sensitive to PTX inhibition (Milman et al., 2006). NARAS also induced vasorelaxation of aortic and mesenteric vessels; however, these relaxant effects were less endothelium-dependent in mesenteric as compared with aortic arteries, and the effect of NARAS in aortic vessels, but not mesenteric vessels, was antagonized by PTX (Milman et al., 2006). Conversely, O-1918 antagonized the effect of NARAS in mesenteric vessels, but not in aortic vessels. Milman et al. (2006) also reported that AbnCBD- and NARAS-induced increases in MAPK activity were attenuated by PTX in HUVEC cells. As a consequence of the similarity between aortic vessels and HUVEC cells, with respect to PTX sensitivity of these ligand-induced effects, the authors suggested that the vascular receptor mediating these effects were one and the same. However, because of the differences between mesenteric and aortic arteries, they suggested the existence of more than one vascular receptor for these ligands. Alternatively, a single receptor coupling to different G-protein subunits and/or signal transduction pathways, depending upon the local milieu, may satisfy these apparent discrepancies in PTX and O-1918 sensitivity of the ‘AbnCBD’ receptor. Additionally, this explanation may also serve to support the identification of GPR18 as the ‘AbnCBD’ receptor.
To add further complexity to the activation of GPR18, a recent study of rat superior cervical ganglion neurons heterologously expressing GPR18 reported that neither NAGly nor AbnCBD resulted in activation of this receptor (Lu et al., 2013). As activation of Gαi-coupled GPCRs is generally associated with a decrease in calcium current via N-channel modulation, yet an increase in calcium current following exposure to NAGly and AbnCBD was observed, these investigators reported that these ligands did not activate GPR18. We chose to examine the properties of GPR18 in HEK293 cells, as they are known to be devoid of N-type voltage-gated calcium channels (Berjukow et al., 1996), and ryanodine receptors (Aoyama et al., 2004). Consequently, any change in calcium mobilization would be a consequence of GPR18 activation, not channel activity. Our findings clearly demonstrate that GPR18 is activated by NAGly, AbnCBD, O-1602, O-1918 and Δ9-THC, resulting in increased calcium mobilization and MAPK activity. Nonetheless, it is feasible that the properties of GPR18 may be different in neuronal cells.
Our findings that PTX totally blocks MAPK activity induced by AbnCBD, NAGly, O-1602 and O-1918, but not Δ9-THC, supports the involvement of multiple signal transduction pathways in GPR18 activation. The attenuation (as opposed to blockade) of Δ9-THC-induced increases in ERK1/2 phosphorylation by PTX suggests involvement of more than Gαi/o coupling. Furthermore, the calcium mobilization studies presented here suggest that GPR18 activation can occur via differential coupling of G-proteins. The complete inhibition of calcium mobilization in the presence of either PTX, or [D-Trp7,9,10]-substance P, suggests a dual pathway of Gαi and Gαq transduction. Attenuation of MAPK by PI3K and PKC inhibitors substantiate the involvement of PKC in GPR18 activation, a known effector of Gαq-coupled receptors. Findings that AbnCBD, NAGly, O-1602, O-1918 and Δ9-THC enhance intracellular calcium and MAPK activity, yet only Δ9-THC activates β-arrestin signalling implies that these agonists are biased. Curiously, at a single concentration the phytocannabinoid CBD, but not synthetic analogues O-1602 or O-1918, engage β-arrestin signalling. As a consequence of an intracellular and plasma membrane localization of GPR18, it is possible that GPR18 mediates cellular events at both of these locations via different signalling pathways. Both CB1 receptors and GPR55 have previously been described as having functional intracellular and plasma membrane locations with unique signalling pathways (Brailoiu et al., 2011; Yu et al., 2013).
Collectively, the results presented herein complement the discoveries that: activation of GPR18 increases MAPK activity, and migration of BV-2, HEK293/GPR18 and endometrial cells (McHugh et al., 2010; 2012), GPR18 mobilizes calcium in the presence of NAGly (Kohno et al., 2006), and NAGly is devoid of β-arrestin activity at GPR18 (Yin et al., 2009). Our findings that O-1918 enhances both GPR18-mediated MAPK activity and calcium mobilization, but not β-arrestin translocation, along with previous reports that O-1918 antagonizes other GPR18-mediated biological events, is suggestive that this ligand is biased. Garnering further support for biased agonism at GPR18 are the findings that NAGly, O-1602 and AbnCBD lead to augmented levels of GPR18-mediated MAPK activity and calcium mobilization, but not β-arrestin signalling. This revelation sheds light on the multiplicity of mechanisms involved in GPR18 signalling, and will ultimately enable identification of additional ligands capable of targeting transduction pathways specific for inhibition or activation of a desired GPR18-mediated biological event. Regardless of whether NAGly is the principal endogenous ligand for GPR18, investigations by several independent laboratories, including ours, strengthen the evidence for activation of GPR18 by NAGly and other cannabinoid ligands. Importantly, activation of GPR18 by Δ9-THC reinforces the classification of GPR18 as a cannabinoid receptor.
Acknowledgments
This research was supported by the National Institutes of Health National Institute on Drug Abuse (Grants R01DA023204, T32DA007237, P30DA013429).
Glossary
- Δ9-THC
Δ9-tetrahydrocannabinol
- AbnCBD
abnormal cannabidiol
- AEA
anandamide
- BI
bisindolymalemide
- CB1 receptor
cannabinoid receptor 1
- CBD
cannabidiol
- GPR18
G-protein coupled receptor 18
- HA
haemagglutinin
- NARAS
N-arachidonoyl-L-serine
- O-1602
5-methyl-4-[ (1R,6R)-3-methyl-6-(1-cyclohexen-1-yl]-1,3-benzenediol
- O-1918
1,3-dimethoxy-5-methyl-2-[ (1R,6R)-3-methyl-6-(1-methylethenyl)-2-cyclohexen-1-yl]-benzene
- PTX
Pertussis toxin
- RT
room temperature
Author contributions
L.C.B. designed and performed experiments, analysed data and wrote the paper; E.B. designed and performed experiments, and analysed data; G.C.B. implemented experiments; H.S. designed experiments and contributed to the writing of the paper; and M.E.A. designed experiments and wrote the paper.
Conflict of interest
None.
References
- Ahn KH, Mahmoud MM, Shim JY, Kendall DA. Distinct roles of beta-arrestin 1 and beta-arrestin 2 in ORG27569-induced biased signaling and internalization of the cannabinoid receptor 1 (CB1) J Biol Chem. 2013;288:9790–9800. doi: 10.1074/jbc.M112.438804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama M, Yamada A, Wang J, Ohya S, Furuzono S, Goto T, et al. Requirement of ryanodine receptors for pacemaker Ca2+ activity in ICC and HEK293 cells. J Cell Sci. 2004;117(Pt 13):2813–2825. doi: 10.1242/jcs.01136. [DOI] [PubMed] [Google Scholar]
- Battista N, Di Tommaso M, Bari M, Maccarrone M. The endocannabinoid system: an overview. Front Behav Neurosci. 2012;6:9. doi: 10.3389/fnbeh.2012.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benovic JL, Pike LJ, Cerione RA, Staniszewski C, Yoshimasa T, Codina J, et al. Phosphorylation of the mammalian beta-adrenergic receptor by cyclic AMP-dependent protein kinase. Regulation of the rate of receptor phosphorylation and dephosphorylation by agonist occupancy and effects on coupling of the receptor to the stimulatory guanine nucleotide regulatory protein. J Biol Chem. 1985;260:7094–7101. [PubMed] [Google Scholar]
- Berjukow S, Doring F, Froschmayr M, Grabner M, Glossmann H, Hering S. Endogenous calcium channels in human embryonic kidney (HEK293) cells. Br J Pharmacol. 1996;118:748–754. doi: 10.1111/j.1476-5381.1996.tb15463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw HB, Rimmerman N, Hu SS, Benton VM, Stuart JM, Masuda K, et al. The endocannabinoid anandamide is a precursor for the signaling lipid N-arachidonoyl glycine by two distinct pathways. BMC Biochem. 2009;10:14. doi: 10.1186/1471-2091-10-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brailoiu GC, Oprea TI, Zhao P, Abood ME, Brailoiu E. Intracellular cannabinoid type 1 (CB1) receptors are activated by anandamide. J Biol Chem. 2011;286:29166–29174. doi: 10.1074/jbc.M110.217463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitkreutz D, Braiman-Wiksman L, Daum N, Denning MF, Tennenbaum T. Protein kinase C family: on the crossroads of cell signaling in skin and tumor epithelium. J Cancer Res Clin Oncol. 2007;133:793–808. doi: 10.1007/s00432-007-0280-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell MD, Hu SS, Viswanathan S, Bradshaw H, Kelly ME, Straiker A. A GPR18-based signalling system regulates IOP in murine eye. Br J Pharmacol. 2013;169:834–843. doi: 10.1111/bph.12136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerione RA, Staniszewski C, Benovic JL, Lefkowitz RJ, Caron MG, Gierschik P, et al. Specificity of the functional interactions of the beta-adrenergic receptor and rhodopsin with guanine nucleotide regulatory proteins reconstituted in phospholipid vesicles. J Biol Chem. 1985;260:1493–1500. [PubMed] [Google Scholar]
- Console-Bram L, Marcu J, Abood ME. Cannabinoid receptors: nomenclature and pharmacological principles. Prog Neuropsychopharmacol Biol Psychiatry. 2012;38:4–15. doi: 10.1016/j.pnpbp.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantz I, Muraoka A, Yang YK, Samuelson LC, Zimmerman EM, Cook H, et al. Cloning and chromosomal localization of a gene (GPR18) encoding a novel seven transmembrane receptor highly expressed in spleen and testis. Genomics. 1997;42:462–466. doi: 10.1006/geno.1997.4752. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hawes BE, Kil E, Green B, O'Neill K, Fried S, Graziano MP. The melanin-concentrating hormone receptor couples to multiple G proteins to activate diverse intracellular signaling pathways. Endocrinology. 2000;141:4524–4532. doi: 10.1210/endo.141.12.7833. [DOI] [PubMed] [Google Scholar]
- Herlitze S, Ruppersberg JP, Mark MD. New roles for RGS2, 5 and 8 on the ratio-dependent modulation of recombinant GIRK channels expressed in Xenopus oocytes. J Physiol. 1999;517(Pt 2):341–352. doi: 10.1111/j.1469-7793.1999.0341t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho WS, Hiley CR. Vasodilator actions of abnormal-cannabidiol in rat isolated small mesenteric artery. Br J Pharmacol. 2003;138:1320–1332. doi: 10.1038/sj.bjp.0705160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein beta gamma subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- Kohno M, Hasegawa H, Inoue A, Muraoka M, Miyazaki T, Oka K, et al. Identification of N-arachidonylglycine as the endogenous ligand for orphan G-protein-coupled receptor GPR18. Biochem Biophys Res Commun. 2006;347:827–832. doi: 10.1016/j.bbrc.2006.06.175. [DOI] [PubMed] [Google Scholar]
- Lauckner JE, Jensen JB, Chen HY, Lu HC, Hille B, Mackie K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc Natl Acad Sci U S A. 2008;105:2699–2704. doi: 10.1073/pnas.0711278105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu VB, Puhl HL, Ikeda SR. N-arachidonyl glycine (NAGly) does not activate G protein-coupled receptor 18 (GPR18) signaling via canonical pathways. Mol Pharmacol. 2013;83:267–282. doi: 10.1124/mol.112.081182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh D. GPR18 in microglia: implications for the CNS and endocannabinoid system signalling. Br J Pharmacol. 2012;167:1575–1582. doi: 10.1111/j.1476-5381.2012.02019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh D, Hu SS, Rimmerman N, Juknat A, Vogel Z, Walker JM, et al. N-arachidonoyl glycine, an abundant endogenous lipid, potently drives directed cellular migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neurosci. 2010;11:44. doi: 10.1186/1471-2202-11-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh D, Page J, Dunn E, Bradshaw HB. Delta(9)-THC and N-arachidonyl glycine are full agonists at GPR18 and induce migration in the human endometrial HEC-1B cells. Br J Pharmacol. 2012;165:2414–2424. doi: 10.1111/j.1476-5381.2011.01497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milman G, Maor Y, Abu-Lafi S, Horowitz M, Gallily R, Batkai S, et al. N-arachidonoyl L-serine, an endocannabinoid-like brain constituent with vasodilatory properties. Proc Natl Acad Sci U S A. 2006;103:2428–2433. doi: 10.1073/pnas.0510676103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai H, Munekata E, Higashijima T. G protein antagonists. A novel hydrophobic peptide competes with receptor for G protein binding. J Biol Chem. 1992;267:16237–16243. [PubMed] [Google Scholar]
- Naor Z, Benard O, Seger R. Activation of MAPK cascades by G-protein-coupled receptors: the case of gonadotropin-releasing hormone receptor. Trends Endocrinol Metab. 2000;11:91–99. doi: 10.1016/s1043-2760(99)00232-5. [DOI] [PubMed] [Google Scholar]
- Obara Y, Ueno S, Yanagihata Y, Nakahata N. Lysophosphatidylinositol causes neurite retraction via GPR55, G13 and RhoA in PC12 cells. PLoS ONE. 2011;6:e24284. doi: 10.1371/journal.pone.0024284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offertaler L, Mo FM, Batkai S, Liu J, Begg M, Razdan RK, et al. Selective ligands and cellular effectors of a G protein-coupled endothelial cannabinoid receptor. Mol Pharmacol. 2003;63:699–705. doi: 10.1124/mol.63.3.699. [DOI] [PubMed] [Google Scholar]
- Oliere S, Joliette-Riopel A, Potvin S, Jutras-Aswad D. Modulation of the endocannabinoid system: vulnerability factor and new treatment target for stimulant addiction. Front Psychiatry. 2013;4:109. doi: 10.3389/fpsyt.2013.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB and CB. Pharmacol Rev. 2010;62:588–631. doi: 10.1124/pr.110.003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pridgeon JW, Klesius PH. G-protein coupled receptor 18 (GPR18) in channel catfish: expression analysis and efficacy as immunostimulant against Aeromonas hydrophila infection. Fish Shellfish Immunol. 2013;35:1070–1078. doi: 10.1016/j.fsi.2013.07.017. [DOI] [PubMed] [Google Scholar]
- Qin Y, Verdegaal EM, Siderius M, Bebelman JP, Smit MJ, Leurs R, et al. Quantitative expression profiling of G-protein-coupled receptors (GPCRs) in metastatic melanoma: the constitutively active orphan GPCR GPR18 as novel drug target. Pigment Cell Melanoma Res. 2011;24:207–218. doi: 10.1111/j.1755-148X.2010.00781.x. [DOI] [PubMed] [Google Scholar]
- Regard JB, Sato IT, Coughlin SR. Anatomical profiling of G protein-coupled receptor expression. Cell. 2008;135:561–571. doi: 10.1016/j.cell.2008.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempel V, Volz N, Glaser F, Nieger M, Brase S, Muller CE. Antagonists for the orphan G protein-coupled receptor GPR55 based on a coumarin scaffold. J Med Chem. 2013;56:4798–4810. doi: 10.1021/jm4005175. [DOI] [PubMed] [Google Scholar]
- Rozengurt E. Mitogenic signaling pathways induced by G protein-coupled receptors. J Cell Physiol. 2007;213:589–602. doi: 10.1002/jcp.21246. [DOI] [PubMed] [Google Scholar]
- Takenouchi R, Inoue K, Kambe Y, Miyata A. N-arachidonoyl glycine induces macrophage apoptosis via GPR18. Biochem Biophys Res Commun. 2012;418:366–371. doi: 10.1016/j.bbrc.2012.01.027. [DOI] [PubMed] [Google Scholar]
- Yin H, Chu A, Li W, Wang B, Shelton F, Otero F, et al. Lipid G protein-coupled receptor ligand identification using beta-arrestin PathHunter assay. J Biol Chem. 2009;284:12328–12338. doi: 10.1074/jbc.M806516200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Deliu E, Zhang XQ, Hoffman NE, Carter RL, Grisanti LA, et al. Differential activation of cultured neonatal cardiomyocytes by plasmalemmal versus intracellular G protein-coupled receptor 55. J Biol Chem. 2013;288:22481–22492. doi: 10.1074/jbc.M113.456178. [DOI] [PMC free article] [PubMed] [Google Scholar]