

Abstract
Bone morphogenetic proteins (BMPs) are members of the TGFβ superfamily of secreted cysteine knot proteins that includes TGFβ1, nodal, activins and inhibins. BMPs were first discovered by Urist in the 1960s when he showed that implantation of demineralized bone into intramuscular tissue of rabbits induced bone and cartilage formation. Since this seminal discovery, BMPs have also been shown to play key roles in several other biological processes, including limb, kidney, skin, hair and neuronal development, as well as maintaining vascular homeostasis. The multifunctional effects of BMPs make them attractive targets for the treatment of several pathologies, including bone disorders, kidney and lung fibrosis, and cancer. This review will summarize current knowledge on the BMP signalling pathway and critically evaluate the potential of recombinant BMPs as pharmacological agents for the treatment of bone repair and tissue fibrosis in patients.
Keywords: bone morphogenetic protein, skeleton, fracture, fibrosis, gremlin, inhibitors, scarring
Introduction
Bone morphogenetic proteins (BMPs) are glycosylated, secreted extracellular matrix-associated molecules that regulate a wide variety of biological processes (Walsh et al., 2010). BMPs are members of the TGFβ superfamily of proteins (for nomenclature see Alexander et al., 2013), and they have been shown to regulate limb and digit formation, kidney development, cancer, angiogenesis and tissue fibrosis. The importance of BMP signalling during development is underpinned by the elaborate regulatory mechanisms controlling BMP signalling intra- and extracellularly. These processes range from epigenetic methylation and miRNA-mediated RNA regulation, protein ubiquitination, pseudo-receptors and secreted extracellular antagonists that bind to BMPs, preventing their engagement with their cognate receptors (Walsh et al., 2010). Dysregulation of the BMP signalling pathway can have drastic consequences during mammalian development. Mutations in BMP receptors are implicated in vascular conditions, such as pulmonary artery hypertension (PAH), skeletal abnormalities, such as brachydactyly, and polyp formation in the colon (reviewed in Miyazono et al., 2010). These data are supported by a wide variety of skeletal and other phenotypes in BMP pathway transgenic and knockout mice. Herein, we will summarize current knowledge on BMP signalling in cells, how BMPs are processed and secreted, and focus on the role of BMPs in bone and cartilage formation. In particular, we will discuss current and potential uses for recombinant human BMPs for the treatment of compound bone fractures and fibrotic diseases of the kidney and lung. Finally, we will briefly discuss new data demonstrating the therapeutic potential of targeting BMP antagonists in PAH.
Intracellular BMP processing and secretion
There are approximately 20 members of the BMP family, and these have been expertly described by colleagues elsewhere (Balemans and Van Hul, 2002; Weiskirchen and Meurer, 2013). BMPs are synthesized as large inactive precursor proteins that contain a signal peptide at the N-terminus and a mature polypeptide at the C-terminus, connected by a pro-domain that regulates proper folding (Xiao et al., 2007). In the case of BMP-4, the precursor protein is cleaved by the proprotein convertase furin at two sites within the prodomain (S1 and S2), a process thought to occur within the Golgi network (Figure 1). Cleavage at the S1 (RXKR) site alone results in a non-covalently associated ligand–prodomain complex that is relatively unstable and targeted for degradation by the proteasome. The mature active BMP peptide is generated by initial cleavage at the S1 site, which is then trafficked to a compartment within the post-trans-Golgi network, where the acidic environment makes the accessibility and subsequent cleavage at the S2 site (RXXR) site possible. This liberates the mature BMP peptide from the prodomain to yield the stable, active, mature BMP protein (Nelsen and Christian, 2009). These active, mature BMP monomers contain seven cysteines, six of which form three intramolecular disulphide bonds, also known as cysteine knots. The remaining seventh cysteine amino acid facilitates the dimerization with another BMP monomer by forming a covalent disulfide bond, establishing a biologically active dimeric ligand for BMP receptor activation (Bragdon et al., 2011). BMP homodimers are the dominant signalling form of each BMP, and these are bound by homodimeric BMP antagonists such as noggin and gremlin, which restrict their activity (discussed below) (Israel et al., 1996; Zhu et al., 2006; Guo and Wu, 2012). A role for BMP-2/7 and BMP-4/7 heterodimers in mesoderm induction and differentiation of bone marrow cells has also been described (Suzuki et al., 1997; Yuan et al., 2011).
Figure 1.
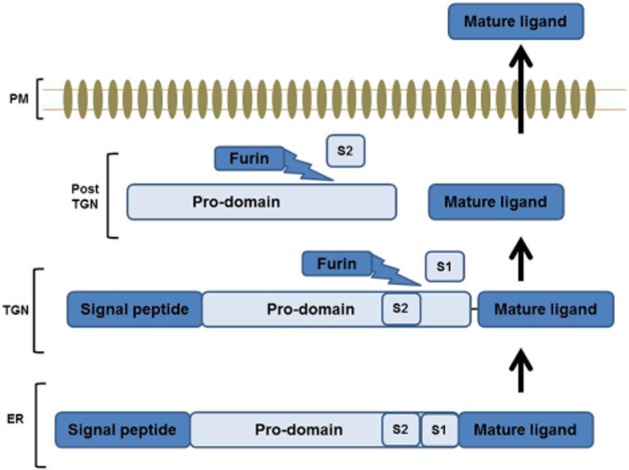
Model of BMP synthesis and site-specific cleavage by proprotein convertases. BMPs are synthesized within the cell as large, inactive dimeric precursor proteins within the ER that require site-specific cleavage by proprotein convertases to produce stable, active dimers. This occurs at two sites located within its pro-domain. Initial cleavage at S1 site is thought to occur in the TGN and results in a pro-domain–ligand complex. This pro-domain–ligand complex is further cleaved at S2 site, which is rendered accessible due to the acidic enviroment within the post-TGN. This releases the mature ligand from the pro-domain, yielding a stable biologically active BMP monomer. ER, endoplasmic reticulum; TGN, trans-Golgi network; CM, cell membrane.
BMP signalling
BMPs elicit their effects through two classes of transmembrane serine/threonine kinase receptors known as type I (BMPR-I) and type II receptors (BMPR-II). (Rosenzweig et al., 1995). There are three type II receptors that BMPs bind to: BMP type II receptor (BMPR2), activin A receptor type II (ActR2) and activin A receptor type IIB (ActR2B). There are also three type I receptors that BMPs preferentially bind to: activin receptor-like kinase (ALK)2, ALK3 (BMPRIA) and ALK6 (BMPRIB) (Nohe et al., 2004). Certain BMPs have been shown to have a higher affinity for certain type I receptors. For example, BMP-4 preferentially binds to ALK3 and ALK6, whereas BMP-6 and -7 preferentially bind to ALK2, but can also engage with ALK3 (Aoki et al., 2001). Once bound, this ligand/receptor complex recruits the constitutively active type II receptor, which phosphorylates the type I receptor on its cytoplasmic domain that is rich in glycine and serine residues (GS domain) (Miyazono et al., 2010). Upon ligand binding, the BMP signal is transmitted from the cell membrane to the nucleus via the canonical sma and mothers against decapentaplegic (Smad)-dependent and/or non-canonical Smad-independent pathways (e.g. MAPK and Akt pathways).
Smad proteins: mediators of BMP signalling
Smad proteins are the vertebrate homologues of the Drosophila melanogaster mothers against decapentaplegic and related Caenorhabditis elegans Sma gene (Riggins et al., 1996). The Smad proteins are grouped based on their activators and functions. Smad1/5/8 are termed as receptor Smads (R-Smads) and are downstream targets of BMP ligands via BMPR1 activation. There is a second class of R-Smads called Smad2/3, which mainly regulates downstream targets of TGFβ signalling through TGFβ2 and ALK5 type I receptors (Massague et al., 2005; Murakami et al., 2009). Recent data suggest that TGFβ can also phosphorylate Smad1/5 in endothelial cells (Liu et al., 2009). This ‘switch’ in Smad protein engagement by TGFβ1 is facilitated by the loss of BMP and activin membrane bound inhibitor (BAMBI) expression and may contribute to endothelial homeostasis (Guillot et al., 2012). Structurally, BAMBI is similar to the type I receptor; however, it lacks an intracellular kinase domain. BMP signalling can also be regulated through BAMBI (Figure 2). As BAMBI is structurally similar to the type I BMP receptor, it competes with the type I receptors for BMP ligand interaction, subsequently trapping BMP ligands and inhibiting signalling (Onichtchouk et al., 1999). BMP-2 has been shown to phosphorylate Smad2/3 in B16 melanoma cells (Murakami et al., 2009), thus demonstrating that the previous model of specific activation of R-Smads by specific TGFβ family members is not as restricted as previously thought.
Figure 2.

Activation and regulation of BMP/Smad-dependent signalling. BMP ligands bind to and activate type I and II serine/threonine kinase receptors (BMPR-I and BMPR-II, respectively), which triggers phosphorylation of the R-Smad1/5/8. Phosphorylated R-Smads form a heteromeric complex with the co-Smad 4, enabling its subsequent translocation to the nucleus where it binds to specific GC rich sequences within the promoters of several target genes in concert with various DNA binding co-factors. This cascade is tightly regulated, through pseudo-receptors such as BAMBI, which quench BMP ligands thereby limiting their availability to interact with their receptors. Extracellular regulation occurs via direct interaction of BMPs with their secreted antagonists, thereby preventing ligand receptor interaction. Intracellularly, this pathway is regulated by the I-Smads, Smad6 and 7. Smad6 prevents the formation of R-Smad and co-Smad complex formation. Smad7 regulates this pathway by forming a complex with Smurf1/2 and competing with R-Smad for type I receptor-mediated activation effectively antagonizing BMP/Smad pathway activation. This pathway is also regulated by phosphatases such as PP1, which dephosphorylates the BMP type I receptor, and PPM1A, which dephosphorylates R-Smads. R-Smad, receptor regulated Smads; I-Smad, inhibitory Smads; co-Smad, co-mediator Smad; ID, inhibitor of differentiation; PAI1, plasminogen activator inhibitor-1; PP1, protein phosphatase 1; PPM1A, metal ion-dependent protein phosphatases 1A; DBC, DNA binding co-factors; PM, plasma membrane.
Once the R-Smads have undergone phosphorylation, interaction with their respective anchor proteins [endofin for R-Smad1/5/8 (Shi et al., 2007) and SARA for R-Smad2/3 (Tsukazaki et al., 1998)] is destabilized, facilitating R-Smad interaction with activated type I receptors to form a heteromeric complex with the common co-mediator Smad, Smad4 (co-Smad) (Xu et al., 2000; Shi et al., 2007). This interaction takes place via the MH2 domain of the R-Smads (Chacko et al., 2001). The MH2 domain is conserved among all types of Smads, including the inhibitory Smads (I-Smads), Smad6/7, which are endogenous inhibitors of this pathway. In contrast, the MH1 domain is conserved in the R-Smads and co-Smad4 only (Miyazono et al., 2010). The MH1 domain is responsible for Smad-mediated DNA binding, interacting with certain DNA binding proteins and antagonizing MH2 functions. The MH2 domain is responsible for BMP receptor recognition [via their Ser-Ser-X-Ser (SSXS) sequence motif], interaction with other Smads, nuclear translocation and DNA binding (Wrana, 2000). Unphosphorylated R-Smads are inactive, as their MH1 and MH2 domains interact with each other, therefore suppressing the functions of each other. Upon receptor activation, Smad-receptor binding occurs, the MH1/MH2 interaction is disrupted, and the Smad is phosphorylated. The Smad nuclear import signal is exposed, which increases R-Smad affinity for co-Smad4, facilitating the formation of an R-Smad/co-Smad complex. This complex then translocates to the nucleus and regulates the transcription of various target genes such as Smad6, ID genes, BAMBI and Smad7 through interaction with either Smad-binding elements (SBEs) or GC-rich sequences found in the promoters of BMP target genes (Figure 2; Miyazono et al., 2010). The affinity between the Smad complex and SBE motif is relatively weak due to promoters of target genes containing only one or more SBEs (Shi et al., 1998; Massague et al., 2005). In vitro, this can be overcome by using concatemers with multiple SBE repeats, which increase the binding affinity for transcriptional activation (Zawel et al., 1998). Physiologically, concatemers rarely occur, so increasing the affinity of Smad binding to target DNA requires the association of Smads with DNA binding cofactors (Massague et al., 2005). One example of R-Smad/co-Smad complex DNA binding cofactor is the forkhead family member, FoxH1. The R-Smad/co-Smad complex binds to FoxH1 to regulate the transcription of Mix2 (Chen et al., 1997). There are, however, a number of other DNA binding cofactors for BMP signalling, including Runx, HOXc8 and CREB-binding protein (Li and Cao, 2006; Miyazono et al., 2010).
Regulation of BMP signalling
The BMP/Smad signalling cascade is tightly regulated by both intra- and extracellular processes: intracellular processes include proteasome-mediated degradation via Smurf proteins, for example, Smurf1, which has been shown to target R-Smad1/5 for degradation (Zhu et al., 1999), and also inhibition via I-Smad6 and 7 action, which act at different points in the pathway. Smad7 resides within the nucleus, and upon BMP/TGFβ receptor activation, it is released into the cytoplasm (Itoh et al., 1998). It has been shown that Smurf1/2 interacts with Smad7 to facilitate inhibition of the Smad pathway via binding to the activated receptor, thereby competitively antagonizing Smad1/5/8 binding (Suzuki et al., 2002). Smad6 specifically inhibits the BMP pathway by interacting with activated R-Smads, preventing the formation of the R-Smad/co-Smad complex, thus inhibiting signal transduction (Hata et al., 1998).
Protein phosphorylation/dephosphorylation is also important in the tight regulation of BMP cell signalling. Protein phosphatase-1 (PP1) regulates BMP signalling by dephosphorylation of the TGFβ/BMP receptor. TGFβ/BMP type 1 receptor stimulation leads to Smad7 interaction with growth arrest and DNA damage protein (GADD34), which is a regulatory/targeting subunit of the type 1 receptor. This then triggers the recruitment of the catalytic subunit of PP1 (PP1c) and subsequent dephosphorylation of the TGFβ type 1 receptor, thereby reducing TGFβ receptor activation (Shi et al., 2004). Dephosphorylation of R-Smads can also occur through the metal ion-dependent protein phosphatases 1A (PPM1A). PPM1A directly interacts with phosphorylated R-Smads, resulting in their inactivation through dephosphorylation (X Lin et al., 2006b). Together, both PP1c and PPM1A act as effective ‘off’ switches for TGFβ/BMP signalling.
The BMP/Smad signalling pathway is also regulated by a family of secreted extracellular antagonists, that directly bind to the BMP ligand preventing their interaction with BMP receptors. These antagonists have been extensively reviewed previously (Balemans and Van Hul, 2002; Walsh et al., 2010; Nakamura and Yanagita, 2012). Briefly, antagonists of BMPs include proteins such as noggin, chordin, gremlin, crossveinless, USAG-1 and follistatin. Most of these proteins are expressed in a highly regulated temporospatial manner during development. The key role of these antagonists and their BMP targets is highlighted by the often lethal developmental defects displayed by mice lacking one or more of these proteins (reviewed in Walsh et al., 2010). Noggin is a potent antagonist of BMP signalling, showing high affinity for BMP-2 and -4, although it can also antagonize BMP-7. The co-crystal of homodimeric noggin bound to homodimeric BMP-7 has provided the field with critical information regarding the structural basis of BMP-BMP antagonist action (Zimmerman et al., 1996; Groppe et al., 2002).
Chordin is expressed in the brains of adult rats, where high levels of BMP-2/4 have also been reported (Mikawa and Sato, 2013). Gremlin was originally identified as a protein capable of inducing secondary axis formation in Xenopus laevis embryos (Hsu et al., 1998). Gremlin has been shown to play a critical role in kidney development, with grem1 homozygous knockout mice born without kidneys as a result of excessive BMP/Smad signalling (Michos et al., 2007). This phenotype was rescued by reducing BMP-4 signalling ‘volume’ in grem1−/−;bmp-4+/− mice (Michos et al., 2007). Like noggin and chordin, gremlin regulates BMP signalling through direct interaction with BMP ligands, thereby blocking ligand–receptor interaction (Wordinger et al., 2008). Gremlin can also regulate BMP signalling intracellularly through intracellular interaction of gremlin with BMP-4 precursor protein, inhibiting the formation and secretion of the mature and active BMP ligand (Sun et al., 2006). Levels of gremlin are increased in fibrotic disease, and pharmacological strategies to inhibit gremlin action will be discussed later in this review.
BMP signalling during bone development
The key role of BMPs in skeletal development has been expertly summarized by others elsewhere. We provide a brief summary of the area below. The vertebrate skeleton consists of bone and cartilage, and contains three main cell types: chondrocytes, osteoblasts and osteoclasts (Karsenty, 2003). Bone formation in vertebrates involves both membranous ossification (involving osteoblast differentiation) and endochondral ossification (involving chondrocyte differentiation, review in Karsenty, 2003; Nishimura et al., 2012). Both of these processes are regulated by BMPs, and BMP-2 and BMP-4 are powerful inducers of osteoblast and chondrocyte differentiation, which induces bone and cartilage formation (Nishimura et al., 2012). BMPs were originally isolated from bone by Marshall Urist in 1965, when he showed that soluble extracts from bone extracellular matrix could stimulate bone formation when injected into rodents (Urist, 1965).
So how do BMPs such as BMP-2 and BMP-4 drive bone formation in vertebrates? The mechanism of BMP activation of their type I/II receptors and the R-Smad1/5/8 canonical signalling pathway is described earlier. This pathway is active in both chondrocytes and osteoblasts, and is highly regulated by a series of intracellular and extracellular mechanisms (reviewed in Walsh et al., 2010). Mature BMPs are secreted from osteoblasts and may either (i) activate their membrane receptors; (ii) be bound and inhibited by one or more of their secreted antagonists; or (iii) bind to extracellular matrix proteins such as collagen and act as a ‘reservoir’ of BMP for neighbouring cells (Miyazono et al., 2010). A series of transcription factors critical to bone and cartilage formation have been identified (reviewed in Nishimura et al., 2012). Runx2, Osterix and Sox9 are key transcription factors that regulate downstream BMP signalling in osteoblasts (Nishimura et al., 2008). The conservation of BMP action and the bone formation process in vertebrates give added value to the interpretation of data from the phenotypes of mouse models displaying manipulations in BMP or BMP antagonist function (Karsenty, 2003). Patients with mutations in the Runx2 gene develop cleidocranial dysplasia, characterized by short stature and defective clavicle (collar bone) formation (Otto et al., 2002), and runx2−/− mice die shortly after birth due to a severe defect in osteogenesis (Komori et al., 1997; Otto et al., 1997).
Apart from the extracellular cysteine-knot containing antagonists such as gremlin and noggin discussed earlier, some BMP proteins themselves act as competitive antagonists that bind to the type I/II receptors. BMP-3 is secreted from osteoblasts and is found in the bone extracellular matrix. BMP-3 binds to the ActR type II and inhibits BMP-2/4 binding (Daluiski et al., 2001; Gamer et al., 2005). Mice lacking BMP-3 display an increased bone mass (Daluiski et al., 2001), whereas transgenic mice overexpressing BMP-3 develop spontaneous fractures, highlighting a key role for BMP-3 in bone development (Gamer et al., 2009). Recent data have shown that BMP-3 can repress osteoblast differentiation from progenitor cells (Kokabu et al., 2012). Inhibin is another circulating antagonist of BMPR2 that also blocks BMP-2/4 action in osteoblasts (Rosen, 2006). Negative feedback for the BMP signal is provided by intracellular molecules such as Smad6 and Tob. Smad6 is a BMP-2 target gene, and increases in Smad6 levels inhibit osteoblast differentiation by triggering Smurf1-dependent proteasomal degradation of the BMP receptor and Smad proteins (Murakami et al., 2003; Horiki et al., 2004; Massague et al., 2005). Another inhibitor of osteoblast function, called Tob, blocks BMP action by binding and inhibiting R-Smads, thus preventing BMP signalling (Yoshida et al., 2000). Other molecules such as CRIM1, BAMBI and endoglin regulate BMP signalling in bone and other tissues (Onichtchouk et al., 1999; Wilkinson et al., 2003; Ishibashi et al., 2010; Pardali et al., 2011). CRIM1 inhibits BMP action via regulation of intracellular BMP processing, whereas BAMBI acts a pseudo-receptor to reduce active BMP binding to type I/II receptors (Onichtchouk et al., 1999; Wilkinson et al., 2003). In contrast, endoglin (also known as CD105) is sometimes referred to as the type III BMP receptor and acts to enhance BMP action at the plasma membrane (Ishibashi et al., 2010).
The highly complex nature of BMP action and regulation emphasizes the critical nature of this pathway in bone (and other tissue) development. The effect of changes in the levels or function of proteins in the BMP pathway can be seen in the wide array of phenotypes observed in patients. Many of these phenotypes have been reviewed elsewhere (Rosen, 2006; Walsh et al., 2010). Mutations in the BMP antagonists noggin and sclerostin cause brachydactyly (lack of digits; Lehmann et al., 2007), and sclerosteosis characterized by fused digits and excessive bone growth (Brunkow et al., 2001). Increases in the level of several BMP antagonists including noggin, chordin and follistatin have been implicated in osteoarthritis (Tardif et al., 2009). Mice overexpressing Grem1 in bone develop osteopenia and increased bone fractures (Gazzerro et al., 2005), whereas mice with bone-specific deletion of Grem1 develop increased bone formation and density (Gazzerro et al., 2007). Changes in the levels of other BMP antagonists such as noggin and twisted gastrulation have also been implicated in bone density changes in mouse models (Wu et al., 2003; Sotillo Rodriguez et al., 2009). All of these data and more underline the principle that tight temporospatial regulation of BMP action is critical for normal bone and cartilage formation in mammals. The ‘volume’ of BMP signalling is a fundamental parameter in determining correct bone formation during development. With this in mind, the next section of this review will discuss the uses of BMPs in modulating bone formation in patients with a range of bone injuries.
Therapeutic uses of BMPs for bone repair
Between 5 and 10% of fractures have impaired healing due to non-union of bone (Gautschi et al., 2007). This can greatly increase patient morbidity due to an extended hospital stay, infection rates concomitant healthcare costs. The gold standard of therapy for non-union fractures is bone autograft, where bone fragments are harvested from the patient's iliac crest and used to heal the fracture (Cook et al., 1994). BMPs have been shown in pre-clinical models to stimulate bone formation (osteoinduction; Einhorn et al., 2003) and angiogenesis (Zhang et al., 2009). Both BMP-2 and BMP-7 are now clinically approved as adjunct therapies for the treatment of non-union fractures (Gautschi et al., 2007). Recombinant human BMP-2 is available from Medtronic (Minneapolis, MN, USA) as InFUSE®, and rhBMP-7 is available from Stryker (Kalamazoo, MI, USA) as OP-1. The efficacy of BMPs has been tested in a number of clinical trials. The BESTT study evaluated the effect of rhBMP-2 on open tibia fractures, as a supplement to standard surgical intervention (Nauth et al., 2009). In this single blind trial, all patients underwent the standard irrigation/mechanical fixation of the fracture, followed by placebo or rhBMP-2 treatment (Govender et al., 2002). RhBMP-2 was delivered via an absorbable type I collagen sponge, and the primary outcome of secondary surgical intervention due to non-union of the fracture was assessed. The study showed that patients treated with rhBMP-2 had accelerated fracture and wound healing, as well as lower infection rates, with few safety concerns (Govender et al., 2002). A follow-up study by Swiontkowski et al. (2006) looking at the effect of rhBMP-2 on open tibial fractures supported the beneficial effects of rhBMP-2 seen above. Others have assessed the effect of rhBMP-2 in parallel with bone allografting and shown benefit in several cases (e.g. Jones et al., 2006, summarized in Nauth et al., 2009). Alternatives to the absorbable collagen sponge carrier have been developed. Boerckel et al. (2011) suggested that a hybrid nanofibre mesh/alginate delivery system yielded greater bone connectivity compared with the collagen sponge. Novel approaches such as using mesenchymal stem cells transfected with BMPs for the treatment of non-union fracture are being explored (Liebergall et al., 2013).
Some caution should be noted regarding the use of BMPs in bone repair. A Cochrane review by Garrison and colleagues collated data from 11 clinical trials and suggested that a lack of robust data on the use of rhBMPs in fracture healing exists. Many of the clinical trials have been limited in terms of unconscious bias risk due to a lack of double blinding, and there has been considerable industry involvement to date. Garrison et al. (2010) concluded that the high cost of rhBMP treatment may be justified only in patients with the most severe fractures. Others have also examined the economics of rhBMP treatment. A single dose of rhBMP costs approximately $5000 in the USA (Nauth et al., 2009). The cost of BMP treatment was weighed against potential savings from a societal/healthcare perspective, with groups in the USA and UK suggesting that rhBMP treatment was only cost-effective when used in severe open tibia fractures and in high-risk patients such as smokers (Jones et al., 2006; Garrison et al., 2007; Ziran et al., 2007). rhBMP-7 is currently used ‘off-label’ for the treatment of non-union fractures (Nauth et al., 2009). Similar to rhBMP-2, studies have shown that rhBMP-7 can accelerate the healing time and reduce the number of secondary interventions when used in conjunction with surgical repair of tibial fractures (Ristiniemi et al., 2007).
BMP-7 may also be useful in cases of osteoarthritis, where BMP-7 induction of extracellular matrix collagen production may oppose the degradation of the articular cartilage via stimulation of chondrocyte function (Fan et al., 2004; Nishida et al., 2004). BMP-7 may also provide a benefit in this situation by reducing pro-inflammatory cytokine release (e.g. IL-1 and IL-6), thereby inhibiting MMP-1 and MMP-13 matrix metalloproteinase expression (Huch et al., 1997; Koepp et al., 1999; Im et al., 2003; Boon et al., 2011). In human patients, rhBMP-7 has entered a Stryker-sponsored phase 2 double-blind randomized dose-finding trial for the treatment of osteoarthritis of the knee. Doses of rhBMP-7 used in this trial ranged from 0.03 to 0.3 mg·mL−1, with appropriate placebo comparators (http://clinicaltrials.gov/ct2/show/NCT01111045). Phase 1 studies by this company suggested limited toxicity to report with rhBMP-7 treatment of age and sex-appropriate (60 years, female) control subjects (Hunter et al., 2010). Results on the phase 2 study for rhBMP-7 are pending. rhBMP-7 was approved by the Food and Drug Administration for use in lumbar spinal fusion in patients where bone autograft was not feasible or likely to be successful, such as in smokers or diabetic patients. Many adverse events identified as a result of rhBMP-7 treatment, such as haematoma, swelling, neurological defects and retrograde ejaculation, may have been underreported. A review of these trials by Dr Nancy Epstein concluded that there is mounting evidence that the use of rhBMP-7 in spinal fusion surgery contributes to major perioperative and post-operative morbidity (Epstein, 2013).
What should we conclude from the wealth of conflicting data reporting on the therapeutic benefit versus side effects of rhBMPs in the treatment of non-union fractures, spinal fusions, and other bone and cartilage-related diseases? Nauth et al. (2009) concluded that the data on the use of rhBMPs have been disappointing so far due to the high doses required, unresolved issues with the carrier molecules and the limitations of the clinical trials completed to date. When the safety concerns identified by Epstein (2013) are also considered, it is clear that more data from well-designed double-blind, placebo controlled, multiple dose and carrier formulation clinical trials are required to allow clinicians to proceed with confidence when utilizing rhBMPs in orthopaedic surgeries and other procedures.
Role of BMPs in tissue fibrosis
BMPs have been implicated in mammalian development, cancer, and fibrosis or tissue scarring. Roles for BMP-2 in heart development, BMP-4 and 7 in neural crest cell maturation and BMP-7 in kidney formation have been demonstrated (summarized in McCormack and O'Dea, 2013). Fibrosis or scarring occurs in organs such as kidney, lung and heart as a result of damage caused by hyperglycaemia, hypoxia and ischaemic insult. Fibrosis is characterized by an increase in the number of fibroblast cells that secrete extracellular matrix proteins such as collagen and fibronectin, which contribute to the damage experienced by the organ or tissue. Scar-forming fibroblasts may derive from the tissue itself (resident fibroblasts), from activation of quiescent cells in the circulation (fibrocytes) or from injured epithelial or endothelial cells that undergo an epithelial/endothelial-mesenchymal transition (EMT/EndMT) to form collagen-secreting myofibroblasts (LeBleu et al., 2013). EMT was originally proposed as a source of myofibroblasts in kidney injury by Iwano et al. (2002), who suggested that approximately one-third of all myofibroblasts in the fibrotic kidney arise from EMT. More recent data using fate mapping suggest that in the fibrotic kidney, 50% of myofibroblasts arise from proliferation of local resident fibroblasts, 35% arise from infiltration of bone marrow-derived cells, 10% arise from endothelial cells via EndMT and 5% arise from epithelial cells via EMT (LeBleu et al., 2013). However, there is a large body of evidence suggesting that EMT does not contribute to renal fibrosis, and that vascular pericytes are the source of the myofibroblast in the kidney (reviewed in Grgic et al., 2012). Why are these details important? Firstly, the degree of tubulointerstitial fibrosis is inversely correlated to renal function and is a good predictor of renal function going forward (Bohle et al., 1994). Secondly, the development of therapeutics to delay, halt or reverse renal fibrosis is at the forefront of many research programmes in both academia and industry. It has been hypothesized that the numbers of scar-forming fibroblasts in a fibrotic tissue can be reduced by pharmacological approaches that aim to reverse EMT or otherwise reduce fibroblast burden in the affected tissue. Manipulation of BMP signalling is at the forefront of many of these strategies, and these data will be summarized below.
Manipulation of BMPs as a strategy to reverse tissue fibrosis
TGFβ1 was identified as the primary fibrotic cytokine that induces EMT in multiple organs including kidney, heart, skin, lung (Okada et al., 1997; Zeisberg et al., 2003b). Exposure of epithelial cells to TGFβ1 leads to a decrease in adherens junction proteins such as E-cadherin and ZO-1, and an increase in α-smooth muscle actin and collagen IV. Regardless of the cellular context, the key inducers of EMT are a series of transcription factors called Snai1/2 and Zeb1, which orchestrate the wave of gene expression changes critical to EMT progression (Peinado et al., 2007). There are a limited number of conflicting reports on the role of BMPs in EMT in different cell types. BMP-4 has been convincingly shown to have a pro-EMT, pro-fibrotic effect in different epithelial cells. BMP-4 induces EMT and enhanced cell migration in airway epithelium (Molloy et al., 2008; McCormack et al., 2013), as well as EMT and invasion in squamous cell carcinoma and other cancer cells (Hamada et al., 2007; Theriault et al., 2007; Xu et al., 2011). BMP-4 also contributes to cardiac hypertrophy in models of pressure overload (Sun et al., 2013). Transgenic overexpression of BMP-4 induced glomerular damage and proteinuria in mice, a phenotype similar to that seen in diabetic nephropathy (DN) (Tominaga et al., 2011). BMP-2 has been suggested to enhance EMT in human skin wounds and in human lung epithelial cells (Yan et al., 2010; McCormack et al., 2013). BMP-2 can also increase the expression of α-SMA in hepatic stellate cells, which are thought to undergo EMT during liver fibrosis (Shen et al., 2003). Others have suggested that BMP-2 possesses anti-fibrotic activity by suppressing TGFβ1-induced EMT in an in vivo model of renal fibrosis (Yang et al., 2009). These authors subsequently showed that BMP-2 attenuated Snail expression, thus reversing TGFβ1-induced EMT (Yang et al., 2011).
In terms of EMT and fibrosis, most of the interest has surrounded BMP-7, as it is this member of the BMP family that appears to have strong anti-fibrotic activity. In the heart, delivery of rhBMP-7 reduced both EndMT and associated cardiac fibrosis induced by pressure overload in mice (Zeisberg et al., 2007). The same report also demonstrated that rhBMP-7 reduced fibrosis in a heart transplant mouse model of chronic organ rejection (Zeisberg et al., 2007). Kang et al. (2010) showed that s.c. delivery of rhBMP-7 reduced vascular calcification in the aorta as a result of vitamin D overload (Kang et al., 2010). Significantly, these authors demonstrated that pretreatment with rhBMP-7 for 7 days could prevent vitamin D-induced vascular calcification (Kang et al., 2010). In an asbestos model of pulmonary fibrosis, administration of rhBMP-7 reduced the severity of fibrosis in these mice (Myllarniemi et al., 2008).
In the liver, BMPs have been implicated in the wound healing response to carbon tetrachloride (CCl4)-induced fibrosis, and Bmpr1a+/− mice displayed retarded healing post-CCl4 treatment (Oumi et al., 2012). Oral administration of adeno-associated virus-rhBMP-7 also suppressed CCl4-induced liver fibrosis in mice (Hao et al., 2012). Intraperitoneal injection of BMP-7 was similarly effective in liver fibrosis in rats (Zhong et al., 2013). The mechanism of BMP-7-mediated repair is suggested to be inhibition of TGFβ1 signalling in hepatic stellate cells due to reductions of extracellular matrix collagen I and III deposition and enhanced hepatocyte regeneration (Hao et al., 2012; Yang et al., 2012). In models of inflammatory bowel disease and colitis, i.v. administration of BMP-7 reduced the expression of TGFβ1 and pro-inflammatory cytokines such as IL-6, causing an attenuation of colitis severity (Maric et al., 2003). Further data from this group identified that BMP-7 administration increased BMP-2 levels and decreased the levels of the BMP antagonist noggin, leading to an overall recovery of BMP signalling that reversed the inflammatory bowel disease phenotype (Maric et al., 2012). These data suggest that BMP-2 may also have a pro-resolution rather than a pro-fibrosis role during fibrosis-associated inflammation in the gut and other tissues.
In the kidney, BMP-7 antagonizes TGFβ1 actions in renal mesangial and tubular epithelial cells. Significantly, loss of BMP-7 is associated with fibrosis associated with DN (Wang et al., 2001; Wang and Hirschberg, 2003; 2004). Importantly, the Hirschberg group also showed that overexpression of BMP-7 in glomerular podocytes attenuated renal fibrosis and improved renal function (Wang et al., 2006). Previous data had shown that the administration of recombinant BMP-7 (OP-1) reduced the severity of ischaemic acute renal injury in mice (Vukicevic et al., 1998). The Kalluri group demonstrated the benefit of BMP-7 administration in a chronic model of nephrotoxic serum nephritis (Zeisberg et al., 2003b), as well as genetic models of renal fibrosis (Zeisberg et al., 2003a) and DN (Sugimoto et al., 2007). These authors also demonstrated a BMP-7-mediated reversal of EMT in their model system, as well as in adult renal fibroblasts, facilitating regeneration of injured kidney (Zeisberg et al., 2005). BMP-7 gene transfer using gold nanoparticles into rabbit keratocytes also inhibits corneal fibrosis in vivo (Tandon et al., 2013).
In contrast, a group from Centocor, Inc., showed that BMP-7 failed to reverse TGFβ1-induced EMT in human proximal tubular epithelial cells (Dudas et al., 2009). Similarly, BMP-7 did not reverse EMT or protect against fibrosis in a mouse model of lung or skin fibrosis (Murray et al., 2008). These groups suggested that recombinant BMP-7 may not be an optimal anti-fibrotic agent for human disease treatment. Despite these disappointing results, the potential for anti-fibrotic therapy based on BMP-7 is still strong. A small peptide mimetic of BMP-7 called THR123, which activates the BMP ALK3 receptor, has shown remarkable activity in reversing renal fibrosis from a diverse range of mouse models of acute and chronic kidney disease (Sugimoto et al., 2012). Some authors in the field have expressed some concerns about the interpretation of these data. For example, issues were raised with the signalling properties of THR123 versus BMP-7, and also whether oral delivery of a peptide such as THR123 can be expected to deliver an effective therapeutic dose in vivo (Whitman et al., 2013). The Kalluri group have responded to these questions and discussions are ongoing as to the therapeutic potential of the THR123 peptide for human kidney disease (Sugimoto et al., 2013).
A small molecule called dorsomorphin was identified by Yu et al. (2008b) as the first small molecular inhibitor of BMP signalling. Using dorsomorphin, the authors identified a role for BMP signalling in iron metabolism in the zebrafish liver (Yu et al., 2008b). Mice expressing a constitutively activated form of the ALK2 receptor develop ectopic endochondral bone formation, mimicking a disease called fibrodysplasia ossificans progressiva (FOP) in humans. Treatment of these mice with a dorsomorphin derivative (LDN-193189) inhibited BMP signalling in C2C12 cells and reduced the severity of the FOP phenotype in mice (Yu et al., 2008a; Boergermann et al., 2010). A recent paper by Sanvitale et al. (2013) described a new class of ALK2 inhibitor, the lead compound of which is called K02288. K02288 inhibits BMP-stimulated Smad1/5/8 phosphorylation, without affecting TGFβ1 signalling, suggesting an impressive degree of specificity (Sanvitale et al., 2013). Both dorsomorphin and its analogues, together with K02288, offer exciting tools for the development of specific, small-molecule inhibitors of BMP signalling in human disease.
Another small molecule called tilerone has been shown to reduce the severity of pulmonary fibrosis in mice, by increasing the expression of BMP-7 (Lepparanta et al., 2013). A secreted molecule called kielin/chordin-like protein (KCP-1) can bind to and inhibit TGFβ1, while enhancing BMP-7 signalling (J Lin et al., 2005; 2006a). Transgenic expression of KCP-1 can attenuate both acute and chronic renal injury in mice (Soofi et al., 2013), and the authors speculate that KCP-1 may have potential as a therapeutic agent if administered in the correct context (Soofi et al., 2013). Therefore, despite the difficulty in translating the anti-fibrotic potential of BMP-7 in animal models to patients, alternative strategies that boost BMP-7 signalling such as THR123 or KCP-1 may provide an indirect route to utilize the anti-fibrotic potential of BMP-7 for the treatment of fibrotic disease in patients.
Targeting BMP antagonists for the treatment of fibrotic disease
One of the proposed mechanisms by which BMP-7 reduced liver fibrosis is by decreasing the expression of the secreted BMP antagonist gremlin (Yang et al., 2012). Increased levels of Grem1 are associated with fibrotic conditions in the kidney, lung, heart liver and eye (Dolan et al., 2005; Lee et al., 2007; Mezzano et al., 2007; Carvajal et al., 2008; Costello et al., 2008; Walsh et al., 2008; Rodrigues-Diez et al., 2012; Yang et al., 2012; Mueller et al., 2013). In parallel with data showing that reduced BMP-7 signalling is associated with diabetic kidney disease, mutations in the BMP receptor type II are implicated in >70% of heritable cases of PAH (Li et al., 2010). These data identify the targeting of Grem1 as a novel therapeutic modality for the treatment of fibrosis in vivo. Supporting this hypothesis, mice lacking one copy of the Grem1 gene (grem1+/−) are partially protected from the early sequelae of DN (Roxburgh et al., 2009). In addition, in vivo delivery of siRNA-mediated targeting of Grem1 demonstrated therapeutic potential for the treatment of DN by restoring BMP-7 levels (Zhang et al., 2010). In the lung, Grem1 is overexpressed in idiopathic pulmonary fibrosis (Koli et al., 2006). Transient adenovirus-mediated overexpression of Grem1 in lung led to epithelial cell activation and a reversible lung fibrosis (Farkas et al., 2011). Grem1 levels are also elevated in response to hypoxia in models of PAH, and haplodeficiency of grem1 increased BMP signalling and reduced vascular remodelling associated with PAH (Costello et al., 2008; Cahill et al., 2012). Given the large body of data demonstrating that elevated levels of Grem1 contribute to tissue fibrosis, pharmacological strategies designed to inhibit Grem1 function in vivo have been tested in a range of disease models. Data from a group in Novartis have shown for the first time that an anti-Grem1 antibody ameliorated PAH in a mouse model (Ciuclan et al., 2013). Pretreatment with the anti-Grem1 monoclonal antibody reduced pulmonary vascular remodelling and right ventricle hypertrophy, and increased BMP signalling in the lung (Ciuclan et al., 2013). These data provide pharmacological proof-of-principle that this approach of targeting Grem1 as a means of increasing BMP signalling is now being explored in other fibrotic conditions of the kidney and other tissues. Of course, other BMP antagonists may also represent important bona fide targets in fibrotic disease. Consistent with this idea, a recent paper from the Yanagita group demonstrated that twisted gastrulation exacerbates podocyte injury via inhibition of BMP-7 signalling (Yamada et al., 2014).
Concluding remarks
This review has summarized a wealth of data suggesting that pharmacological manipulation of the BMP pathway holds great potential for the treatment of human diseases of bone, kidney fibrosis, cancer, etc. The identification of small molecules that specifically target the BMP pathway creates the potential for screening these compounds in a range of in vitro and in vivo models of disease where BMP actions are implicated. The natural progression of this work is the drive towards clinical trials for the small-molecule inhibitors of BMP signalling in various diseases. We look forward to monitoring the evolution of this exciting field, which will hopefully generate improved targeted therapies for patients suffering from bone disorders as well as fibrosis in the kidney, lung and other tissues.
Acknowledgments
We apologize to those colleagues whose important work could not be cited due to space restrictions or author oversight. We thank Professor Finian Martin (UCD Dublin) for comments on the manuscript prior to submission. Work in the Brazil laboratory was funded by Diabetes UK, BBSRC, Northern Ireland Kidney Research Fund (NIKRF) and DEL Northern Ireland. I.H.A.A. is funded by a DEL Northern Ireland PhD fellowship.
Glossary
- ALK,
activin receptor-like kinase
- BMP
bone morphogenetic protein
- DN
diabetic nephropathy
- EMT
epithelial-mesenchymal transition
- Smad
sma and mothers against decapentaplegic
Conflict of interest
D.B. collaborates with scientists in Astra Zeneca, Mölndal, Gothenburg, Sweden, on a BBSRC CASE PhD studentship.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, Peters JA, Harmar AJ CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14:Catalytic receptors. Br J Pharmacol. 2013;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki H, Fujii M, Imamura T, Yagi K, Takehara K, Kato M, et al. Synergistic effects of different bone morphogenetic protein type I receptors on alkaline phosphatase induction. J Cell Sci. 2001;114((Pt 8)):1483–1489. doi: 10.1242/jcs.114.8.1483. [DOI] [PubMed] [Google Scholar]
- Balemans W, Van Hul W. Extracellular regulation of BMP signaling in vertebrates: a cocktail of modulators. Dev Biol. 2002;250:231–250. [PubMed] [Google Scholar]
- Boerckel JD, Kolambkar YM, Dupont KM, Uhrig BA, Phelps EA, Stevens HY, et al. Effects of protein dose and delivery system on BMP-mediated bone regeneration. Biomaterials. 2011;32:5241–5251. doi: 10.1016/j.biomaterials.2011.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boergermann JH, Kopf J, Yu PB, Knaus P. Dorsomorphin and LDN-193189 inhibit BMP-mediated Smad, p38 and Akt signalling in C2C12 cells. Int J Biochem Cell Biol. 2010;42:1802–1807. doi: 10.1016/j.biocel.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohle A, Strutz F, Muller GA. On the pathogenesis of chronic renal failure in primary glomerulopathies: a view from the interstitium. Exp Nephrol. 1994;2:205–210. [PubMed] [Google Scholar]
- Boon MR, van der Horst G, van der Pluijm G, Tamsma JT, Smit JW, Rensen PC. Bone morphogenetic protein 7: a broad-spectrum growth factor with multiple target therapeutic potency. Cytokine Growth Factor Rev. 2011;22:221–229. doi: 10.1016/j.cytogfr.2011.08.001. [DOI] [PubMed] [Google Scholar]
- Bragdon B, Moseychuk O, Saldanha S, King D, Julian J, Nohe A. Bone morphogenetic proteins: a critical review. Cell Signal. 2011;23:609–620. doi: 10.1016/j.cellsig.2010.10.003. [DOI] [PubMed] [Google Scholar]
- Brunkow ME, Gardner JC, Van Ness J, Paeper BW, Kovacevich BR, Proll S, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001;68:577–589. doi: 10.1086/318811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill E, Costello CM, Rowan SC, Harkin S, Howell K, Leonard MO, et al. Gremlin plays a key role in the pathogenesis of pulmonary hypertension. Circulation. 2012;125:920–930. doi: 10.1161/CIRCULATIONAHA.111.038125. [DOI] [PubMed] [Google Scholar]
- Carvajal G, Droguett A, Burgos ME, Aros C, Ardiles L, Flores C, et al. Gremlin: a novel mediator of epithelial mesenchymal transition and fibrosis in chronic allograft nephropathy. Transplant Proc. 2008;40:734–739. doi: 10.1016/j.transproceed.2008.02.064. [DOI] [PubMed] [Google Scholar]
- Chacko BM, Qin B, Correia JJ, Lam SS, de Caestecker MP, Lin K. The L3 loop and C-terminal phosphorylation jointly define Smad protein trimerization. Nat Struct Biol. 2001;8:248–253. doi: 10.1038/84995. [DOI] [PubMed] [Google Scholar]
- Chen X, Weisberg E, Fridmacher V, Watanabe M, Naco G, Whitman M. Smad4 and FAST-1 in the assembly of activin-responsive factor. Nature. 1997;389:85–89. doi: 10.1038/38008. [DOI] [PubMed] [Google Scholar]
- Ciuclan L, Sheppard K, Dong L, Sutton D, Duggan N, Hussey M, et al. Treatment with anti-gremlin 1 antibody ameliorates chronic hypoxia/SU5416-induced pulmonary arterial hypertension in mice. Am J Pathol. 2013;183:1461–1473. doi: 10.1016/j.ajpath.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook SD, Dalton JE, Tan EH, Whitecloud TS, 3rd, Rueger DC. In vivo evaluation of recombinant human osteogenic protein (rhOP-1) implants as a bone graft substitute for spinal fusions. Spine. 1994;19:1655–1663. doi: 10.1097/00007632-199408000-00002. [DOI] [PubMed] [Google Scholar]
- Costello CM, Howell K, Cahill E, McBryan J, Konigshoff M, Eickelberg O, et al. Lung-selective gene responses to alveolar hypoxia: potential role for the bone morphogenetic antagonist gremlin in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2008;295:L272–L284. doi: 10.1152/ajplung.00358.2007. [DOI] [PubMed] [Google Scholar]
- Daluiski A, Engstrand T, Bahamonde ME, Gamer LW, Agius E, Stevenson SL, et al. Bone morphogenetic protein-3 is a negative regulator of bone density. Nat Genet. 2001;27:84–88. doi: 10.1038/83810. [DOI] [PubMed] [Google Scholar]
- Dolan V, Murphy M, Sadlier D, Lappin D, Doran P, Godson C, et al. Expression of gremlin, a bone morphogenetic protein antagonist, in human diabetic nephropathy. Am J Kidney Dis. 2005;45:1034–1039. doi: 10.1053/j.ajkd.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Dudas PL, Argentieri RL, Farrell FX. BMP-7 fails to attenuate TGF-beta1-induced epithelial-to-mesenchymal transition in human proximal tubule epithelial cells. Nephrol Dial Transplant. 2009;24:1406–1416. doi: 10.1093/ndt/gfn662. [DOI] [PubMed] [Google Scholar]
- Einhorn TA, Majeska RJ, Mohaideen A, Kagel EM, Bouxsein ML, Turek TJ, et al. A single percutaneous injection of recombinant human bone morphogenetic protein-2 accelerates fracture repair. J Bone Joint Surg Am. 2003;85-A:1425–1435. doi: 10.2106/00004623-200308000-00002. [DOI] [PubMed] [Google Scholar]
- Epstein NE. Complications due to the use of BMP/INFUSE in spine surgery: the evidence continues to mount. Surg Neurol Int. 2013;4(Suppl. 5):S343–S352. doi: 10.4103/2152-7806.114813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z, Chubinskaya S, Rueger DC, Bau B, Haag J, Aigner T. Regulation of anabolic and catabolic gene expression in normal and osteoarthritic adult human articular chondrocytes by osteogenic protein-1. Clin Exp Rheumatol. 2004;22:103–106. [PubMed] [Google Scholar]
- Farkas L, Farkas D, Gauldie J, Warburton D, Shi W, Kolb M. Transient overexpression of gremlin results in epithelial activation and reversible fibrosis in rat lungs. Am J Respir Cell Mol Biol. 2011;44:870–878. doi: 10.1165/rcmb.2010-0070OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamer LW, Nove J, Levin M, Rosen V. BMP-3 is a novel inhibitor of both activin and BMP-4 signaling in Xenopus embryos. Dev Biol. 2005;285:156–168. doi: 10.1016/j.ydbio.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Gamer LW, Cox K, Carlo JM, Rosen V. Overexpression of BMP3 in the developing skeleton alters endochondral bone formation resulting in spontaneous rib fractures. Dev Dyn. 2009;238:2374–2381. doi: 10.1002/dvdy.22048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison KR, Donell S, Ryder J, Shemilt I, Mugford M, Harvey I, et al. Clinical effectiveness and cost-effectiveness of bone morphogenetic proteins in the non-healing of fractures and spinal fusion: a systematic review. Health Technol Assess. 2007;11:1–150. doi: 10.3310/hta11300. iii–iv. [DOI] [PubMed] [Google Scholar]
- Garrison KR, Shemilt I, Donell S, Ryder JJ, Mugford M, Harvey I, et al. Bone morphogenetic protein (BMP) for fracture healing in adults. Cochrane Database Syst Rev. 2010;(6) doi: 10.1002/14651858.CD006950.pub2. CD006950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautschi OP, Frey SP, Zellweger R. Bone morphogenetic proteins in clinical applications. ANZ J Surg. 2007;77:626–631. doi: 10.1111/j.1445-2197.2007.04175.x. [DOI] [PubMed] [Google Scholar]
- Gazzerro E, Pereira RC, Jorgetti V, Olson S, Economides AN, Canalis E. Skeletal overexpression of gremlin impairs bone formation and causes osteopenia. Endocrinology. 2005;146:655–665. doi: 10.1210/en.2004-0766. [DOI] [PubMed] [Google Scholar]
- Gazzerro E, Smerdel-Ramoya A, Zanotti S, Stadmeyer L, Durant D, Economides AN, et al. Conditional deletion of gremlin causes a transient increase in bone formation and bone mass. J Biol Chem. 2007;282:31549–31557. doi: 10.1074/jbc.M701317200. [DOI] [PubMed] [Google Scholar]
- Govender S, Csimma C, Genant HK, Valentin-Opran A, Amit Y, Arbel R, et al. Recombinant human bone morphogenetic protein-2 for treatment of open tibial fractures: a prospective, controlled, randomized study of four hundred and fifty patients. J Bone Joint Surg Am. 2002;84-A:2123–2134. doi: 10.2106/00004623-200212000-00001. [DOI] [PubMed] [Google Scholar]
- Grgic I, Duffield JS, Humphreys BD. The origin of interstitial myofibroblasts in chronic kidney disease. Pediatr Nephrol. 2012;27:183–193. doi: 10.1007/s00467-011-1772-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groppe J, Greenwald J, Wiater E, Rodriguez-Leon J, Economides AN, Kwiatkowski W, et al. Structural basis of BMP signalling inhibition by the cystine knot protein Noggin. Nature. 2002;420:636–642. doi: 10.1038/nature01245. [DOI] [PubMed] [Google Scholar]
- Guillot N, Kollins D, Gilbert V, Xavier S, Chen J, Gentle M, et al. BAMBI regulates angiogenesis and endothelial homeostasis through modulation of alternative TGFβ signaling. PLoS ONE. 2012;7:e39406. doi: 10.1371/journal.pone.0039406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Wu G. The signaling and functions of heterodimeric bone morphogenetic proteins. Cytokine Growth Factor Rev. 2012;23:61–67. doi: 10.1016/j.cytogfr.2012.02.001. [DOI] [PubMed] [Google Scholar]
- Hamada S, Satoh K, Hirota M, Kimura K, Kanno A, Masamune A, et al. Bone morphogenetic protein 4 induces epithelial-mesenchymal transition through MSX2 induction on pancreatic cancer cell line. J Cell Physiol. 2007;213:768–774. doi: 10.1002/jcp.21148. [DOI] [PubMed] [Google Scholar]
- Hao ZM, Cai M, Lv YF, Huang YH, Li HH. Oral administration of recombinant adeno-associated virus-mediated bone morphogenetic protein-7 suppresses CCl(4)-induced hepatic fibrosis in mice. Mol Ther. 2012;20:2043–2051. doi: 10.1038/mt.2012.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata A, Lagna G, Massague J, Hemmati-Brivanlou A. Smad6 inhibits BMP/Smad1 signaling by specifically competing with the Smad4 tumor suppressor. Genes Dev. 1998;12:186–197. doi: 10.1101/gad.12.2.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiki M, Imamura T, Okamoto M, Hayashi M, Murai J, Myoui A, et al. Smad6/Smurf1 overexpression in cartilage delays chondrocyte hypertrophy and causes dwarfism with osteopenia. J Cell Biol. 2004;165:433–445. doi: 10.1083/jcb.200311015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu DR, Economides AN, Wang X, Eimon PM, Harland RM. The Xenopus dorsalizing factor gremlin identifies a novel family of secreted proteins that antagonize BMP activities. Mol Cell. 1998;1:673–683. doi: 10.1016/s1097-2765(00)80067-2. [DOI] [PubMed] [Google Scholar]
- Huch K, Wilbrink B, Flechtenmacher J, Koepp HE, Aydelotte MB, Sampath TK, et al. Effects of recombinant human osteogenic protein 1 on the production of proteoglycan, prostaglandin E2, and interleukin-1 receptor antagonist by human articular chondrocytes cultured in the presence of interleukin-1beta. Arthritis Rheum. 1997;40:2157–2161. doi: 10.1002/art.1780401209. [DOI] [PubMed] [Google Scholar]
- Hunter DJ, Pike MC, Jonas BL, Kissin E, Krop J, McAlindon T. Phase 1 safety and tolerability study of BMP-7 in symptomatic knee osteoarthritis. BMC Musculoskelet Disord. 2010;11:232. doi: 10.1186/1471-2474-11-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im HJ, Pacione C, Chubinskaya S, Van Wijnen AJ, Sun Y, Loeser RF. Inhibitory effects of insulin-like growth factor-1 and osteogenic protein-1 on fibronectin fragment- and interleukin-1beta-stimulated matrix metalloproteinase-13 expression in human chondrocytes. J Biol Chem. 2003;278:25386–25394. doi: 10.1074/jbc.M302048200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi O, Ikegame M, Takizawa F, Yoshizawa T, Moksed MA, Iizawa F, et al. Endoglin is involved in BMP-2-induced osteogenic differentiation of periodontal ligament cells through a pathway independent of Smad-1/5/8 phosphorylation. J Cell Physiol. 2010;222:465–473. doi: 10.1002/jcp.21968. [DOI] [PubMed] [Google Scholar]
- Israel DI, Nove J, Kerns KM, Kaufman RJ, Rosen V, Cox KA, et al. Heterodimeric bone morphogenetic proteins show enhanced activity in vitro and in vivo. Growth Factors. 1996;13:291–300. doi: 10.3109/08977199609003229. [DOI] [PubMed] [Google Scholar]
- Itoh S, Landstrom M, Hermansson A, Itoh F, Heldin CH, Heldin NE, et al. Transforming growth factor beta1 induces nuclear export of inhibitory Smad7. J Biol Chem. 1998;273:29195–29201. doi: 10.1074/jbc.273.44.29195. [DOI] [PubMed] [Google Scholar]
- Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones AL, Bucholz RW, Bosse MJ, Mirza SK, Lyon TR, Webb LX, et al. Recombinant human BMP-2 and allograft compared with autogenous bone graft for reconstruction of diaphyseal tibial fractures with cortical defects. A randomized, controlled trial. J Bone Joint Surg Am. 2006;88:1431–1441. doi: 10.2106/JBJS.E.00381. [DOI] [PubMed] [Google Scholar]
- Kang YH, Jin JS, Yi DW, Son SM. Bone morphogenetic protein-7 inhibits vascular calcification induced by high vitamin D in mice. Tohoku J Exp Med. 2010;221:299–307. doi: 10.1620/tjem.221.299. [DOI] [PubMed] [Google Scholar]
- Karsenty G. The complexities of skeletal biology. Nature. 2003;423:316–318. doi: 10.1038/nature01654. [DOI] [PubMed] [Google Scholar]
- Koepp HE, Sampath KT, Kuettner KE, Homandberg GA. Osteogenic protein-1 (OP-1) blocks cartilage damage caused by fibronectin fragments and promotes repair by enhancing proteoglycan synthesis. Inflamm Res. 1999;48:199–204. doi: 10.1007/s000110050446. [DOI] [PubMed] [Google Scholar]
- Kokabu S, Gamer L, Cox K, Lowery J, Tsuji K, Raz R, et al. BMP3 suppresses osteoblast differentiation of bone marrow stromal cells via interaction with Acvr2b. Mol Endocrinol. 2012;26:87–94. doi: 10.1210/me.2011-1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koli K, Myllarniemi M, Vuorinen K, Salmenkivi K, Ryynanen MJ, Kinnula VL, et al. Bone morphogenetic protein-4 inhibitor gremlin is overexpressed in idiopathic pulmonary fibrosis. Am J Pathol. 2006;169:61–71. doi: 10.2353/ajpath.2006.051263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- LeBleu VS, Taduri G, O'Connell J, Teng Y, Cooke VG, Woda C, et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013;19:1047–1053. doi: 10.1038/nm.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, O'Meara SJ, O'Brien C, Kane R. The role of gremlin, a BMP antagonist, and epithelial-to-mesenchymal transition in proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci. 2007;48:4291–4299. doi: 10.1167/iovs.07-0086. [DOI] [PubMed] [Google Scholar]
- Lehmann K, Seemann P, Silan F, Goecke TO, Irgang S, Kjaer KW, et al. A new subtype of brachydactyly type B caused by point mutations in the bone morphogenetic protein antagonist NOGGIN. Am J Hum Genet. 2007;81:388–396. doi: 10.1086/519697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepparanta O, Tikkanen JM, Bespalov MM, Koli K, Myllarniemi M. Bone morphogenetic protein-inducer tilorone identified by high-throughput screening is antifibrotic in vivo. Am J Respir Cell Mol Biol. 2013;48:448–455. doi: 10.1165/rcmb.2012-0201OC. [DOI] [PubMed] [Google Scholar]
- Li W, Dunmore BJ, Morrell NW. Bone morphogenetic protein type II receptor mutations causing protein misfolding in heritable pulmonary arterial hypertension. Proc Am Thorac Soc. 2010;7:395–398. doi: 10.1513/pats.201002-024AW. [DOI] [PubMed] [Google Scholar]
- Li X, Cao X. BMP signaling and skeletogenesis. Ann N Y Acad Sci. 2006;1068:26–40. doi: 10.1196/annals.1346.006. [DOI] [PubMed] [Google Scholar]
- Liebergall M, Schroeder J, Mosheiff R, Gazit Z, Yoram Z, Rasooly L, et al. Stem cell-based therapy for prevention of delayed fracture union: a randomized and prospective preliminary study. Mol Ther. 2013;21:1631–1638. doi: 10.1038/mt.2013.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Patel SR, Cheng X, Cho EA, Levitan I, Ullenbruch M, et al. Kielin/chordin-like protein, a novel enhancer of BMP signaling, attenuates renal fibrotic disease. Nat Med. 2005;11:387–393. doi: 10.1038/nm1217. [DOI] [PubMed] [Google Scholar]
- Lin J, Patel SR, Wang M, Dressler GR. The cysteine-rich domain protein KCP is a suppressor of transforming growth factor beta/activin signaling in renal epithelia. Mol Cell Biol. 2006a;26:4577–4585. doi: 10.1128/MCB.02127-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Duan X, Liang YY, Su Y, Wrighton KH, Long J, et al. PPM1A functions as a Smad phosphatase to terminate TGFbeta signaling. Cell. 2006b;125:915–928. doi: 10.1016/j.cell.2006.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu IM, Schilling SH, Knouse KA, Choy L, Derynck R, Wang XF. TGFbeta-stimulated Smad1/5 phosphorylation requires the ALK5 L45 loop and mediates the pro-migratory TGFbeta switch. EMBO J. 2009;28:88–98. doi: 10.1038/emboj.2008.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maric I, Poljak L, Zoricic S, Bobinac D, Bosukonda D, Sampath KT, et al. Bone morphogenetic protein-7 reduces the severity of colon tissue damage and accelerates the healing of inflammatory bowel disease in rats. J Cell Physiol. 2003;196:258–264. doi: 10.1002/jcp.10275. [DOI] [PubMed] [Google Scholar]
- Maric I, Kucic N, Turk Wensveen T, Smoljan I, Grahovac B, Zoricic Cvek S, et al. BMP signaling in rats with TNBS-induced colitis following BMP7 therapy. Am J Physiol Gastrointest Liver Physiol. 2012;302:G1151–G1162. doi: 10.1152/ajpgi.00244.2011. [DOI] [PubMed] [Google Scholar]
- Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- McCormack N, O'Dea S. Regulation of epithelial to mesenchymal transition by bone morphogenetic proteins. Cell Signal. 2013;25:2856–2862. doi: 10.1016/j.cellsig.2013.09.012. [DOI] [PubMed] [Google Scholar]
- McCormack N, Molloy EL, O'Dea S. Bone morphogenetic proteins enhance an epithelial-mesenchymal transition in normal airway epithelial cells during restitution of a disrupted epithelium. Respir Res. 2013;14:36. doi: 10.1186/1465-9921-14-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezzano S, Droguett A, Burgos ME, Aros C, Ardiles L, Flores C, et al. Expression of gremlin, a bone morphogenetic protein antagonist, in glomerular crescents of pauci-immune glomerulonephritis. Nephrol Dial Transplant. 2007;22:1882–1890. doi: 10.1093/ndt/gfm145. [DOI] [PubMed] [Google Scholar]
- Michos O, Goncalves A, Lopez-Rios J, Tiecke E, Naillat F, Beier K, et al. Reduction of BMP4 activity by gremlin 1 enables ureteric bud outgrowth and GDNF/WNT11 feedback signalling during kidney branching morphogenesis. Development. 2007;134:2397–2405. doi: 10.1242/dev.02861. [DOI] [PubMed] [Google Scholar]
- Mikawa S, Sato K. Chordin expression in the adult rat brain. Neuroscience. 2013;258C:16–33. doi: 10.1016/j.neuroscience.2013.11.006. [DOI] [PubMed] [Google Scholar]
- Miyazono K, Kamiya Y, Morikawa M. Bone morphogenetic protein receptors and signal transduction. J Biochem. 2010;147:35–51. doi: 10.1093/jb/mvp148. [DOI] [PubMed] [Google Scholar]
- Molloy EL, Adams A, Moore JB, Masterson JC, Madrigal-Estebas L, Mahon BP, et al. BMP4 induces an epithelial-mesenchymal transition-like response in adult airway epithelial cells. Growth Factors. 2008;26:12–22. doi: 10.1080/08977190801987166. [DOI] [PubMed] [Google Scholar]
- Mueller KA, Tavlaki E, Schneider M, Jorbenadze R, Geisler T, Kandolf R, et al. Gremlin-1 identifies fibrosis and predicts adverse outcome in patients with heart failure undergoing endomyocardial biopsy. J Card Fail. 2013;19:678–684. doi: 10.1016/j.cardfail.2013.09.001. [DOI] [PubMed] [Google Scholar]
- Murakami G, Watabe T, Takaoka K, Miyazono K, Imamura T. Cooperative inhibition of bone morphogenetic protein signaling by Smurf1 and inhibitory Smads. Mol Biol Cell. 2003;14:2809–2817. doi: 10.1091/mbc.E02-07-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M, Kawachi H, Ogawa K, Nishino Y, Funaba M. Receptor expression modulates the specificity of transforming growth factor-beta signaling pathways. Genes Cells. 2009;14:469–482. doi: 10.1111/j.1365-2443.2009.01283.x. [DOI] [PubMed] [Google Scholar]
- Murray LA, Hackett TL, Warner SM, Shaheen F, Argentieri RL, Dudas P, et al. BMP-7 does not protect against bleomycin-induced lung or skin fibrosis. PLoS ONE. 2008;3:e4039. doi: 10.1371/journal.pone.0004039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myllarniemi M, Lindholm P, Ryynanen MJ, Kliment CR, Salmenkivi K, Keski-Oja J, et al. Gremlin-mediated decrease in bone morphogenetic protein signaling promotes pulmonary fibrosis. Am J Respir Crit Care Med. 2008;177:321–329. doi: 10.1164/rccm.200706-945OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura J, Yanagita M. Bmp modulators in kidney disease. Discov Med. 2012;13:57–63. [PubMed] [Google Scholar]
- Nauth A, Ristiniemi J, McKee MD, Schemitsch EH. Bone morphogenetic proteins in open fractures: past, present, and future. Injury. 2009;40(Suppl. 3):S27–S31. doi: 10.1016/S0020-1383(09)70008-7. [DOI] [PubMed] [Google Scholar]
- Nelsen SM, Christian JL. Site-specific cleavage of BMP4 by furin, PC6, and PC7. J Biol Chem. 2009;284:27157–27166. doi: 10.1074/jbc.M109.028506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida Y, Knudson CB, Knudson W. Osteogenic Protein-1 inhibits matrix depletion in a hyaluronan hexasaccharide-induced model of osteoarthritis. Osteoarthritis Cartilage. 2004;12:374–382. doi: 10.1016/j.joca.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Nishimura R, Hata K, Ikeda F, Ichida F, Shimoyama A, Matsubara T, et al. Signal transduction and transcriptional regulation during mesenchymal cell differentiation. J Bone Miner Metab. 2008;26:203–212. doi: 10.1007/s00774-007-0824-2. [DOI] [PubMed] [Google Scholar]
- Nishimura R, Hata K, Matsubara T, Wakabayashi M, Yoneda T. Regulation of bone and cartilage development by network between BMP signalling and transcription factors. J Biochem. 2012;151:247–254. doi: 10.1093/jb/mvs004. [DOI] [PubMed] [Google Scholar]
- Nohe A, Keating E, Knaus P, Petersen NO. Signal transduction of bone morphogenetic protein receptors. Cell Signal. 2004;16:291–299. doi: 10.1016/j.cellsig.2003.08.011. [DOI] [PubMed] [Google Scholar]
- Okada H, Danoff TM, Kalluri R, Neilson EG. Early role of Fsp1 in epithelial-mesenchymal transformation. Am J Physiol. 1997;273((4 Pt 2)):F563–F574. doi: 10.1152/ajprenal.1997.273.4.F563. [DOI] [PubMed] [Google Scholar]
- Onichtchouk D, Chen YG, Dosch R, Gawantka V, Delius H, Massague J, et al. Silencing of TGF-beta signalling by the pseudoreceptor BAMBI. Nature. 1999;401:480–485. doi: 10.1038/46794. [DOI] [PubMed] [Google Scholar]
- Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- Otto F, Kanegane H, Mundlos S. Mutations in the RUNX2 gene in patients with cleidocranial dysplasia. Hum Mutat. 2002;19:209–216. doi: 10.1002/humu.10043. [DOI] [PubMed] [Google Scholar]
- Oumi N, Taniguchi KA, Kanai AM, Yasunaga M, Nakanishi T, Sato K. A crucial role of bone morphogenetic protein signaling in the wound healing response in acute liver injury induced by carbon tetrachloride. Int J Hepatol. 2012;2012:476820. doi: 10.1155/2012/476820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardali E, van der Schaft DW, Wiercinska E, Gorter A, Hogendoorn PC, Griffioen AW, et al. Critical role of endoglin in tumor cell plasticity of Ewing sarcoma and melanoma. Oncogene. 2011;30:334–345. doi: 10.1038/onc.2010.418. [DOI] [PubMed] [Google Scholar]
- Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- Riggins GJ, Thiagalingam S, Rozenblum E, Weinstein CL, Kern SE, Hamilton SR, et al. Mad-related genes in the human. Nat Genet. 1996;13:347–349. doi: 10.1038/ng0796-347. [DOI] [PubMed] [Google Scholar]
- Ristiniemi J, Flinkkila T, Hyvonen P, Lakovaara M, Pakarinen H, Jalovaara P. RhBMP-7 accelerates the healing in distal tibial fractures treated by external fixation. J Bone Joint Surg Br. 2007;89:265–272. doi: 10.1302/0301-620X.89B2.18230. [DOI] [PubMed] [Google Scholar]
- Rodrigues-Diez R, Lavoz C, Carvajal G, Rayego-Mateos S, Rodrigues Diez RR, Ortiz A, et al. Gremlin is a downstream profibrotic mediator of transforming growth factor-beta in cultured renal cells. Nephron Exp Nephrol. 2012;122:62–74. doi: 10.1159/000346575. [DOI] [PubMed] [Google Scholar]
- Rosen V. BMP and BMP inhibitors in bone. Ann N Y Acad Sci. 2006;1068:19–25. doi: 10.1196/annals.1346.005. [DOI] [PubMed] [Google Scholar]
- Rosenzweig BL, Imamura T, Okadome T, Cox GN, Yamashita H, ten Dijke P, et al. Cloning and characterization of a human type II receptor for bone morphogenetic proteins. Proc Natl Acad Sci U S A. 1995;92:7632–7636. doi: 10.1073/pnas.92.17.7632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roxburgh SA, Kattla JJ, Curran SP, O'Meara YM, Pollock CA, Goldschmeding R, et al. Allelic depletion of grem1 attenuates diabetic kidney disease. Diabetes. 2009;58:1641–1650. doi: 10.2337/db08-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanvitale CE, Kerr G, Chaikuad A, Ramel MC, Mohedas AH, Reichert S, et al. A new class of small molecule inhibitor of BMP signaling. PLoS ONE. 2013;8:e62721. doi: 10.1371/journal.pone.0062721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Huang GJ, Gong YW. Effect of transforming growth factor beta and bone morphogenetic proteins on rat hepatic stellate cell proliferation and trans-differentiation. World J Gastroenterol. 2003;9:784–787. doi: 10.3748/wjg.v9.i4.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Sun C, He B, Xiong W, Shi X, Yao D, et al. GADD34-PP1c recruited by Smad7 dephosphorylates TGFbeta type I receptor. J Cell Biol. 2004;164:291–300. doi: 10.1083/jcb.200307151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Chang C, Nie S, Xie S, Wan M, Cao X. Endofin acts as a Smad anchor for receptor activation in BMP signaling. J Cell Sci. 2007;120((Pt 7)):1216–1224. doi: 10.1242/jcs.03400. [DOI] [PubMed] [Google Scholar]
- Shi Y, Wang YF, Jayaraman L, Yang H, Massague J, Pavletich NP. Crystal structure of a Smad MH1 domain bound to DNA: insights on DNA binding in TGF-beta signaling. Cell. 1998;94:585–594. doi: 10.1016/s0092-8674(00)81600-1. [DOI] [PubMed] [Google Scholar]
- Soofi A, Zhang P, Dressler GR. Kielin/chordin-like protein attenuates both acute and chronic renal injury. J Am Soc Nephrol. 2013;24:897–905. doi: 10.1681/ASN.2012070759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo Rodriguez JE, Mansky KC, Jensen ED, Carlson AE, Schwarz T, Pham L, et al. Enhanced osteoclastogenesis causes osteopenia in twisted gastrulation-deficient mice through increased BMP signaling. J Bone Miner Res. 2009;24:1917–1926. doi: 10.1359/JBMR.090507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto H, Grahovac G, Zeisberg M, Kalluri R. Renal fibrosis and glomerulosclerosis in a new mouse model of diabetic nephropathy and its regression by bone morphogenic protein-7 and advanced glycation end product inhibitors. Diabetes. 2007;56:1825–1833. doi: 10.2337/db06-1226. [DOI] [PubMed] [Google Scholar]
- Sugimoto H, LeBleu VS, Bosukonda D, Keck P, Taduri G, Bechtel W, et al. Activin-like kinase 3 is important for kidney regeneration and reversal of fibrosis. Nat Med. 2012;18:396–404. doi: 10.1038/nm.2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto H, LeBleu VS, Bosukonda D, Keck P, Taduri G, Bechtel W, et al. Reply to Regarding the mechanism of action of a proposed peptide agonist of the bone morphogenetic protein receptor activin-like kinase 3. Nat Med. 2013;19:810–811. doi: 10.1038/nm.3081. [DOI] [PubMed] [Google Scholar]
- Sun B, Huo R, Sheng Y, Li Y, Xie X, Chen C, et al. Bone morphogenetic protein-4 mediates cardiac hypertrophy, apoptosis, and fibrosis in experimentally pathological cardiac hypertrophy. Hypertension. 2013;61:352–360. doi: 10.1161/HYPERTENSIONAHA.111.00562. [DOI] [PubMed] [Google Scholar]
- Sun J, Zhuang FF, Mullersman JE, Chen H, Robertson EJ, Warburton D, et al. BMP4 activation and secretion are negatively regulated by an intracellular gremlin-BMP4 interaction. J Biol Chem. 2006;281:29349–29356. doi: 10.1074/jbc.M603833200. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Kaneko E, Maeda J, Ueno N. Mesoderm induction by BMP-4 and -7 heterodimers. Biochem Biophys Res Commun. 1997;232:153–156. doi: 10.1006/bbrc.1997.6219. [DOI] [PubMed] [Google Scholar]
- Suzuki C, Murakami G, Fukuchi M, Shimanuki T, Shikauchi Y, Imamura T, et al. Smurf1 regulates the inhibitory activity of Smad7 by targeting Smad7 to the plasma membrane. J Biol Chem. 2002;277:39919–39925. doi: 10.1074/jbc.M201901200. [DOI] [PubMed] [Google Scholar]
- Swiontkowski MF, Aro HT, Donell S, Esterhai JL, Goulet J, Jones A, et al. Recombinant human bone morphogenetic protein-2 in open tibial fractures. A subgroup analysis of data combined from two prospective randomized studies. J Bone Joint Surg Am. 2006;88:1258–1265. doi: 10.2106/JBJS.E.00499. [DOI] [PubMed] [Google Scholar]
- Tandon A, Sharma A, Rodier JT, Klibanov AM, Rieger FG, Mohan RR. BMP7 gene transfer via gold nanoparticles into stroma inhibits corneal fibrosis in vivo. PLoS ONE. 2013;8:e66434. doi: 10.1371/journal.pone.0066434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardif G, Pelletier JP, Boileau C, Martel-Pelletier J. The BMP antagonists follistatin and gremlin in normal and early osteoarthritic cartilage: an immunohistochemical study. Osteoarthritis Cartilage. 2009;17:263–270. doi: 10.1016/j.joca.2008.06.022. [DOI] [PubMed] [Google Scholar]
- Theriault BL, Shepherd TG, Mujoomdar ML, Nachtigal MW. BMP4 induces EMT and Rho GTPase activation in human ovarian cancer cells. Carcinogenesis. 2007;28:1153–1162. doi: 10.1093/carcin/bgm015. [DOI] [PubMed] [Google Scholar]
- Tominaga T, Abe H, Ueda O, Goto C, Nakahara K, Murakami T, et al. Activation of bone morphogenetic protein 4 signaling leads to glomerulosclerosis that mimics diabetic nephropathy. J Biol Chem. 2011;286:20109–20116. doi: 10.1074/jbc.M110.179382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukazaki T, Chiang TA, Davison AF, Attisano L, Wrana JL. SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell. 1998;95:779–791. doi: 10.1016/s0092-8674(00)81701-8. [DOI] [PubMed] [Google Scholar]
- Urist MR. Bone: formation by autoinduction. Science. 1965;150:893–899. doi: 10.1126/science.150.3698.893. [DOI] [PubMed] [Google Scholar]
- Vukicevic S, Basic V, Rogic D, Basic N, Shih MS, Shepard A, et al. Osteogenic protein-1 (bone morphogenetic protein-7) reduces severity of injury after ischemic acute renal failure in rat. J Clin Invest. 1998;102:202–214. doi: 10.1172/JCI2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DW, Roxburgh SA, McGettigan P, Berthier CC, Higgins DG, Kretzler M, et al. Co-regulation of gremlin and notch signalling in diabetic nephropathy. Biochim Biophys Acta. 2008;1782:10–21. doi: 10.1016/j.bbadis.2007.09.005. [DOI] [PubMed] [Google Scholar]
- Walsh DW, Godson C, Brazil DP, Martin F. Extracellular BMP-antagonist regulation in development and disease: tied up in knots. Trends Cell Biol. 2010;20:244–256. doi: 10.1016/j.tcb.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Wang S, Hirschberg R. BMP7 antagonizes TGF-beta-dependent fibrogenesis in mesangial cells. Am J Physiol Renal Physiol. 2003;284:F1006–F1013. doi: 10.1152/ajprenal.00382.2002. [DOI] [PubMed] [Google Scholar]
- Wang S, Hirschberg R. Bone morphogenetic protein-7 signals opposing transforming growth factor beta in mesangial cells. J Biol Chem. 2004;279:23200–23206. doi: 10.1074/jbc.M311998200. [DOI] [PubMed] [Google Scholar]
- Wang S, de Caestecker M, Kopp J, Mitu G, Lapage J, Hirschberg R. Renal bone morphogenetic protein-7 protects against diabetic nephropathy. J Am Soc Nephrol. 2006;17:2504–2512. doi: 10.1681/ASN.2006030278. [DOI] [PubMed] [Google Scholar]
- Wang SN, Lapage J, Hirschberg R. Loss of tubular bone morphogenetic protein-7 in diabetic nephropathy. J Am Soc Nephrol. 2001;12:2392–2399. doi: 10.1681/ASN.V12112392. [DOI] [PubMed] [Google Scholar]
- Weiskirchen R, Meurer SK. BMP-7 counteracting TGF-beta1 activities in organ fibrosis. Front Biosci. 2013;18:1407–1434. doi: 10.2741/4189. [DOI] [PubMed] [Google Scholar]
- Whitman M, Rosen V, Brivanlou AH, Groppe JC, Sebald W, Mueller T. Regarding the mechanism of action of a proposed peptide agonist of the bone morphogenetic protein receptor activin-like kinase 3. Nat Med. 2013;19:809–810. doi: 10.1038/nm.3080. [DOI] [PubMed] [Google Scholar]
- Wilkinson L, Kolle G, Wen D, Piper M, Scott J, Little M. CRIM1 regulates the rate of processing and delivery of bone morphogenetic proteins to the cell surface. J Biol Chem. 2003;278:34181–34188. doi: 10.1074/jbc.M301247200. [DOI] [PubMed] [Google Scholar]
- Wordinger RJ, Zode G, Clark AF. Focus on molecules: gremlin. Exp Eye Res. 2008;87:78–79. doi: 10.1016/j.exer.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrana JL. Regulation of Smad activity. Cell. 2000;100:189–192. doi: 10.1016/s0092-8674(00)81556-1. [DOI] [PubMed] [Google Scholar]
- Wu XB, Li Y, Schneider A, Yu W, Rajendren G, Iqbal J, et al. Impaired osteoblastic differentiation, reduced bone formation, and severe osteoporosis in noggin-overexpressing mice. J Clin Invest. 2003;112:924–934. doi: 10.1172/JCI15543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao YT, Xiang LX, Shao JZ. Bone morphogenetic protein. Biochem Biophys Res Commun. 2007;362:550–553. doi: 10.1016/j.bbrc.2007.08.045. [DOI] [PubMed] [Google Scholar]
- Xu L, Chen YG, Massague J. The nuclear import function of Smad2 is masked by SARA and unmasked by TGFbeta-dependent phosphorylation. Nat Cell Biol. 2000;2:559–562. doi: 10.1038/35019649. [DOI] [PubMed] [Google Scholar]
- Xu T, Yu CY, Sun JJ, Liu Y, Wang XW, Pi LM, et al. Bone morphogenetic protein-4-induced epithelial-mesenchymal transition and invasiveness through Smad1-mediated signal pathway in squamous cell carcinoma of the head and neck. Arch Med Res. 2011;42:128–137. doi: 10.1016/j.arcmed.2011.03.003. [DOI] [PubMed] [Google Scholar]
- Yamada S, Nakamura J, Asada M, Takase M, Matsusaka T, Iguchi T, et al. Twisted gastrulation, a BMP antagonist, exacerbates podocyte injury. PLoS ONE. 2014;9:e89135. doi: 10.1371/journal.pone.0089135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C, Grimm WA, Garner WL, Qin L, Travis T, Tan N, et al. Epithelial to mesenchymal transition in human skin wound healing is induced by tumor necrosis factor-alpha through bone morphogenic protein-2. Am J Pathol. 2010;176:2247–2258. doi: 10.2353/ajpath.2010.090048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Chen SL, Lu XJ, Shen CY, Liu Y, Chen YP. Bone morphogenetic protein 7 suppresses the progression of hepatic fibrosis and regulates the expression of gremlin and transforming growth factor beta1. Mol Med Rep. 2012;6:246–252. doi: 10.3892/mmr.2012.892. [DOI] [PubMed] [Google Scholar]
- Yang YL, Liu YS, Chuang LY, Guh JY, Lee TC, Liao TN, et al. Bone morphogenetic protein-2 antagonizes renal interstitial fibrosis by promoting catabolism of type I transforming growth factor-beta receptors. Endocrinology. 2009;150:727–740. doi: 10.1210/en.2008-0090. [DOI] [PubMed] [Google Scholar]
- Yang YL, Ju HZ, Liu SF, Lee TC, Shih YW, Chuang LY, et al. BMP-2 suppresses renal interstitial fibrosis by regulating epithelial-mesenchymal transition. J Cell Biochem. 2011;112:2558–2565. doi: 10.1002/jcb.23180. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Tanaka S, Umemori H, Minowa O, Usui M, Ikematsu N, et al. Negative regulation of BMP/Smad signaling by Tob in osteoblasts. Cell. 2000;103:1085–1097. doi: 10.1016/s0092-8674(00)00211-7. [DOI] [PubMed] [Google Scholar]
- Yu PB, Deng DY, Lai CS, Hong CC, Cuny GD, Bouxsein ML, et al. BMP type I receptor inhibition reduces heterotopic [corrected] ossification. Nat Med. 2008a;14:1363–1369. doi: 10.1038/nm.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu PB, Hong CC, Sachidanandan C, Babitt JL, Deng DY, Hoyng SA, et al. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat Chem Biol. 2008b;4:33–41. doi: 10.1038/nchembio.2007.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S, Pan Q, Liu W, Wu B, Han X, Bi Z. Recombinant BMP 4/7 fusion protein induces differentiation of bone marrow stem cells. J Cell Biochem. 2011;112:3054–3060. doi: 10.1002/jcb.23230. [DOI] [PubMed] [Google Scholar]
- Zawel L, Dai JL, Buckhaults P, Zhou S, Kinzler KW, Vogelstein B, et al. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell. 1998;1:611–617. doi: 10.1016/s1097-2765(00)80061-1. [DOI] [PubMed] [Google Scholar]
- Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- Zeisberg M, Bottiglio C, Kumar N, Maeshima Y, Strutz F, Muller GA, et al. Bone morphogenic protein-7 inhibits progression of chronic renal fibrosis associated with two genetic mouse models. Am J Physiol Renal Physiol. 2003a;285:F1060–F1067. doi: 10.1152/ajprenal.00191.2002. [DOI] [PubMed] [Google Scholar]
- Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, et al. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003b;9:964–968. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- Zeisberg M, Shah AA, Kalluri R. Bone morphogenic protein-7 induces mesenchymal to epithelial transition in adult renal fibroblasts and facilitates regeneration of injured kidney. J Biol Chem. 2005;280:8094–8100. doi: 10.1074/jbc.M413102200. [DOI] [PubMed] [Google Scholar]
- Zhang F, Qiu T, Wu X, Wan C, Shi W, Wang Y, et al. Sustained BMP signaling in osteoblasts stimulates bone formation by promoting angiogenesis and osteoblast differentiation. J Bone Miner Res. 2009;24:1224–1233. doi: 10.1359/JBMR.090204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Shi Y, Wada J, Malakauskas SM, Liu M, Ren Y, et al. In vivo delivery of gremlin siRNA plasmid reveals therapeutic potential against diabetic nephropathy by recovering bone morphogenetic protein-7. PLoS ONE. 2010;5:e11709. doi: 10.1371/journal.pone.0011709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, Wang X, Wang S, Yang L, Gao H, Yang C. The anti-fibrotic effect of bone morphogenic protein-7(BMP-7) on liver fibrosis. Int J Med Sci. 2013;10:441–450. doi: 10.7150/ijms.5765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Kavsak P, Abdollah S, Wrana JL, Thomsen GH. A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature. 1999;400:687–693. doi: 10.1038/23293. [DOI] [PubMed] [Google Scholar]
- Zhu W, Kim J, Cheng C, Rawlins BA, Boachie-Adjei O, Crystal RG, et al. Noggin regulation of bone morphogenetic protein (BMP) 2/7 heterodimer activity in vitro. Bone. 2006;39:61–71. doi: 10.1016/j.bone.2005.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman LB, De Jesus-Escobar JM, Harland RM. The Spemann organizer signal noggin binds and inactivates bone morphogenetic protein 4. Cell. 1996;86:599–606. doi: 10.1016/s0092-8674(00)80133-6. [DOI] [PubMed] [Google Scholar]
- Ziran BH, Hendi P, Smith WR, Westerheide K, Agudelo JF. Osseous healing with a composite of allograft and demineralized bone matrix: adverse effects of smoking. Am J Orthop. 2007;36:207–209. [PubMed] [Google Scholar]