

Abstract
BACKGROUND AND PURPOSE
Cardiac rupture is a catastrophic complication that occurs after acute myocardial infarction (MI) and, at present, there are no effective pharmacological strategies for preventing this condition. Here we investigated the effect of the angiotensin II receptor blocker olmesartan (Olm) on post-infarct cardiac rupture and its underlying mechanisms of action.
EXPERIMENTAL APPROACH
C57Bl/6 mice with MI were treated with Olm, aldosterone (Aldo) or vehicle. Cultured neonatal cardiomyocytes and fibroblasts were exposed to normoxia or anoxia and treated with angiotensin II (Ang II), RNH6270 (active ingredient of Olm) or Aldo.
KEY RESULTS
The mortality rate and incidence of cardiac rupture in MI mice during the first week in the Olm-treated group were significantly lower than in the vehicle-treated group. Olm or RNH6270 reduced myeloperoxidase staining in the infarcted myocardium, decreased apoptosis in cultured cardiomyocytes and fibroblasts, as assessed by Hoechst staining and TUNEL assay, attenuated the accumulation of p53 and phosphorylated p53 and cleaved caspase 3 induced by MI or Ang II, as assessed by Western blotting, and up-regulated growth differentiation factor-15 (GDF-15). In cultured cardiomyocytes and fibroblasts, treatment with Ang II, Aldo or anoxia significantly down-regulated the expression of GDF-15.
CONCLUSIONS AND IMPLICATIONS
Olm prevents cardiac rupture through inhibition of apoptosis and inflammation, which is attributable to the down-regulation of p53 activity and up-regulation of GDF-15. Our findings suggest that early administration of an AT1 receptor anatagonist to patients with acute MI is a potential preventive approach for cardiac rupture.
Keywords: cardiac rupture, olmesartan, p53, GDF-15, aldosterone, myocardial infarction
Introduction
Cardiac rupture is a catastrophic complication for patients with acute myocardial infarction (MI). Despite significant advances in reperfusion strategies such as percutaneous coronary intervention, mortality from cardiac rupture remains high, indicating the need to develop effective preventive approaches. Over the past two decades the incidence of cardiac rupture, globally, after acute MI is approximately 6% and the mortality in patients with cardiac rupture can be as high as 58% (Becker et al., 1996; Lopez-Sendon et al., 2010; Shamshad et al., 2010). In patients with ST segment elevated MI, ventricle rupture occurs within 10 days (Gao et al., 2012). Similarly in mice with MI, cardiac rupture usually happens within the first 10 days and peaks on days 3 to 5 after acute MI (Gao et al., 2012; Xuan et al., 2012). Therefore, the acute MI model of mice has become an ideal tool for clarifying the mechanisms of cardiac rupture and searching for a preventive approach.
Inflammation and apoptosis are two important factors influencing cardiac rupture. Excessive inflammation plays an important role in the development of cardiac rupture. The vulnerable myocardium after MI, which consists of necrotic tissue and inflammatory cells, is susceptible to wall stress and consequently cardiac rupture. Intensive interstitial neutrophil infiltration in ruptured myocardial wall is found in patients with MI (Zidar et al., 2005), and myeloperoxidase (MPO) knockout mice are resistant to MI-induced cardiac rupture (Askari et al., 2003). In contrast, growth differentiation factor-15 (GDF-15), an anti-inflammatory factor, has been demonstrated to prevent cardiac rupture in mice with MI (Kempf et al., 2011). In terms of apoptosis, it was reported that deletion of pro-apoptotic p53 in mice can prevent cardiac rupture due to MI (Matsusaka et al., 2006). Based on these findings, it is plausible to speculate that anti-inflammatory as well as anti-apoptotic drugs can both help prevent cardiac rupture.
Both the circulating renin–angiotensin–aldosterone system (RAAS) and intra-cardiac renin–angiotensin system (RAS) are known to be activated in patients with MI (Sun, 2010). Also angiotensin II (Ang II) has been demonstrated to stimulate inflammation and apoptosis in MI mice (Leri et al., 1998; Zhao et al., 2004; Acosta et al., 2009), and excessive production of aldosterone (Aldo) exacerbates the progress of cardiac rupture in MI mice (He et al., 2011), implying that the activation of RAAS may lead to cardiac rupture; but it remains unclear whether pharmacological inhibition of Ang II, by modulating GDF-15 and p53, can prevent cardiac rupture.
The Ang II receptor antagonist olmesartan medoxomil (Olm) has been found to have a 12 500-fold greater affinity for the Ang II type 1 receptor (AT1 receptor) than for the Ang II type 2 receptor (AT2 receptor) and is used to treat hypertension, coronary heart disease and heart failure. Olm has a strong anti-inflammatory effect (Fliser et al., 2004) and inhibits the production of Aldo and reduces the risk of cardiovascular death (Savarese et al., 2013), but it is not known whether it can prevent post-MI cardiac rupture. In this study, we tested the hypothesis that Olm can prevent cardiac rupture by modulating p53 and GDF-15.
Methods
All procedures were performed in accordance with our institutional guidelines for animal research that conforms to the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85–23, revised 1996), and this study was approved by the Ethical Committee of Nanfang Hospital, Southern Medical University. Mice were kept in standard housing conditions with a light/dark cycle of 12 h and free access to food and water. The dose and concentration of Aldo, Olm and PD123319 were determined according to previous reports (Kanamori et al., 2007; Abadir et al., 2011; He et al., 2011). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Animal models of MI
C57BL/6 male mice (aged 8–12 weeks, weighing 20–25 g) were used to generate MI models by left coronary artery ligation as described elsewhere (Xuan et al., 2012). Mice were anaesthetized with a mixture of xylazine (5 mg·kg−1, injected i.p.) and ketamine (100 mg·kg−1, injected i.p.), and the depth of anaesthesia was assessed by monitoring the pedal withdrawal reflex. The operated mice that survived for 12 h were randomized to receive treatment with Olm (10 mg·kg−1·day−1, Daiichi Sankyo company, Tokyo, Japan) or vehicle for 1 to 7 days. Sham-operated mice were also treated vehicle or Olm. Based on the daily food intake of the mice, Olm was mixed into their food, which was available ad libitum. The food–drug mixture was freshly made every day. A group of MI mice were given Aldo (1.44 mg·kg−1·day−1, i.p.) dissolved in DMSO (Sigma Chemicals, St. Louis, MO, USA) for 3 days, and the mice in the vehicle-treated group received the same volume of DMSO. At the indicated time points, the mice were killed by an overdose of anaesthetic, pentobarbital sodium (150 mg·kg−1, i.p.), and cervical dislocation, and their hearts were extracted for further analysis. For histological examinations, hearts were fixed in 10% formalin, whereas for molecular analysis the hearts were snap-frozen in liquid nitrogen and stored at −80°C until used. All studies involving drug/molecular target nomenclature conform to BJP's Concise Guide to Pharmacology (Alexander et al., 2013).
Invasive assessment of haemodynamics and echocardiographic measurements
Both the haemodynamic assessment and echocardiographic measurement were performed 3 days after surgery (details in Supporting Information).
Cell culture
The isolation and culturing of neonatal rat ventricular cardiomyocytes and fibroblasts were carried out as described previously (Xuan et al., 2012). In the Ang II treated groups, cells were treated with 10−6 M Ang II (Sigma Chemicals), whereas for the RNH6270 treatment group, 10−6 M RNH6270 (Daiichi Sankyo company) dissolved in DMSO was added to the cells 20 min before the addition of Ang II. We designed two control groups: blank and vehicle group (treated with DMSO). Cells were harvested 24 h after the addition of Ang II (or vehicle). In the Aldo-treated groups, cells were treated with 10−7 M Aldo (Sigma Chemicals) for 24 h and DMSO was added to the cells as a control treatment.
The effects of anoxia on GDF-15 were also analysed in cells. Cultured cells were divided into three groups: (i) control group: cardiomyocytes and fibroblasts were incubated with culture medium for 24 h in normoxic conditions; (ii) anoxia group: cells were incubated with anaerobic-simulated ischaemia buffer for 24 h; and (iii) anoxia + RNH6270 group: cells were treated with RNH6270 for 20 min followed by anoxic conditions for 24 h.
PCR
Total RNA was extracted from cultured cells and mouse heart tissues (total RNA isolation system, Omega, Norcross, GA, USA). Reverse transcription was carried out in 20 μL of reaction mixture containing 1 μg of total RNA. Digital images for GDF-15, periostin, GADPH and β-actin bands separated on ethidium bromide-stained agarose gels were obtained. Quantitative analysis was performed using Image J software (National Institutes of Health, Bethesda, MD, USA). The mRNA expressions of GDF-15 and β-actin in mouse heart were determined using the Quantitect SYBR Green real-time PCR method. Primer sequences are listed in Supporting Information Table S1.
Western blotting
The following antibodies were used for the Western blotting analysis: anti-GDF-15 (Santa Cruz, CA, USA), anti-p53 (Santa Cruz), anti-p-p53 (phospho S15; Abcam, New Territories, Hong Kong), anti-caspase 3 or anti-cleaved caspase 3 (Abcam, Cambridge, MA, USA), anti-GAPDH or anti-β-actin (ZSGB-Bio, Peking, China). Proteins were obtained from whole heart or cells. Samples containing equal amounts of protein were separated by 10% SDS-PAGE and transferred onto PVDF membranes. The membranes were blocked with 5% skimmed milk at room temperature for 2 h and then incubated overnight at 4°C with the primary antibody. After being incubated with IRDye 800CW goat anti-rabbit or goat anti-mouse secondary antibody (LI-COR, Lincoln, NE, USA) for 1 h at room temperature, the blots were detected in the Odyssey Infrared Imaging System (LI-COR) and quantified by densitometry using the Image J Analysis software (National Institutes of Health).
Immunohistochemistry
The heart tissues from different groups were fixed in 4% paraformaldehyde and embedded in paraffin; 4 μm sections were prepared for immunostaining analysis. After the antigens were retrieved in citrate buffer at pH 6.0, the sections were incubated with rabbit anti-MPO antibodies (Abcam) or anti-periostin antibodies (Abcam) overnight at 4°C. The tissue sections were scanned for intensity and area of staining; the staining intensity was scored as: 0 (negative), 1 (weak), 2 (medium) or 3 (strong). The extent of staining was scored as 0 (0%), 1 (1–25%), 2 (26–50%), 3 (51–75%) or 4 (76–100%), according to the percentages of positively stained areas in relation to the whole visual field. The sum of the staining intensity and extent score was used as the final staining score (0–7) for MPO or periostin.
Hoechst stain and TUNEL assay
After the cultured cardiomyocytes or fibroblasts had been treated with Ang II or RNH6270 for 24 h, the culture medium was removed and the cells were then washed with PBS, three times. Hoechst reagent (Beyotime Chemicals, Haimen, China) was added to the cells for 30 min at room temperature in a dark place. The photographs were taken under a fluorescent inverted microscope (Olympus, Hicksville, NY, USA).
Apoptosis in rat cultured neonatal cardiomyocytes and fibroblasts, and mouse myocardium was determined using TUNEL assay (for details see Supporting Information).
Statistical analysis
All data are expressed as mean ± SEM, and P < 0.05 was considered to be statistically significant. Statistical differences were evaluated by one-way anova followed by Bonferroni's multiple comparison exact probability test. The overall survival of MI mice was evaluated using Kaplan–Meier survival analysis. All analyses were performed using SPSS 13.0 software (SPSS Inc, Chicago, IL, USA).
Results
Olm prevents cardiac rupture and acute heart failure in MI mice
Since cardiac ruptures in mice mostly happen within the first week after MI, we observed the influence of Olm on the survival rate of MI mice for one week. The survival of mice was checked twice a day to ensure that once a dead mouse was found, an autopsy was performed immediately to confirm the reason of death. We found that the overall survival rate of mice treated with Olm was significantly higher than in mice in the vehicle group (88% vs. 48%, P = 0.001; Figure 1A). There was clear ST segment elevation after left coronary artery ligation in all the MI mice (Figure 1B). Autopsy on dead mice showed that cardiac rupture and acute heart failure (AHF) were the principal causes of death during the first 7 days of MI (Figure 1C and D). In 53 vehicle-treated mice, 28 died (52.8%), among which mice with cardiac rupture accounted for 53.6% (15/28), slits of cardiac rupture and blood clots around the heart were found through autopsy (Figure 1C); mice with AHF accounted for 60.7% (17/28), for their lung weight/body weight ratio was twice as high as that of the sham-operated group (Figure 1D). Mice with AHF but without cardiac rupture were considered to have died of AHF. As a result, mice that died of AHF represented 21.4% (6/28) of the dead mice. However, among the 30 mice receiving Olm treatment, there was no cardiac rupture and only three died of AHF. Using Kaplan–Meier analysis, we found that Olm significantly lowered the incidence of cardiac rupture and AHF.
Figure 1.
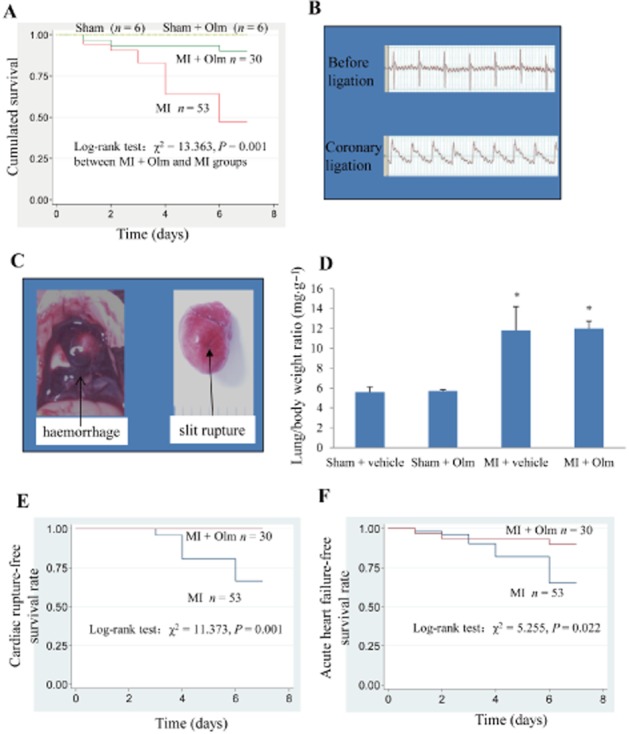
Effects of Olm treatment on survival and cardiac rupture of MI mice. (A) One week survival rate of four groups in response to MI or sham with/without Olm treatment. (B) ECG before and after ligation of the MI mice. After ligation, the ST segment was significantly elevated, which was the sign of success for MI surgery. (C) Examples of autopsy pictures from a dead mouse. Haemorrhage (blood clots) in the chest and obvious rupture slits in the left ventricle free wall were identified. (D) Lung weight/body weight ratio of mice that died from AHF accompanied with/without cardiac rupture following MI and sham-operated mice killed 1 week after surgery. *P < 0.01 versus the corresponding sham group, n = 6 in both sham + vehicle and sham + Olm groups; n = 16 in MI + vehicle group (16 mice died from AHF of a total 53 mice), n = 3 in MI + Olm group (three mice died from AHF of a total 30 mice). (E) Cardiac rupture-free survival rate during the first week after MI. (F) AHF-free survival rate during the first week after MI. Dose of Olm was 10 mg·kg−1·day−1.
Effects of Olm on left ventricular haemodynamics
Since the improvement in haemodynamics and the lowering of ventricular wall stress could be one reason for the decreased incidence of cardiac rupture in Olm-treated mice, we analysed the influence of Olm on the left ventricular haemodynamics and cardiac systolic function of MI mice (Supporting Information Figure S1A–G). On the day before the peak day of mortality (day 4), we found there was no significant difference in left ventricular fractional shortening (LVFS) between the MI + vehicle and MI + Olm groups (Supporting Information Figure S1B). We also examined the LV systolic pressure, LV end-diastolic pressure, maximum rate of rise of LV pressure (dp/dt max), descending rate of LV pressure (dp/dt min) and LV contractility, and no significant difference was found between the two groups (Supporting Information Figure S1C–G). These findings suggest that the dose of Olm employed in this study is not sufficient to change the LV haemodynamics in the acute phase of MI. When the observation period was prolonged to 6 weeks, we found that the LVFS of MI mice in the Olm-treated group was markedly higher than that in the untreated group (12.3 ± 0.9% in MI group, 32 ± 3% in MI ± Olm group, P < 0.001. Supporting Information Figure S2).
Effects of Olm on myocardial expression of MPO and periostin in MI mice
Knowing that inflammation is considered as one major mechanism for cardiac rupture, the question arose as to whether the mechanism for Olm preventing cardiac rupture is related to inhibition of inflammation. Hence, we determined the MPO expression levels in the cardiac tissues of the sham-operated and MI groups of mice on days 2, 4, 8 after MI. The result shows that the MPO expression in the infarct area peaked on day 2, but was markedly reduced on days 4 and 8 (Figure 2 and Supporting Information Figure S3). However, the MPO expression in the Olm-treated group was already significantly lower than the vehicle-treated group on day 2 (P < 0.05, Figure 2A and B), while the levels of MPO remained high even 3 days after MI in mice treated with Aldo (Supporting Information Figure S4).
Figure 2.
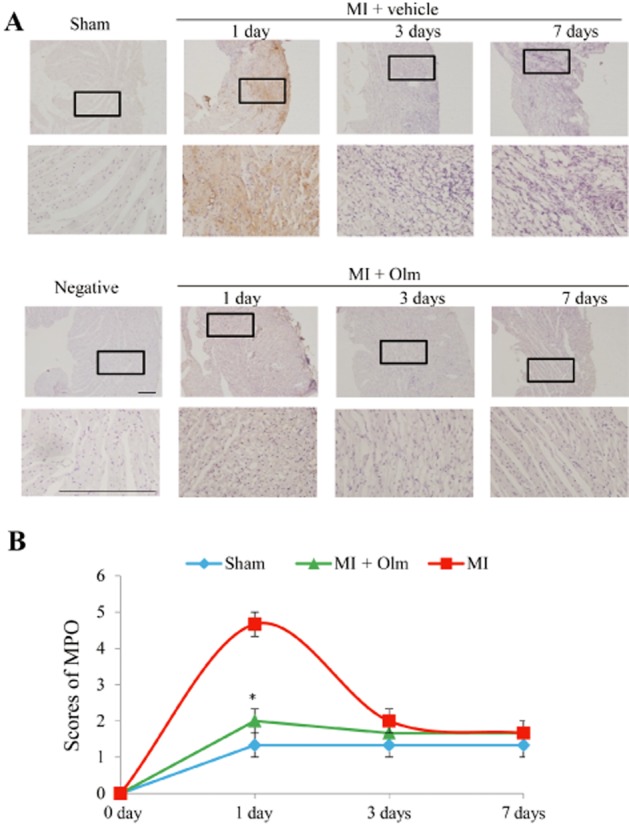
Effect of Olm on MPO expression in mice with MI. (A) Examples of pictures showing a time course of immunohistochemical MPO staining in response to MI for 1 day, 3 days and 7 days, respectively (upper panels), and effect of Olm treatment (10 mg·kg−1·day−1) on MPO expression at the corresponding time points (lower panels). Scale bar = 100 μm. (B) Semi-quantitative analysis of MPO expression using a score system in each group. *P < 0.05 versus MI + vehicle group. n = 3 per group.
A lack of periostin has been shown to increase the incidence of cardiac rupture in mice after acute MI (Shimazaki et al., 2008), thus we examined whether Olm influences myocardial periostin expression. The result shows that periostin mRNA and protein both were up-regulated time-dependently after MI, Olm treatment did not influence its expression level (Supporting Information Figure S5A–C). In cell culture experiments, anoxia for 6–24 h did not increase the periostin mRNA expression in cardiomyocytes, but significantly up-regulated the expression of periostin in fibroblasts (Supporting Information Figure S5D). Similar to the in vivo experiments, Olm had no significant effect on the expression level of periostin in the cardiomyocytes or fibroblasts in vitro.
Olm inhibits apoptosis of cardiac cells
The myocardial cell apoptosis 3 days after MI or sham operation detected by TUNEL assay was significantly increased in MI mice compared with the corresponding sham group, but the amplitude of this increase was markedly smaller in the MI + Olm group but larger in the MI + Aldo group (Figure 3A and B). Western blotting of whole-heart tissues showed that the expression levels of p53 and phosphorylated p53 (p-p53) 1 day after MI were significantly lower in the Olm-treated mice than in vehicle-treated mice (Supporting Information Figure S6A and B). Expression levels of p-p53 and cleaved caspase 3 protein in both infarcted and remote myocardial tissues were markedly up-regulated in MI mice, but the amplitude of the increase was significantly smaller in the MI + Olm group mice (Figure 3C–E) but larger in the MI + Aldo group (Figure 3F and G).
Figure 3.
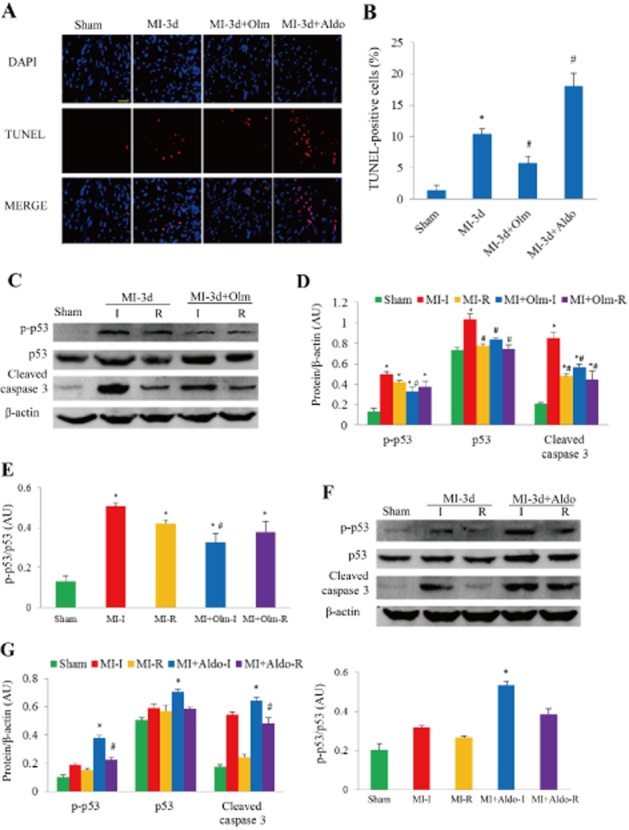
Olm inhibited apoptosis and the apoptotic signal in vivo in response to MI for 3 days. (A) Representative images of TUNEL assay in different groups. The nucleus was labelled with DAPI (blue staining), apoptosis was labelled with TUNEL reaction mixture (red staining). The merged pink staining indicates apoptotic nucleus. Scale bar = 100 μm. (B) Quantitative analysis of apoptotic cells. (C) Western blots of myocardial p53, p-p53 and cleaved caspase 3 in response to MI treated with/without Olm or sham. (D) Semi-quantitative analysis of p53, p-p53 and cleaved caspase 3 expression normalized to β-actin in each group. (E) p-p53/p53 ratio in each group. (F) Western blots of myocardial p53, p-p53 and cleaved caspase 3 in response to MI treated with/without Aldo or sham. (G) Semi-quantitative analysis of p53, p-p53 and cleaved caspase 3 expression normalized to β-actin and p-p53/p53 ratio. I, infarct myocardial tissue; R, remote myocardial tissue. *P < 0.05 versus sham, #P < 0.05 versus MI group, n = 6 in each group. Dose of Olm was 10 mg·kg−1·day−1, Aldo was 1.44 mg·kg−1·day−1.
Ang II (1 μM) stimulation for 24 h in both cultured cardiomyocytes and fibroblasts significantly increased cell apoptosis as assessed by Hoechst staining (Figure 4A), while RNH6270, the active ingredient of Olm, markedly suppressed Ang II-induced apoptosis (P < 0.05, Figure 4A and B). Similar results were obtained using the TUNEL assay (Figure 4C and D). In cultured cardiomyocytes and fibroblasts, Ang II stimulation for 24 h significantly up-regulated p-p53 and cleaved casepase 3, while co-treatment with RNH6270 suppressed their up-regulation in both cardiomyocytes and fibroblasts (Figure 5A and B). Stimulation with Aldo or anoxia also markedly up-regulated p-p53 and cleaved caspase 3, as assessed by Western blotting (Figure 5C and D), while co-treatment with RNH6270 attenuated the effect of anoxia (Figure 5C and D). The AT2 receptor antagonist PD123319 had no significant effect on Ang II-induced up-regulation of p-p53 and cleaved caspase 3 (Supporting Information Figure S7A and B).
Figure 4.
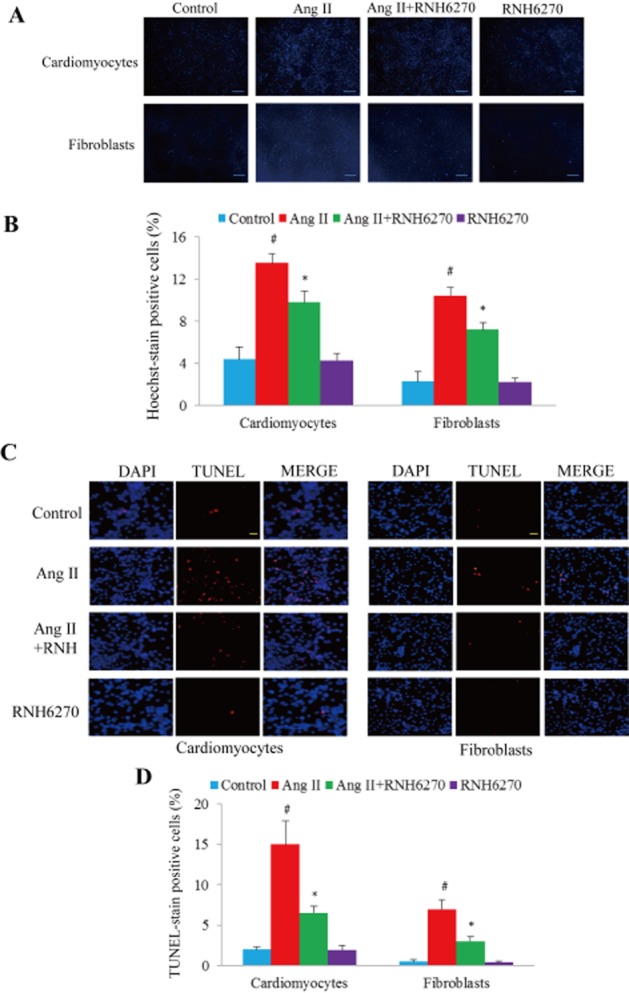
Effect of RNH6270 on apoptosis in cultured cells. (A) Hoechst stain of cultured neonatal rat cardiomyocytes and fibroblasts treated with Ang II (1 μM) or RNH6270 (1 μM) for 24 h. Scale bar = 100 μm. (B) Semi-quantitative analysis of positive apoptotic cells in each group. (C) Representative pictures showing apoptosis measured by TUNEL assay. The merged pink staining indicates apoptotic nucleus. Scale bar = 100 μm. (D) Quantitative analysis of apoptotic cells. The positive rate of TUNEL-labelled nuclei was calculated from four different and randomly selected areas under confocal microscopy for each slide. *P < 0.05 versus Ang II + RNH6270 group. #P < 0.05 versus control group, n = 4 (culture plates) per group.
Figure 5.
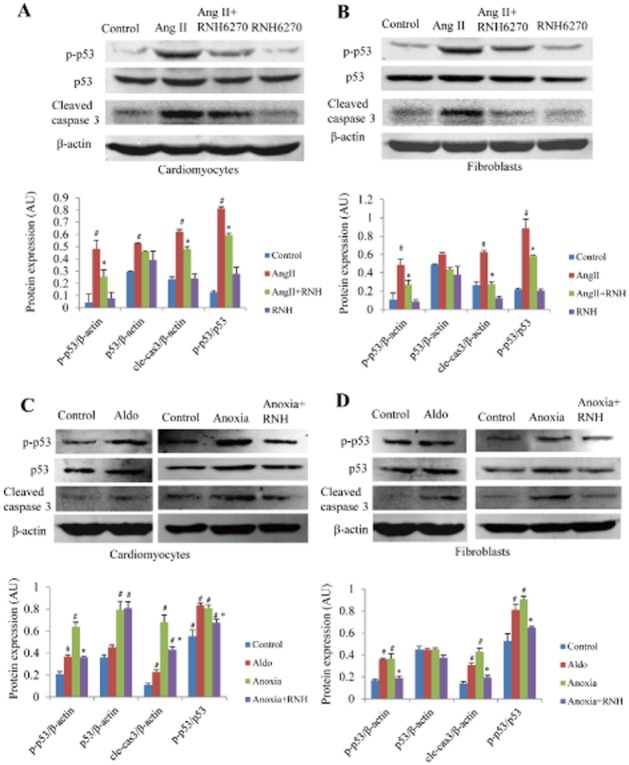
RNH6270 inhibited the apoptotic signal in vitro. Western blots of p53, p-p53 and cleaved caspase 3 induced by Ang II in cultured neonatal rat cardiomyocytes (A) and fibroblasts (B) were performed, and the expression of each protein was normalized to β-actin and the p-p53/p53 ratios were calculated. Effect of Aldo or anoxia on protein expression of p53, p-p53 and cleaved caspase 3 was also investigated in neonatal rat cultured cardiomyocytes (C) and fibroblasts (D). #P < 0.05 versus control group, *P < 0.05 versus Ang II or Aldo or anoxia-treated group. Experiments were repeated three times. Concentrations of Ang II and RNH6270 in cell culture experiments were both 1 μM, whereas that of Aldo was 0.1 μM.
Olm up-regulates cardiac GDF-15
Both mRNA and protein of GDF-15 in whole-heart tissues were also significantly up-regulated in response to MI for 1 day or for 3 days (Supporting Information Figure S8A–D). GDF-15 in both infarct and remote myocardial tissues was significantly up-regulated at 1 and 3 days after MI (Figure 6A and B), and treatment with Olm further increased GDF-15 expression (Figure 6A). In contrast, Aldo treatment for 3 days significantly down-regulated the expression of GDF-15 (P < 0.05, Figure 6B). In cultured cardiomyocytes and fibroblasts, stimulation with Aldo, anoxia or Ang II also markedly down-regulated GDF-15 as indicated by Western blot (Figure 6C–E), while co-treatment with RNH6270 attenuated this effect (Figure 6C–E).
Figure 6.
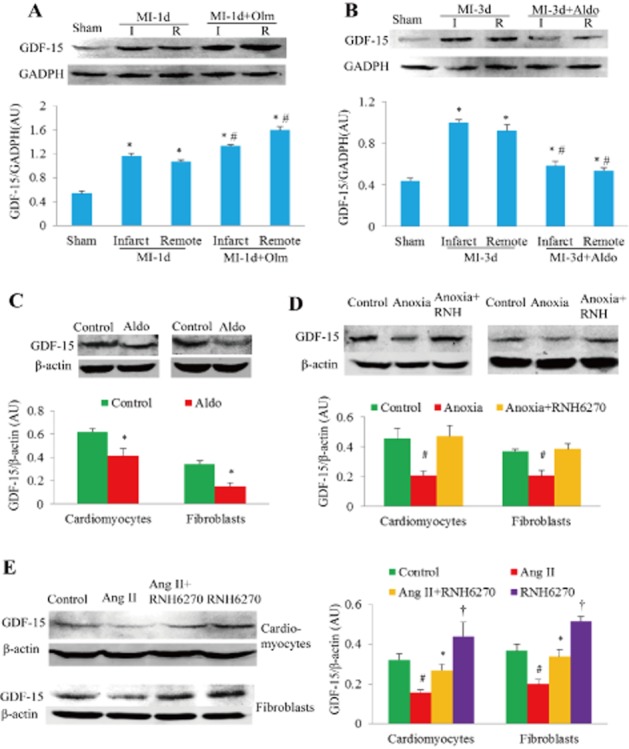
Effects of Olm or RNH6270 on the expression of GDF-15. (A) Myocardial GDF-15 expression 1 day after MI or sham-operation was detected by Western blotting. Semi-quantitative analysis showed that Olm increased GDF-15 expression significantly. (B) Aldo blunted GDF-15 protein up-regulation at 3 days after MI. For panels A and B, *P < 0.05 versus sham group, #P < 0.05 versus the corresponding MI group treated with vehicle, n = 6 in each group. I, infarct myocardial tissue; R, remote myocardial tissue. Dose of Olm and Aldo in vivo was 10 mg·kg−1·day−1 and 1.44 mg·kg−1·day−1 respectively. (C) Aldo also reduced the expression of GDF-15 protein in neonatal rat cardiomyocytes and fibroblasts. *P < 0.05 versus control group. (D) GDF-15 protein levels in response to anoxia with/without pretreatment with RNH6270 for 24 h in both neonatal rat cardiomyocytes and fibroblasts. #P < 0.05 versus control group or anoxia + RNH6270 group. (E) GDF-15 protein levels in response to Ang II with/without pretreatment with RNH6270 for 24 h in both cardiomyocytes and fibroblasts of neonatal rat. #P < 0.05 versus control group, *P < 0.05 versus Ang II-treated group. †P < 0.05 versus control group. For panels C–E, experiments were repeated three times. Concentrations of Ang II, RNH6270 and Aldo in the cell culture experiments were 1 μM, 1 μM and 0.1 μM respectively.
Discussion
As shown in Figure 7, in this study we found that Olm prevents cardiac rupture in MI mice, and that this is attributable, at least in part, to a reduction in apoptosis induced by down-regulating p53 and p-p53 as well as the attenuation of inflammation evoked by up-regulating GDF-15 and inhibiting the production of Aldo. How Olm reduces the incidence of AHF remains unclear and needs to be clarified in future studies; in the present study we focused on why Olm prevents cardiac rupture.
Figure 7.
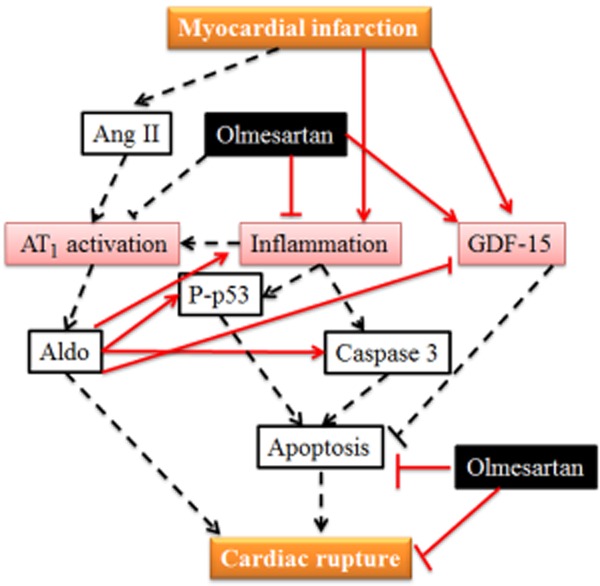
Illustration of the mechanisms by which Olm could inhibit cardiac rupture. MI activates Ang II–Ang II receptor type 1(AT1)– Aldo system and promotes myocardial inflammation, which leads to cell apoptosis. GDF-15 is also increased, which has anti-apoptotic and anti-inflammatory effects. Olm prevents cardiac rupture through inhibition of apoptosis and inflammation, which is attributable, at least partially, to the down-regulation of p53 activity and up-regulation of GDF-15. Solid lines stand for the work done in this study, dotted lines represent the well-known facts or evidence from the literature. ↑, activation; ⊥, inhibition.
Inflammation, apoptosis and blunted fibrotic healing are closely associated with the risk of cardiac rupture. Coincidently, RAS activation is known to be involved in all of these pathophysiological processes, but it is still unclear whether pharmacological interventions targeting Ang II would reduce cardiac rupture. Genetic evidence has shown that the incidence of cardiac rupture in AT2 receptor gene knockout mice is significantly higher than in the wild type mice (Ichihara et al., 2002), which indicates that maintaining AT2 receptor activation to a certain degree could help to prevent cardiac rupture. Since Olm is a highly selective AT1 receptor blocker, we used it to study the preventive effect of blocking AT1 receptors on cardiac rupture. Olm markedly reduced post-MI cardiac rupture in mice, and this was found to be associated with its strong AT1 receptor blocking effect as well as its amplifying effect on Ang II-mediated activation of AT2 receptors.
The preventive effect of Olm on cardiac rupture might be also associated with its anti-inflammatory effect. We found that 24 h after MI there was a significant increase in MPO in the infarct area, although this decreased markedly a few days afterwards; such excessive inflammation after acute MI would promote degradation of the extracellular matrix and blunt fibrotic healing process, finally leading to cardiac rupture. As a molecular marker of inflammation, MPO is released upon leukocyte activation and accumulates in the infarct area, whereas in MPO knockout mice cardiac rupture was demonstrated to be delayed (Askari et al., 2003). In support of these findings, in other cardiovascular diseases, Olm has been shown to have a strong anti-inflammatory effect. Olm significantly reduces vascular micro-inflammation in patients with essential hypertension (Fliser et al., 2004) and attenuates cardiac inflammatory reactions following MI (Sandmann et al., 2006).
The preventive effect of Olm on cardiac rupture might also be related to its anti-apoptotic effect on cardiomyocytes. It was reported that Ang II induces apoptosis in cardiomyocytes and fibroblasts through activation of p53 (Leri et al., 1998); p53 is a well-known pro-apoptotic factor contributing to post-MI cardiac rupture (Matsusaka et al., 2006). According to these results, we could reasonably hypothesize that inhibition of Ang II-induced p53 accumulation and p53 activation could prevent cardiac rupture. In the present study we found that Olm markedly reduced p53 activation in myocardium at 24 h after MI and also prevented cardiac rupture at 3 to 7 days after MI, findings that support the above hypothesis.
Recently, Aldo has been considered to be one of the major factors that cause post-MI cardiac rupture. Previous reports demonstrated that Aldo exerts direct toxic actions on myocardium causing cardiac rupture and increased mortality after MI in mice and induces apoptosis of cardiomyocytes (De Silva et al., 2009; He et al., 2011). Olm inhibits the generation of Aldo by blocking AT1 receptors, which could be another mechanism by which Olm prevents cardiac rupture. We also found that Aldo down-regulated the expression of GDF-15, a cytokine that prevents cardiac rupture.
GDF-15, a TGFβ-related cytokine, is induced in the infarcted heart of mice and humans and it has been shown to protect mice from cardiac rupture after MI by inhibiting the activation of chemokine-triggered leukocyte integrin (Kempf et al., 2011). Activated leukocytes such as macrophages in myocardial tissues after MI may also contribute to the increase in GDF-15 (Fairlie et al., 1999). As a newly identified anti-inflammatory cytokine, GDF-15 has been shown to attenuate endothelial cell apoptosis in response to a high-glucose stimulus (Li et al., 2013). Interestingly, we found that Olm up-regulated GDF-15 expression in MI mice, which could be another mechanism by which Olm prevents post-MI cardiac rupture.
Fibrosis is believed to occur during a persistent tissue repair process. Recent studies indicate that Ang II promotes cardiac fibrosis (Westermann et al., 2012; Duerrschmid et al., 2013). Thus drugs that inhibit the angiotensin pathway appear to be effective in reducing cardiac fibrosis in various animal models (Shibasaki et al., 2005; Iwamoto et al., 2010). However, prevention of cardiac fibrosis in the early phase of MI might accelerate cardiac rupture (Ichihara et al., 2002; Shimazaki et al., 2008). However, we found that instead of accelerating, Olm actually prevented cardiac rupture. One possible reason for this finding could be that, by blocking AT1 receptors, Olm promotes AT2 receptor activation and, therefore, maintains the recruitment of fibroblastic cells to the infarct region, which is essential for the cardiac healing process. Additionally, we investigated the effect of Olm on the expression of periostin, an extracellular matrix protein that is essential for cardiac healing after MI (Shimazaki et al., 2008). It was reported that more than 80% of periostin knockout mice died of post-MI cardiac rupture within the first 10 days, in contrast to the 40% of wild-type littermates (Shimazaki et al., 2008). In this study, we found that Olm did not change myocardial periostin expression in mice during the first week after MI. These results indicate that Olm does not inhibit the cardiac healing process in the early phase of MI.
Olm is used to treat a variety of cardiomyopathies. Our novel finding that Olm prevents post-MI cardiac rupture suggests that AT1 receptor blockers could be a valuable potential preventive approach for this catastrophic complication of MI.
Limitations: although neonatal rat cardiomyocytes and even cardiomyocyte cell line such as H9C2 cell line have been extensively used to clarify the underlying mechanisms of the phenomenon discovered in adult animals, neonatal cells behave differently from adult cells, thus it would be better to use the same type of cells to investigate both the phenotypes and mechanisms. In addition, it is necessary to perform clinical trials or carry out related meta-analysis before the findings in this study are translated to the clinical setting.
Acknowledgments
We thank Biying Zhou (Department of Pathophysiology, School of Basic Medical Science, Southern Medical University) for creating murine MI models. She is blind to the pharmacological treatments employed in this study.
Glossary
- AHF
acute heart failure
- Aldo
aldosterone
- Ang II
angiotensin II
- AT1 receptor
angiotensin II type 1 receptor
- AT2 receptor
angiotensin II type 2 receptor
- GDF-15
growth differentiation factor-15
- LVFS
left ventricular fractional shortening
- MI
myocardial infarction
- MPO
myeloperoxidase
- Olm
olmesartan medoxomil
- RAS
renin–angiotensin system
- RAAS
renin–angiotensin–aldosterone system
Author contributions
Yulin Liao, Masafumi Kitakaze, Baihe Chen conceived and designed the study; Baihe Chen, Yulin Liao, Di Lu, Yujuan Fu, Xiaobo Huang, Shiping Cao, Jianping Bin, Qiaobing Huang, Dingli Xu analysed and interpreted the results; Baihe Chen, Yulin Liao, Di Lu, Yujuan Fu, Xiaobo Huang, Shiping Cao performed the experiments and collected the data; Yulin Liao, Jingwen Zhang, Baihe Chen drafted, edited and revised the paper before submission; and all authors read and approved the final version of the paper.
Source of funding
This work was supported by grants from the National Natural Science Foundation of China (81170146, 31271513 to Y.L.), the Team Program of Natural Science Foundation of Guangdong Province, China (S2011030003134, to Y.L. and B.J.).
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12736
Table S1 Sequences of primers used for real-time PCR.
Figure S1 Left ventricular (LV) haemodynamics and fractional shortening (LVFS) in mice with myocardial ischaemia (MI) for 3 days. (A) Representative recording of LV pressure and change rate. (B) Results of LV fractional shortening (LVFS), n = 6, 5, 10 in sham, MI and MI + Olm group respectively. (C) LV systolic pressure (LVSP). (D) Maximum rate of LV pressure (dp/dt max). (E) LV contractility. (F) LV end-diastolic pressure (LVEDP). (G) Minimum rate of LV pressure (dp/dt min). NS, not significant, for panels C–F, n = 5 in each group. MI, myocardial infarction; Olm, olmesartan.
Figure S2 Left ventricular fractional shortening (LVFS) in mice with myocardial ischaemia (MI) for 6 weeks. *P < 0.05 versus sham; #P < 0.05 versus MI group. Data are shown as the mean ± SEM, n = 12 in each group.
Figure S3 MPO stain in murine heart cross sections. Whole view of myeloperoxidase (MPO) stain of mouse hearts in sham, MI and MI + Olm groups. Scale bar = 1 mm. The area indicated by black box was magnified in Figure 2A.
Figure S4 Immunohistochemical detection of MPO in myocardial infarction (MI) mice treated with/without aldosterone. (A) Examples of pictures show MPO staining in the heart cross section for whole view (40×, upper panels), magnification 100× (middle panels, black boxes were magnified in lower panels) and 400× (lower panels). Aldosterone (Aldo, 1.44 mg·kg−1·d−1) or olmesartan (Olm,10 mg·kg−1·d−1) treatment was given in MI mice for 3 days. (B) Semi-quantitative analysis of MPO expression using a score system in each group. *P < 0.05 versus MI-3d group, n = 5 per group.
Figure S5 Time course of periostin expression in vivo and in vitro. (A) Dynamic changes of periostin gene expression in sham, MI and Olm-treated groups and each group was exposed to ischaemia for 1 day, 3 days and 7 days respectively. Olm had no effects on periostin expression after MI. (B) Immunohistochemical stain of periostin in sham, MI and Olm-treated groups, each group was treated with ischaemia for 1 day, 3 days and 7 days (Scale bar = 0.1 mm). (C) Scores of periostin corresponding to the results of immunohistochemical stain, P > 0.05 MI versus Olm-treated groups, n = 3 in each group. (D) Changes of periostin gene expression in cultured cardiomyocytes and fibroblasts when being normoxia and anoxia for 6 h, 12 h and 24 h respectively. Ang II (10−6M) and RNH6270 (10−6M) were added to the culture medium and the treated groups were divided into five groups: normoxia, anoxia, anoxia + Ang II, anoxia + Ang II + RNH6270, anoxia + RNH6270. There showed no differences among anoxia-treated groups at every time spot in cardiomyocytes or fibroblasts.
Figure S6 Olmesartan (Olm) inhibited apoptotic signal. (A) Western blots of myocardial p53 and p-p53 in mice at 24 h after myocardial infarction (MI) or sham. β-actin served as the loading control. (B) Semi-quantitative analysis of p53 and p-p53 expression in each group. #P < 0.05 versus MI group, n = 4 in sham group, n = 6 in MI or MI + Olm group. AU, arbitrary unit.
Figure S7 Angiotensin receptor 2 antagonist PD123319 did not affect the apoptosis signal induced by angiotensin II (Ang II). (A) Representative Western blots of p-p53, p53 and cleaved caspase 3 in neonatal rat cardiomyocytes and fibroblasts treated with angiotensin II (Ang II, 1 μM) or PD123319 (1 μM) for 24 h. (B) Results of semi-quantitation. *P < 0.05 versus control, n = 4 (culture plates) per group. AU, arbitrary unit.
Figure S8 Effects of olmesartan (Olm) and aldosterone (Aldo) on myocardial GDF-15 expression in mice subjected to myocardial infarction (MI). Result of routine PCR (A) and real-time quantitative PCR (B) for GDF-15 expression in response to sham or MI treated with/without Olm. (C) Western blotting of GDF-15 expression in response to sham or MI treated with/without Olm. (D) Western blots of GDF-15 in MI mice for 3 days treated with/without Aldo. *P < 0.05 versus sham group, #P < 0.05 versus MI group, n = 4 in sham groups, n = 6 in MI, MI + Olm and MI + Aldo groups.
References
- Abadir PM, Walston JD, Carey RM, Siragy HM. Angiotensin II type-2 receptors modulate inflammation through signal transducer and activator of transcription proteins 3 phosphorylation and TNFalpha production. J Interferon Cytokine Res. 2011;31:471–474. doi: 10.1089/jir.2010.0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta E, Pena O, Naftolin F, Avila J, Palumbo A. Angiotensin II induces apoptosis in human mural granulosa-lutein cells, but not in cumulus cells. Fertil Steril. 2009;91(5 Suppl):1984–1989. doi: 10.1016/j.fertnstert.2008.04.026. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, Peters JA, Harmar AJ CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askari AT, Brennan ML, Zhou X, Drinko J, Morehead A, Thomas JD, et al. Myeloperoxidase and plasminogen activator inhibitor 1 play a central role in ventricular remodeling after myocardial infarction. J Exp Med. 2003;197:615–624. doi: 10.1084/jem.20021426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker RC, Gore JM, Lambrew C, Weaver WD, Rubison RM, French WJ, et al. A composite view of cardiac rupture in the United States National Registry of Myocardial Infarction. J Am Coll Cardiol. 1996;27:1321–1326. doi: 10.1016/0735-1097(96)00008-3. [DOI] [PubMed] [Google Scholar]
- De Silva DS, Wilson RM, Hutchinson C, Ip PC, Garcia AG, Lancel S, et al. Fenofibrate inhibits aldosterone-induced apoptosis in adult rat ventricular myocytes via stress-activated kinase-dependent mechanisms. Am J Physiol Heart Circ Physiol. 2009;296:H1983–H1993. doi: 10.1152/ajpheart.00002.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerrschmid C, Crawford JR, Reineke E, Taffet GE, Trial J, Entman ML, et al. TNF receptor 1 signaling is critically involved in mediating angiotensin-II-induced cardiac fibrosis. J Mol Cell Cardiol. 2013;57:59–67. doi: 10.1016/j.yjmcc.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairlie WD, Moore AG, Bauskin AR, Russell PK, Zhang HP, Breit SN. MIC-1 is a novel TGF-beta superfamily cytokine associated with macrophage activation. J Leukoc Biol. 1999;65:2–5. doi: 10.1002/jlb.65.1.2. [DOI] [PubMed] [Google Scholar]
- Fliser D, Buchholz K, Haller H EUropean Trial on Olmesartan and Pravastatin in Inflammation and Atherosclerosis (EUTOPIA) Investigators. Antiinflammatory effects of angiotensin II subtype 1 receptor blockade in hypertensive patients with microinflammation. Circulation. 2004;110:1103–1107. doi: 10.1161/01.CIR.0000140265.21608.8E. [DOI] [PubMed] [Google Scholar]
- Gao XM, White DA, Dart AM, Du XJ. Post-infarct cardiac rupture: recent insights on pathogenesis and therapeutic interventions. Pharmacol Ther. 2012;134:156–179. doi: 10.1016/j.pharmthera.2011.12.010. [DOI] [PubMed] [Google Scholar]
- He BJ, Joiner ML, Singh MV, Luczak ED, Swaminathan PD, Koval OM, et al. Oxidation of CaMKII determines the cardiotoxic effects of aldosterone. Nat Med. 2011;17:1610–1618. doi: 10.1038/nm.2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichihara S, Senbonmatsu T, Price E, Jr, Ichiki T, Gaffney FA, Inagami T. Targeted deletion of angiotensin II type 2 receptor caused cardiac rupture after acute myocardial infarction. Circulation. 2002;106:2244–2249. doi: 10.1161/01.cir.0000033826.52681.37. [DOI] [PubMed] [Google Scholar]
- Iwamoto M, Hirohata S, Ogawa H, Ohtsuki T, Shinohata R, Miyoshi T, et al. Connective tissue growth factor induction in a pressure-overloaded heart ameliorated by the angiotensin II type 1 receptor blocker olmesartan. Hypertens Res. 2010;33:1305–1311. doi: 10.1038/hr.2010.189. [DOI] [PubMed] [Google Scholar]
- Kanamori H, Takemura G, Li Y, Okada H, Maruyama R, Aoyama T, et al. Inhibition of Fas-associated apoptosis in granulation tissue cells accompanies attenuation of postinfarction left ventricular remodeling by olmesartan. Am J Physiol Heart Circ Physiol. 2007;292:H2184–H2194. doi: 10.1152/ajpheart.01235.2006. [DOI] [PubMed] [Google Scholar]
- Kempf T, Zarbock A, Widera C, Butz S, Stadtmann A, Rossaint J, et al. GDF-15 is an inhibitor of leukocyte integrin activation required for survival after myocardial infarction in mice. Nat Med. 2011;17:581–588. doi: 10.1038/nm.2354. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leri A, Claudio PP, Li Q, Wang X, Reiss K, Wang S, et al. Stretch-mediated release of angiotensin II induces myocyte apoptosis by activating p53 that enhances the local renin-angiotensin system and decreases the Bcl-2-to-Bax protein ratio in the cell. J Clin Invest. 1998;101:1326–1342. doi: 10.1172/JCI316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Yang L, Qin W, Zhang G, Yuan J, Wang F. Adaptive induction of growth differentiation factor 15 attenuates endothelial cell apoptosis in response to high glucose stimulus. PLoS ONE. 2013;8:e65549. doi: 10.1371/journal.pone.0065549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Sendon J, Gurfinkel EP, Lopez de Sa E, Agnelli G, Gore JM, Steg PG, et al. Factors related to heart rupture in acute coronary syndromes in the Global Registry of Acute Coronary Events. Eur Heart J. 2010;31:1449–1456. doi: 10.1093/eurheartj/ehq061. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsusaka H, Ide T, Matsushima S, Ikeuchi M, Kubota T, Sunagawa K, et al. Targeted deletion of p53 prevents cardiac rupture after myocardial infarction in mice. Cardiovasc Res. 2006;70:457–465. doi: 10.1016/j.cardiores.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Sandmann S, Li J, Fritzenkotter C, Spormann J, Tiede K, Fischer JW, et al. Differential effects of olmesartan and ramipril on inflammatory response after myocardial infarction in rats. Blood Press. 2006;15:116–128. doi: 10.1080/08037050600586593. [DOI] [PubMed] [Google Scholar]
- Savarese G, Costanzo P, Cleland JG, Vassallo E, Ruggiero D, Rosano G, et al. A meta-analysis reporting effects of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers in patients without heart failure. J Am Coll Cardiol. 2013;61:131–142. doi: 10.1016/j.jacc.2012.10.011. [DOI] [PubMed] [Google Scholar]
- Shamshad F, Kenchaiah S, Finn PV, Soler-Soler J, McMurray JJ, Velazquez EJ, et al. Fatal myocardial rupture after acute myocardial infarction complicated by heart failure, left ventricular dysfunction, or both: the VALsartan In Acute myocardial iNfarcTion Trial (VALIANT) Am Heart J. 2010;160:145–151. doi: 10.1016/j.ahj.2010.02.037. [DOI] [PubMed] [Google Scholar]
- Shibasaki Y, Nishiue T, Masaki H, Tamura K, Matsumoto N, Mori Y, et al. Impact of the angiotensin II receptor antagonist, losartan, on myocardial fibrosis in patients with end-stage renal disease: assessment by ultrasonic integrated backscatter and biochemical markers. Hypertens Res. 2005;28:787–795. doi: 10.1291/hypres.28.787. [DOI] [PubMed] [Google Scholar]
- Shimazaki M, Nakamura K, Kii I, Kashima T, Amizuka N, Li M, et al. Periostin is essential for cardiac healing after acute myocardial infarction. J Exp Med. 2008;205:295–303. doi: 10.1084/jem.20071297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y. Intracardiac renin-angiotensin system and myocardial repair/remodeling following infarction. J Mol Cell Cardiol. 2010;48:483–489. doi: 10.1016/j.yjmcc.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann D, Becher PM, Lindner D, Savvatis K, Xia Y, Frohlich M, et al. Selective PDE5A inhibition with sildenafil rescues left ventricular dysfunction, inflammatory immune response and cardiac remodeling in angiotensin II-induced heart failure in vivo. Basic Res Cardiol. 2012;107:308–320. doi: 10.1007/s00395-012-0308-y. [DOI] [PubMed] [Google Scholar]
- Xuan W, Wu B, Chen C, Chen B, Zhang W, Xu D, et al. Resveratrol improves myocardial ischemia and ischemic heart failure in mice by antagonizing the detrimental effects of fractalkine. Crit Care Med. 2012;40:3026–3033. doi: 10.1097/CCM.0b013e31825fd7da. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Ishibashi M, Hiasa K, Tan C, Takeshita A, Egashira K. Essential role of vascular endothelial growth factor in angiotensin II-induced vascular inflammation and remodeling. Hypertension. 2004;44:264–270. doi: 10.1161/01.HYP.0000138688.78906.6b. [DOI] [PubMed] [Google Scholar]
- Zidar N, Jeruc J, Balazic J, Stajer D. Neutrophils in human myocardial infarction with rupture of the free wall. Cardiovasc Pathol. 2005;14:247–250. doi: 10.1016/j.carpath.2005.04.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Sequences of primers used for real-time PCR.
Figure S1 Left ventricular (LV) haemodynamics and fractional shortening (LVFS) in mice with myocardial ischaemia (MI) for 3 days. (A) Representative recording of LV pressure and change rate. (B) Results of LV fractional shortening (LVFS), n = 6, 5, 10 in sham, MI and MI + Olm group respectively. (C) LV systolic pressure (LVSP). (D) Maximum rate of LV pressure (dp/dt max). (E) LV contractility. (F) LV end-diastolic pressure (LVEDP). (G) Minimum rate of LV pressure (dp/dt min). NS, not significant, for panels C–F, n = 5 in each group. MI, myocardial infarction; Olm, olmesartan.
Figure S2 Left ventricular fractional shortening (LVFS) in mice with myocardial ischaemia (MI) for 6 weeks. *P < 0.05 versus sham; #P < 0.05 versus MI group. Data are shown as the mean ± SEM, n = 12 in each group.
Figure S3 MPO stain in murine heart cross sections. Whole view of myeloperoxidase (MPO) stain of mouse hearts in sham, MI and MI + Olm groups. Scale bar = 1 mm. The area indicated by black box was magnified in Figure 2A.
Figure S4 Immunohistochemical detection of MPO in myocardial infarction (MI) mice treated with/without aldosterone. (A) Examples of pictures show MPO staining in the heart cross section for whole view (40×, upper panels), magnification 100× (middle panels, black boxes were magnified in lower panels) and 400× (lower panels). Aldosterone (Aldo, 1.44 mg·kg−1·d−1) or olmesartan (Olm,10 mg·kg−1·d−1) treatment was given in MI mice for 3 days. (B) Semi-quantitative analysis of MPO expression using a score system in each group. *P < 0.05 versus MI-3d group, n = 5 per group.
Figure S5 Time course of periostin expression in vivo and in vitro. (A) Dynamic changes of periostin gene expression in sham, MI and Olm-treated groups and each group was exposed to ischaemia for 1 day, 3 days and 7 days respectively. Olm had no effects on periostin expression after MI. (B) Immunohistochemical stain of periostin in sham, MI and Olm-treated groups, each group was treated with ischaemia for 1 day, 3 days and 7 days (Scale bar = 0.1 mm). (C) Scores of periostin corresponding to the results of immunohistochemical stain, P > 0.05 MI versus Olm-treated groups, n = 3 in each group. (D) Changes of periostin gene expression in cultured cardiomyocytes and fibroblasts when being normoxia and anoxia for 6 h, 12 h and 24 h respectively. Ang II (10−6M) and RNH6270 (10−6M) were added to the culture medium and the treated groups were divided into five groups: normoxia, anoxia, anoxia + Ang II, anoxia + Ang II + RNH6270, anoxia + RNH6270. There showed no differences among anoxia-treated groups at every time spot in cardiomyocytes or fibroblasts.
Figure S6 Olmesartan (Olm) inhibited apoptotic signal. (A) Western blots of myocardial p53 and p-p53 in mice at 24 h after myocardial infarction (MI) or sham. β-actin served as the loading control. (B) Semi-quantitative analysis of p53 and p-p53 expression in each group. #P < 0.05 versus MI group, n = 4 in sham group, n = 6 in MI or MI + Olm group. AU, arbitrary unit.
Figure S7 Angiotensin receptor 2 antagonist PD123319 did not affect the apoptosis signal induced by angiotensin II (Ang II). (A) Representative Western blots of p-p53, p53 and cleaved caspase 3 in neonatal rat cardiomyocytes and fibroblasts treated with angiotensin II (Ang II, 1 μM) or PD123319 (1 μM) for 24 h. (B) Results of semi-quantitation. *P < 0.05 versus control, n = 4 (culture plates) per group. AU, arbitrary unit.
Figure S8 Effects of olmesartan (Olm) and aldosterone (Aldo) on myocardial GDF-15 expression in mice subjected to myocardial infarction (MI). Result of routine PCR (A) and real-time quantitative PCR (B) for GDF-15 expression in response to sham or MI treated with/without Olm. (C) Western blotting of GDF-15 expression in response to sham or MI treated with/without Olm. (D) Western blots of GDF-15 in MI mice for 3 days treated with/without Aldo. *P < 0.05 versus sham group, #P < 0.05 versus MI group, n = 4 in sham groups, n = 6 in MI, MI + Olm and MI + Aldo groups.