

Abstract
T cells which are genetically modified to express single-chain chimeric antigen receptors (CARs) have shown promise in early cancer immunotherapy clinical trials. Unfortunately, two recent deaths in cancer patients treated with CAR-T cells have created some uncertainty on how to best mitigate patient risk, while continuing to advance this very promising therapeutic avenue. In order to address these concerns, the Recombinant DNA Advisory Committee (RAC) held a symposium, the objectives of which were to first review the reported treatment-associated toxicities and second, to discuss methods for improving safety and efficacy. This report highlights the issues raised as part of this discussion with a specific focus on protocols infusing CAR-T cells. Because this was not a consensus conference, the opinions described should not be construed to represent those of any individual RAC member, the RAC as a body, conference participants, NIH or the FDA.
Keywords: Chimeric antigen receptor, T-cell therapy, gene therapy, CD19, adverse events
Evolution of CARs
Following appropriate preconditioning regimens that deplete circulating lymphocytes, the adoptive transfer of tumor infiltrating lymphocytes (TILs) into patients with metastatic melanoma leads to objective clinical responses in select individuals (Supplemental references (SR) 1,2). Building on this approach, recent studies suggest that autologous T cells, genetically engineered to express tumor associated antigen (TAA)-specific TCRs, can replace TILs in certain clinical settings. (1) Importantly, tumor regressions are observed even under conditions of widely metastatic and bulky disease, and in patients who have failed both prior surgical extirpation and medical therapy. Unfortunately, the potential for widespread use of adoptive transfer strategies using TILs or T cells with recombinant TCRs is limited (SR#3).
In order to overcome the historic problems associated with the cellular targeting of HLA restricted TAA, Eshhar et al. developed a strategy to redirect T-cell specificity using Chimeric Antigen Receptors (CARs) or T-bodies (Figure 1). First generation vectors for the production of CAR-T cells contain the heavy and light chain immunoglobulin (Ig) variable regions, fused as a single chain to the epsilon, gamma or zeta signaling sequences of the T cell receptor (TCR) or the signaling region of the Fcγ domain. (2-8) T cells expressing such first generation CARs recognize surface TAAs, independent of HLA restriction, but cannot recognize intracellular TAAs (Table 1). Early clinical experiences with 1st generation CAR-T cells demonstrated that (a) the survival of adoptively transferred CAR-T cells was limited in cancer patients and (b) few objective anti-tumor responses were observed. (8-10)
Figure 1. Evolution of CAR-T cell Design.
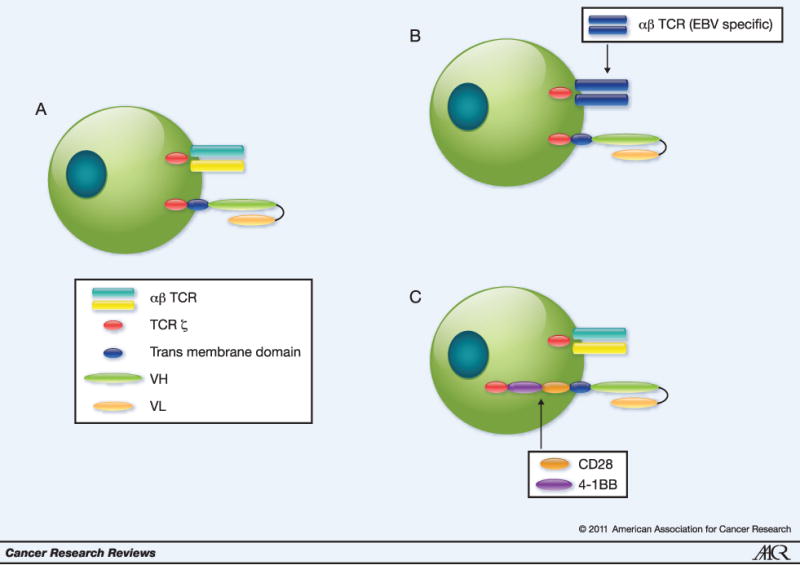
A. Initial CAR-T cells were composed of an antigen specific scFv linked to the TCR zeta chain. These CARs were expressed in non-clonal T cell populations with diverse specificities of their endogenous TCRs. B. In subsequent strategies, investigators have attempted to prolong gene-modified T cell survival by expressing 1st generation CARs in T cells whose endogenous TCRs recognize specific antigens of persisting viruses, e.g. EBV, allowing for the induction of the costimulatory cascade upon cognate TCR ligation. C. Because of difficulty isolating TA-specific T cells from most types of solid tumors, other investigators have employed a modified CAR design in which the 4-1BB/CD28 intracellular signaling domains are linked to the transmembrane region. In these CARs, scFv antigen recognition results in T cell activation through the TCR zeta chain leading to prolonged survival secondary to appropriate costimulation.
Table 1. Limitations of Gene Modified T cells by Antigen Recognition Moiety.
| Gene Modified T cell Type | ||
|---|---|---|
| αβ TCR | CAR | |
| Limitations | ||
| Susceptible to loss/down regulation of HLA expression on tumor cells | + | - |
| Susceptible to dysfunction of the tumor cell antigen processing machinery | + | - |
| Cell surface target antigen restriction | - | + |
| Potential for vector recombination with endogenous TCR | + | - |
To enhance survival and/or increase proliferation of transferred CAR-T cells, investigators have incorporated signaling moieties from costimulatory molecules including CD28 (B7-1), CD134 (OX40), and CD137 (4-1BB), alone (2nd generation) or in sequence (3rd generation). (11-15) A recent study comparing simultaneously infused CD19-specific CAR-T cells with or without the CD28 signaling sequence indeed demonstrates improved survival of CD28-modified cells..(16) Additionally, combining CD28 with 4-1BB signaling sequences further promotes engraftment of CAR-T cells. (17, 18) As an alternative to incorporating costimulatory moieties into the vector, some investigators express first generation CARs in T cells specific to endogenous viral antigens e.g. Epstein Barr virus (EBV). In this setting, antigen recognition by the CAR provides TCR ζ signaling, while recognition of processed EBV peptides in the context of appropriate HLA molecules by the physiologic αβTCR, allows for intrinsic costimulation.(19)
Focus Points for Future Consideration
Reported serious adverse events (SAE) following CAR-T cell transfer
At least 40 trials using CAR-T cells for treatment of cancer have been registered with the NIH Office of Biotechnology Activities (OBA) and undergone NIH RAC review and at least 120 subjects have been dosed across all trials (Supplemental Table 1). (20) Overall, most SAEs that were viewed as possibly related to transfer of CAR-T cells have been mild, self-limited and occurred shortly after infusion (Supplemental Table 2). Unfortunately, 2 patients died shortly after adoptive transfer of CAR-T cells. (21, 22)
Following RAC review of the death in the HER2-CAR-T cell trial at its December 2009 meeting [http://oba.od.nih.gov/rdna_rac/rac_past_meetings_2000.html] the Committee recommended holding a safety symposium to review clinical trials employing CAR-T cells and to then formulate a report to both facilitate subsequent RAC and Institutional Biosafety Committee (IBC) reviews of these trials and to assist investigators in designing future studies. A planning committee, consisting of RAC members, leading investigators in this field, and OBA staff, identified key components of these trials based on their potential impact on patient safety and their role in therapy. These issues were discussed in a panel format and are summarized under the heading “Points to Consider in the Design and Implementation of Clinical Trials using CAR-T cells.” A paradigm for considering how to incorporate these considerations into clinical trial design is provided in Table 2 and Supplemental Tables 3-5.
Table 2. Principles Applicable to all Trials Using CAR-T cells.
|
The potential for malignancy secondary to insertion mutagenesis is unknown. Although it has not been seen in the CAR trials to date, the long-term follow-up is small. An analysis by OBA in 2007 of adverse events in trials using retroviral and lentiviral vectors in terminally differentiated cells found 12 malignancies, none of which could be directly attributed to the vector. (SR# 16, 17).
Points to Consider in the Design and Implementation of Clinical Trials using CAR-T cells
1. Unwanted On-Target Effects of CAR-T Cells
Recognition of antigen expressed on non-tumor cells by CAR-T cells is emerging as the major risk factor of CAR-T-cell transfer. Such recognition may become manifest as: (a) immediate toxicity and (b) late or sustained toxicity resulting from long term depletion of cells with important homeostatic functions.
A. Mitigating the potential for early toxicity
A feared complication after infusion of CAR-T cells is their massive activation leading to the research participant's death. This is especially of concern with CARs directed against untested and/or endogenously prevalent TAAs, as T cells with high avidity receptors can respond to cells that express their targets at levels that are currently too low for detection by conventional means. One approach to limit such toxicity is an inter-patient (and sometimes intra-patient) dose-escalation scheme. Alternatively one could conduct the initial dose escalation with 1st generation CAR-T cells without conditioning or cytokine supplementation under the expectation that stimulation of these adoptively transferred T cells may be suboptimal, although this is not yet proven. Once the 1st generation CAR-T cells are shown to be safe, 2nd (or perhaps 3rd) generation CAR-T cells, with a potential for more pronounced responses and prolonged persistence, could be explored in combination with supplementary treatments. (23) An important caveat to this statement is that it is currently unclear whether lack of toxicity using 1st generation CAR-T cells will translate into a similar safety profile for 2nd or 3rd generation CAR-T cells combined with lymphodepletion and/or cytokine support. As an alternative approach, if 2nd or 3rd generation CARs targeting a new antigen are evaluated without previous experience with a 1st generation CAR, a very conservative dose escalation strategy should be adopted (see: Starting dose of CAR-T cells in phase I trials).
The question of whether the co-expression of conditional suicide genes might safeguard against some of the potential side effects of non-tumor cell recognition by CAR-T cells was discussed. (24) As exemplified by the fatal SAE using HER2-CAR-T cells, the CAR-T cells may act within minutes after engagement of their target antigen and symptoms are not expected until significant damage has occurred. Therefore, inclusion of a suicide gene, whose benefit will necessarily take time, was deemed to have limited potential for preventing acute toxicity although it may have utility in modulating late toxicities.
A third approach to reduce risks of immediate toxicity is splitting of the T-cell dose over two or more days as is being tested in recipients of CD19-specific T cells. (21) Using this scheme it would be desirable to monitor cytokines or chemokines in the serum as potential indicators for toxicity after the first of the split doses. A fourth approach using a very conservative dose escalation strategy is discussed below under the heading: Starting dose of CAR-T cells in Phase I trials. Importantly, in all trials, pre-clinical studies should carefully test for expression of the TCR's target antigen in healthy tissues. Furthermore, the use of CAR T cells against antigens that are widely expressed on non-tumor cells essential for important physiological functions should, to the extent feasible, not be chosen as targets unless there is convincing preclinical data that their expression are at such low levels that on-target toxicity is unlikely to occur.
B. Mitigating the potential for late toxicity
Another risk of CAR-T cells is the long term depletion of cells, which are important for normal human function. For example, CAR-T cells for treatment of B-cell malignancies have targeted CD19 and CD20, markers expressed on normal B cells. (25) Treatment with CAR-T cells directed against either of these antigens has the potential to deplete the patients' B cells.(26) Furthermore, unlike the α-CD20 monoclonal antibody (mAb), Rituxan™, which has a defined half-life, these CAR-T cells may potentially survive and function for the life of the patient. The lack of CD19 or CD20 expression on most plasma cells allows the maintenance of physiologic antibody levels in the majority of patients following depletion of CD19+ or CD20+ cells (SR#4). In addition, it is expected that B cells would rapidly be replaced from lymphoid progenitors once the CAR-T cells die or become functionally impaired. Finally, even under conditions that would allow for long-term persistence of functioning CD19- or CD20-specific CAR-T cells, resulting in continuous depletion of B cells, this could be managed by Ig transfer and would thus be preferable to a fatal cancer.
A related concern is the potential for CAR-T cells to negatively impact organ function secondary to low levels of target antigen expression in a non-tumor site e.g. vascular endothelial growth factor receptor 2 (VEGFR2)-specific CARs targeting the tumor vasculature (SR#5,6). VEGFR2 is expressed on endothelial cells during physiological angiogenesis, vasculogenesis, arteriogenesis, and lymphangiogenesis and is needed for processes such as wound healing and embryogenesis (SR#7). As such, VEGFR2-specific persisting CAR-T cells could interfere with formation of indispensable vasculature. While there is no universal strategy for mitigating these late CAR-T-cell associated toxicities, the resultant conditions could potentially be managed medically. Alternatively, risks of long-term toxicity could be mitigated by the insertion of suicide genes into the CAR-T cells.
2. Effects of Costimulatory Signaling
Results to date demonstrate that while target cell lysis by 1st generation CAR-T cells is independent of costimulatory signaling, their proliferation, production of cytokines and up-regulation of other effector cell molecules depends on and/or is improved by co-stimulation. (27) Furthermore, based on what is understood regarding natural functions of CD28 and 4-1BB in vivo, it is anticipated that CAR-T cells bearing these domains might be more resistant to activation-induced cell death (SR#8). Therefore, the majority of participants concluded that the incorporation of CD28 and/or 4-1BB into CAR constructs offers an important potential for therapeutic benefit. Evaluations of other cosignaling moieties, which may enhance the proliferation and/or survival of CAR-T cells are appropriate. In addition to modifying the CAR to improve T cell engraftment it may also be possible to a priori identify T-cell sub-populations with the capacity for sustained proliferation. In fact, there is evidence that the starting population of T cells used for CAR modification may affect their ability to survive and expand (SR#9). For example, in non-human primates, effector CD8+ T cells generated ex vivo from central rather than effector memory populations, enjoy enhanced survival following adoptive transfer (SR#10). Because the ratios of different T-cell subsets varies between patients, selective use of T-cell subsets prior to genetic manipulation may provide a more discriminating path to predict the long-term outcome of CAR-T cell transfer. Finally, clinical experience with the expression of CARs in non-T cell populations e.g. NK cells, is limited. (28) Therefore, special care must be taken with CAR use in T-cell subsets and non-T cell lymphocyte populations.
3. Systemic Conditioning
Adoptive transfer of CAR-T cells can be further modified by auxiliary therapies such as partial or complete myeloablation to improve preservation of the transferred T-cell populations. The rationale behind lymphodepletion prior to T-cell infusion is multifactorial and includes, but is not limited to, the elimination of “cytokine sinks,” creation of space for the expansion of adoptively transferred cells, and removal of suppressor cell populations. In fact, depletion of lymphocytes in melanoma patients infused with ex vivo expanded TILs is requisite for treatment efficacy and recent studies suggest a correlation between the degree of lymphocyte depletion and objective response rates (SR#11). Despite the recognized risks of myelosuppression, the majority of the RAC meeting participants felt that administration of CAR-T cells that do not carry an endogenous TCR to a persisting virus is unlikely to result in significant clinical benefit without prior conditioning.
4. Cytokine supplements
Recombinant soluble cytokines such as IL-2 are administered upon CAR-T cell transfer with the expectation that they will promote the expansion and survival of transferred cells. If and to what degree cytokines may exacerbate the potential toxicity of 2nd or 3rd generation CAR-T cells is uncertain. It is also unclear if 2nd and 3rd generation CAR-T cells need to be supplemented with IL-2 either at all or at the high and potentially toxic doses used for TIL transfer (SR#12). For the initial evaluation of CAR-T cells to novel targets, dosing without addition of cytokines or with injection of low to moderate amounts of cytokines may be prudent and would facilitate differentiation of SAEs induced by CAR-T cells alone from those caused by the potential synergy between these drugs. Furthermore, the design of later Phase studies employing 2nd and 3rd generation CAR-T cells should consider incorporating treatment arms with and without cytokine support. Ongoing studies exploring the use of CAR-T cells genetically modified to secrete cytokines such as IL-15 or IL-12 may eliminate the need for systemic cytokine administration. (29) If and to what degree this approach will improve the potential benefit of CAR-T cells or instead pose additional risks, is currently unknown.
5. CARs expressed in T cells targeting persisting viruses
Some groups have combined the advantages of CAR-T cells with those of traditional antigen-specific T cells (SR#13). Specifically, T cells to persisting viruses such as EBV have been enriched in vitro and then genetically modified by insertion of a transgene encoding a TAA-specific CAR. In a neuroblastoma trial, 11 individuals were treated simultaneously with EBV-CTLs expressing a GD2-specific first generation CAR and activated T lymphocytes expressing the same CAR. Only CAR EBV-CTLs showed significant in vivo persistence, and four of eight patients with evaluable tumors had evidence of tumor necrosis or regressions, including a sustained complete remission.” Lymphodepletion did not improve CAR-T cell persistence or clinical outcome. (19) Potential deleterious effects due to virus-specific CAR-T cells without a costimulatory endodomain are anticipated to be similar to those of other T cells expressing 2nd or 3rd generation CARs.
6. Starting dose of CAR-T cells in Phase I trials
Phase I trials are primarily designed to assess safety and feasibility rather than efficacy; biological activity and proof of concept are usually secondary aims. Phase I trials for CAR-T cells typically start with reduced cell doses which are then gradually escalated. The starting dose should be adjusted depending on the type of CAR, i.e., T cells with a 2nd or 3rd generation CARs should start at a lower dose than those with a 1st generation CAR. Similarly, transfer of CAR-T cells into partially pre-conditioned patients should commence at a lower dose than transfer into non- myeloablated patients. Although one would expect that immediate toxicity due to transfer of CAR-T cells may directly correlate with numbers of injected cells, late adverse events may be independent of the injected dose due to construct-dependent dynamic changes in cell numbers.
Providing reliable guidelines for starting doses of CAR-T cells is currently not possible. To date, clinical trials have employed a fairly wide range of starting doses and some investigators dosed according to weight, others according to body surface area (BSA) and still others employed flat dosing schedules (Supplemental Table 1). The use of unadjusted dosing should be avoided and a more uniform dosing scheme such as that based on cells/kg should be considered. An open question is whether cells/kg should be based on an ideal body weight or actual weight given that weight increases due to obesity may not justify an increase in dose (SR#14).
7. Ethical Considerations
One of the ethical dilemmas facing investigators is the requirement in early phase research to design studies that are safe while at the same time hoping to show biological activity or possibly even benefit to the individual subject, many of whom have few if any other therapeutic or even palliative options. In response to the unexpected death in the trial infusing HER2-specific CAR-T cells, initial starting doses in a number of trials administering HER2-specific CAR-T cells, were lowered to 104 cells/m2. There was general agreement among the RAC members that a lower starting dose of 104 cells/m2 would likely be safe but would unlikely have potential benefit to the subject. Therefore, the risk-benefit calculus was such that any benefit realized from the research would most likely be a societal benefit, i.e. an increase in generalizable knowledge with minimized risk but little potential benefit for the patient-subject. It will be a challenge to design early phase studies which appropriately balance accurate disclosure of the risks and benefits posed by this research and appropriate informed consent among a seriously ill patient-subject population with few therapeutic options. While the expectation of clinical benefit must be discouraged in clinical trials, especially those at the earliest stages of research, selecting a dose that would be relatively safe but have potential biologic activity is an appropriate goal. Whether the acceptable level of risk should be adjusted in relation to the disease prognosis for a given patient cohort remains a subject worthy of future debate.
Summary
CAR-T cells have shown some benefit in cancer patients.(19, 26, 30) The major challenge for achieving therapeutic benefit by CAR-T cell transfer remains lack of sustained engraftment and loss of T cell functions. These hurdles may be overcome in part by incorporation of costimulatory domains into CAR constructs or by modifying EBV-specific T cells. Additional studies are needed to assess the performance of EBV-specific CAR-T cells in cancer patients of diverse ages. Results to date (excluding those based on EBV-specific T cells) suggest that partial myeloablation is required for survival of CAR-T cells. Mechanisms that cause loss of transferred 2nd and 3rd generation CAR-T cells remain poorly understood and may relate to the differentiation status of the transduced T cell subsets. It is unclear if and at what doses cytokines are needed to improve the clinical outcome of 2nd and 3rd generation CAR-T cells. Until this issue is clarified, the upfront use of high doses of systemic cytokines should be carefully justified.
The major immediate risk factor for CAR-T cells remains their activity against non-tumor cells rather than genotoxicity from the gene transfer event. Long term risk factors, e.g. sustained depletion of normal cell populations or exacerbated expansion of CAR-T cells cannot yet be assessed. While the risk of insertional mutagenesis remains a concern, this has neither been observed clinically nor in recent preclinical studies that have attempted to recreate insertional mutagenesis (SR#15). However, as the field moves towards the use of less differentiated CAR-T cells, the risk of insertional mutagenesis may increase.
Supplementary Material
Acknowledgments
The authors acknowledge the following staff from the Office of Biotechnology Activities, Dr. R. Jambou, Dr. E. Rosenthal, Dr. M. O'Reilly, Ms. M. Montgomery and Ms. L. Gargiulo, for their contributions in bringing together this symposium and in assembling the data on trials and adverse events. The authors also thank Dr. Richard Junghans for helpful discussions.
Footnotes
Conflict of Interest: Dr. Strome is the cofounder and a major stockholder in Gliknik, Inc., a biotechnology company. He also receives royalties through the Mayo Clinic College of Medicine for the licensure of IP relating to 4-1BB (CD137) and B7-H1 (PD-L1).
References
- 1.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer Regression in Patients After Transfer of Genetically Engineered Lymphocytes. Science. 2006;314:126–9. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hwu P, Shafer GE, Treisman J, Schindler DG, Gross G, Cowherd R, et al. Lysis of ovarian cancer cells by human lymphocytes redirected with a chimeric gene composed of an antibody variable region and the Fc receptor gamma chain. J Exp Med. 1993;178:361–6. doi: 10.1084/jem.178.1.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:720–4. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stancovski I, Schindler DG, Waks T, Yarden Y, Sela M, Eshhar Z. Targeting of T lymphocytes to Neu/HER2-expressing cells using chimeric single chain Fv receptors. J Immunol. 1993;151:6577–82. [PubMed] [Google Scholar]
- 5.Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:10024–8. doi: 10.1073/pnas.86.24.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moritz D, Wels W, Mattern J, Groner B. Cytotoxic T lymphocytes with a grafted recognition specificity for ERBB2-expressing tumor cells. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:4318–22. doi: 10.1073/pnas.91.10.4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pinthus JH, Waks T, Kaufman-Francis K, Schindler DG, Harmelin A, Kanety H, et al. Immuno-Gene Therapy of Established Prostate Tumors Using Chimeric Receptor-redirected Human Lymphocytes. Cancer Research. 2003;63:2470–6. [PubMed] [Google Scholar]
- 8.Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A Phase I Study on Adoptive Immunotherapy Using Gene-Modified T Cells for Ovarian Cancer. Clinical Cancer Research. 2006;12:6106–15. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park JR, DiGiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, et al. Adoptive Transfer of Chimeric Antigen Receptor Re-directed Cytolytic T Lymphocyte Clones in Patients with Neuroblastoma. Mol Ther. 2007;15:825. doi: 10.1038/sj.mt.6300104. [DOI] [PubMed] [Google Scholar]
- 10.Lamers CHJ, Sleijfer S, Vulto AG, Kruit WHJ, Kliffen M, Debets R, et al. Treatment of Metastatic Renal Cell Carcinoma With Autologous T-Lymphocytes Genetically Retargeted Against Carbonic Anhydrase IX: First Clinical Experience. J Clin Oncol. 2006;24:e20–2. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 11.Zhao Y, Wang QJ, Yang S, Kochenderfer JN, Zheng Z, Zhong X, et al. A Herceptin-Based Chimeric Antigen Receptor with Modified Signaling Domains Leads to Enhanced Survival of Transduced T Lymphocytes and Antitumor Activity. J Immunol. 2009;183:5563–74. doi: 10.4049/jimmunol.0900447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murphy A, Westwood JA, Teng MWL, Moeller M, Darcy PK, Kershaw MH. Gene Modification Strategies to Induce Tumor Immunity. Immunity. 2005;22:403. doi: 10.1016/j.immuni.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 13.Finney HM, Akbar AN, Lawson ADG. Activation of Resting Human Primary T Cells with Chimeric Receptors: Costimulation from CD28, Inducible Costimulator, CD134, and CD137 in Series with Signals from the TCR{zeta} Chain. J Immunol. 2004;172:104–13. doi: 10.4049/jimmunol.172.1.104. [DOI] [PubMed] [Google Scholar]
- 14.Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCR[zeta] /CD28 receptor. Nat Biotech. 2002;20:70. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 15.Friedmann-Morvinski D, Bendavid A, Waks T, Schindler D, Eshhar Z. Redirected primary T cells harboring a chimeric receptor require costimulation for their antigen-specific activation. Blood. 2005;105:3087–93. doi: 10.1182/blood-2004-09-3737. [DOI] [PubMed] [Google Scholar]
- 16.Savoldo B, Ramos C, Bollard C, Liu E, Mims M, Keating M, et al. Simultaneous Comparison of T Cells Expressing 1st or 2nd Generation CARs in Human Subjects with B-Cell Malignancies: Contribution of Costimulatory Endodomains. Abstract from the annual meeting of the American Society of Gene and Cell Therapy. 2010 [Google Scholar]
- 17.Wang J, Jensen M, Lin Y, Sui X, Chen E, Lindgren CG, et al. Optimizing Adoptive Polyclonal T Cell Immunotherapy of Lymphomas, Using a Chimeric T Cell Receptor Possessing CD28 and CD137 Costimulatory Domains. Human Gene Therapy. 2007;18:712–25. doi: 10.1089/hum.2007.028. [DOI] [PubMed] [Google Scholar]
- 18.Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proceedings of the National Academy of Sciences. 2009;106:3360–65. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jena B, Dotti G, Cooper LJN. Redirecting T-cell specificity by introducing a tumor-specific chimeric antigen receptor. Blood. doi: 10.1182/blood-2010-01-043737. blood-2010-2001-043737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brentjens R, Yeh R, Bernal Y, Riviere I, Sadelain M. Treatment of Chronic Lymphocytic Leukemia With Genetically Targeted Autologous T Cells: Case Report of an Unforeseen Adverse Event in a Phase I Clinical Trial. Mol Ther. 18:666. doi: 10.1038/mt.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol Ther. 18:843. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Junghans R. Strategy escalation: an emerging paradigm for safe clinical development of T cell gene therapies. J Transl Med. 2010;8:55. doi: 10.1186/1479-5876-8-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Straathof KC, Pule MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, et al. An inducible caspase 9 safety switch for T-cell therapy. Blood. 2005;105:4247–54. doi: 10.1182/blood-2004-11-4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bryder D, Sigvardsson M. Shaping up a lineage--lessons from B lymphopoesis. Current Opinion in Immunology. 22:148. doi: 10.1016/j.coi.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 26.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically-engineered to recognize CD19. Blood. doi: 10.1182/blood-2010-04-281931. blood-2010-2004-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N. CD28 Costimulation Provided through a CD19-Specific Chimeric Antigen Receptor Enhances In vivo Persistence and Antitumor Efficacy of Adoptively Transferred T Cells. Cancer Research. 2006;66:10995–1004. doi: 10.1158/0008-5472.CAN-06-0160. [DOI] [PubMed] [Google Scholar]
- 28.Altvater B, Landmeier S, Pscherer S, Temme J, Schweer K, Kailayangiri S, et al. 2B4 (CD244) Signaling by Recombinant Antigen-specific Chimeric Receptors Costimulates Natural Killer Cell Activation to Leukemia and Neuroblastoma Cells. Clinical Cancer Research. 2009;15:4857–66. doi: 10.1158/1078-0432.CCR-08-2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 24:1160. doi: 10.1038/leu.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brentjens RJ, Riviere I, Frattini M, Wang X, Taylor C, Olszewska M, et al. Marked Regression of Adenopathy Following Infusion of Autologous T Cells Genetically Targeted to the CD19 Antigen in a Patient with Bulky CLL. Abstract from the annual meeting of the American Society of Gene and Cell Therapy. 2010 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.