

Abstract
The reversible covalent attachment of chemical probes to proteins has long been sought as a means to visualize and manipulate proteins. Here we demonstrate the full reversibility of post-translational custom pantetheine modification of E. coli acyl carrier protein (ACP) for visualization and functional studies. We utilize this iterative enzymatic methodology in vitro for reversible labeling variants and apply these tools to Nuclear Magnetic Resonance (NMR) structural studies of protein-substrate interactions.
Post-translational protein modification is important for adding functions to proteins that can be exploited for therapeutics1, protein engineering2, affinity design3,4, and enzyme immobilization5, among other applications6. Acyl carrier protein (ACP) labeling with 4’-phosphopantetheine (ppant), conjugated to a tag of choice by its transferase (PPTase) represents one of the most flexible covalent protein labeling methods, as illustrated by the application of ACP tagged peptides7, for bio-gel formation8, and ACP-dependent protein immobilization9. Labeling ACP and ACP fusion proteins with ppant analogs is also successfully leveraged for visualization10,11, isolation12, functional13, and structural14,15 studies of carrier protein-dependent biosynthetic enzymes16. Yet further advancement of these tools is hampered by an inability to easily reverse ppant attachment. Indeed, naturally occurring ACPs, often isolated in holo- form (including a native ppant modification), cannot be further modified directly with another ppant tag. To overcome these difficulties, we use AcpH (acyl carrier protein hydrolase), a phosphodiesterase from Pseudomonas aeruginosa17 and Sfp, a PPTase from Bacillus subtilis18 to swap different ppant-conjugated small molecules on free ACP and ACP fusion proteins (Fig. 1). This reversible tagging system offers the ability to connect synthetic and biological chemistry with ease and provides uniformly labeled, high quality ACP and ACP fusion proteins, as demonstrated here through fluorescent labeling and solution-phase protein NMR.
Figure 1. Reversible labeling of E. coli ACP.
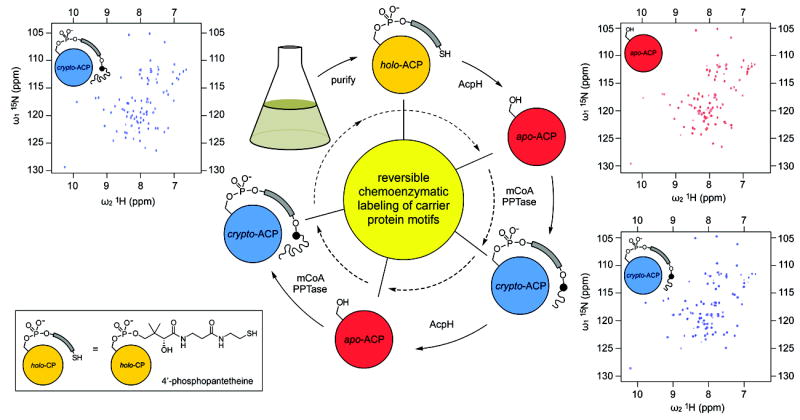
Recombinant ACP-15N is isolated in the holo- state (top, yellow). ACP-15N is prepared for covalent labeling by treatment with AcpH to generate exclusively apo-ACP-15N (red). Protein purity and modification homogeneity is confirmed by 2D-NMR (top right). Labeling with acyl pantetheine analogs to the crypto-ACP-15N (blue), or labeled form, proceeds via PPTase and mCoA modification that is analyzed by NMR (bottom right, top left). Modification is quantitatively reversed by AcpH, returning labeled proteins to the apo- form.
For evaluation of iterative labeling, we began with fluorescent ACP labeling directly in cellular lysate (Supplementary Fig. 1a) from E. coli strain DK554, which overexpresses native fatty acid ACP (AcpP) in predominantly apo- form19. Treatment of this lysate with coumarin-CoA20 and Sfp generated a blue-fluorescent band upon excitation of sodium-dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) samples at 254 nm (Supplementary Fig. 1b) that co-migrated with a coumarin-labeled ACP standard. Subsequent treatment of coumarin-labeled lysate with recombinant AcpH uniformly removed the coumarin-ppant from ACP, as demonstrated by disappearance of the blue band (Supplementary Fig. 1b). Subsequent treatment of the sample with Sfp and rhodamine-CoA21 generated a new red-fluorescent SDS-PAGE band upon excitation at 532 nm (Supplementary Fig. 1b,c); this label can also be removed (Fig. 2a, Supplementary Fig. 2) with AcpH.
Figure 2. Gel Detection of Reversible ACP Labeling.
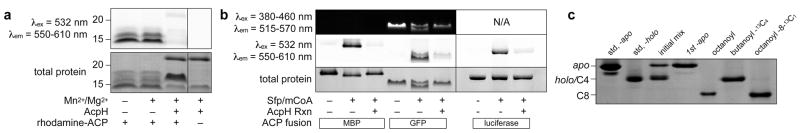
(a) Analysis of rhodamine-labeled crypto-ACP confirms AcpH’s ablity to remove rhodamine-pantetheine (separate lane of same gel indicated by demarcation). (b) Reversibly labeling fusion-ACPs: MBP-PaACP, GFP-ACP, and Luciferase-ACP. Apo- ACP fusions are labeled with rhodamine-CoA (mCoA) and Sfp, and subsequently unlabeled with AcpH (separate gels indicated by demarcation). (c) Acyl-pantetheine analogs were installed on ACP-15N used for NMR analysis. ACP standards for apo- and holo- allow labeling evaluation. Initial ACP-15N apo and holo mixture is readily converted to full apo- with AcpH. “One-Pot” Sfp and AcpH methodology allows conversion of one protein sample to octanoyl-, butanoyl-13C4-, and octanoyl-8-13C1-ACP-15N. Full length gels are presented in Supplementary Information (Supplementary Fig. 2).
After demonstrating the compatibility of AcpH in removing various ppant analogs, we sought to demonstrate the flexibility of our technique by evaluating interaction with ACP fusion proteins. AcpH removed rhodamine-ppant from ACP attached to three different fusion partners, an N-terminal maltose binding protein (MBP) (Fig. 2b, Supplementary Fig. 3a), a C-terminal green fluorescent protein (GFP) (Fig. 2b, Supplementary Fig. 3b)), and an N-terminal bacterial luciferase fusion (Lux-ACP) (Fig. 2b, Supplementary Fig. 4). The luciferase-ACP fusion’s activity did not change notably following label manipulation (Supplementary Fig. 5). We additionally found that labeling of the ACP-GFP fusion with rhodamine-CoA generates an observable Förster resonance energy transfer (FRET) signal (Supplementary Fig. 6), lending itself to observing AcpH or PPTase activity in a simple and scalable assay format.
Recent studies of fatty acid and polyketide pathways focus on the extent and function of substrate sequestration by ACP, where the growing acyl chain is covalently attached via a thioester linkage to the terminus of post-translationally added 4’-phosphopantetheine22. Biosynthetic intermediates with varied chemical structures participate in intermolecular interactions with ACP that modulate substrate dynamics and ACP structure9,15. The nature of these ACP-substrate interactions depends on the chemical structure of the biosynthetic intermediate and can vary with respect to chain length and oxidation state23. Furthermore, observations of this phenomenon appear to vary depending on the analytical method used. X-ray crystallography of hexanoyl-, heptanoyl-, and decanoyl-ACPs from E. coli fatty acid biosynthesis all show the acyl chain clearly buried in ACP, while two crystal forms of butanoyl-ACP have the acyl chain in different positions both inside and outside of the protein24,25. NMR studies have shown that short-chain polyketide analogs protrude into solution when appended to S. coelicolor actinorhodin ACP (actACP), while saturated acyl chains of 4 to 8 carbons associate more closely with this polyketide ACP15. Variations in substrate dynamics must clearly play a role in the catalytic processivity of these synthases, and we hypothesize that the dynamics of substrate binding serves a critical function in substrate specificity.
In order to evaluate substrate dynamics with respect to substrate identity, it is necessary to perform multiple studies on the same protein with varying acyl substrates covalently attached. Given the labor and expense of preparing uniformly labeled, isotope-enriched proteins for nuclear magnetic resonance (NMR) structural studies, we investigated the use of AcpH as a means to recycle 15N enriched ACP. We reversibly labeled a single sample of 15N enriched E. coli ACP with several acyl pantetheine analogs and characterized the dynamics of the appended acyl-ACP-15N species by NMR spectroscopy. By incorporating fatty-acyl pantetheines with 13C labels within the alkyl chain, we directly observed intramolecular interactions with the pendant acyl chain using NOE measurements.
We evaluated ACP-15N at each labeling conformation via gel (Fig. 2c and Supplementary Figs. 7 and 8) and NMR analysis (Supplementary Figs. 9-14). We obtained an initial apo and holo mixture following E. coli expression, requiring full conversion to the apo- form (Fig. 2c) using AcpH. Subsequent conversion to octanoyl-ACP-15N (Fig. 2c) utilized the chemo-enzymatic synthesis of octanoyl-CoA14 with Sfp labeling. After NMR evaluation, we converted the ACP-15N back to the apo- form with AcpH for subsequent relabeling. We acquired 15N-1H heteronuclear single quantum coherence (HSQC) spectra of all three ACP-15N species (apo-, octanoyl-, and regenerated apo-). Comparing apo-ACP-15N to the octanoyl-ACP-15N (Fig. 3a), we observed chemical shift perturbations characteristic of acyl chain sequestration in the hydrophobic binding pocket26. Conversion from this acylated form back to the apo- form by AcpH provided uniformly unlabeled apo-ACP-15N, as confirmed by an HSQC spectrum of the regenerated protein that identically matched that of the original (Fig. 3b). This validated the feasibility of reversible ACP labeling, as it demonstrated that the regenerated apo-ACP-15N is properly folded and ready for subsequent modification.
Figure 3. HSQC spectra of recycled ACP-15N in various acyl states overlayed with apo-ACP-15N.
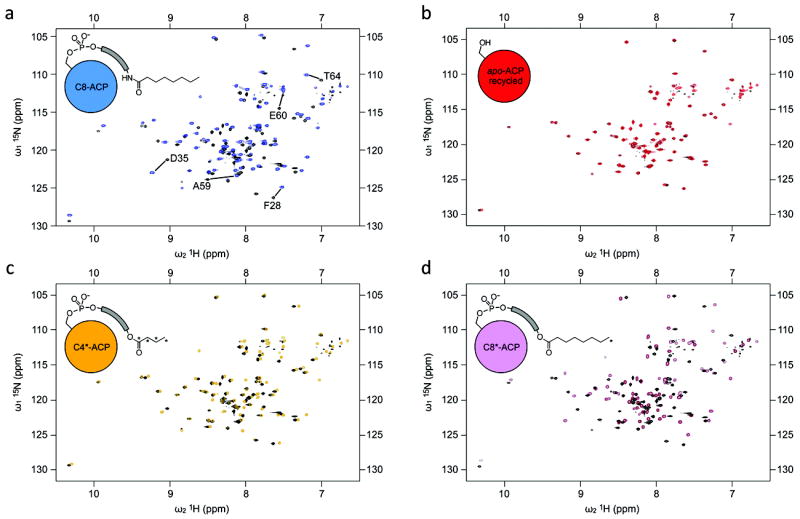
All spectra were collected on the same protein sample. (a) 15N-1H -HSQC of the originally prepared apo-ACP-15N (black) is overlayed with the HSQC of octanoyl-ACP-15N (blue). (b) HSQC of regenerated apo-ACP-15N (red) overlayed with the original apo-ACP-15N preparation (black). (c) HSQC of butanoyl-13C4-ACP-15N (orange) is overlayed with the original apo-ACP-15N (black). (d) HSQC of octanoyl-8-13C1-ACP-15N (lavender) is overlayed with the original apo-ACP-15N (black). Full spectra are available in Supplementary Information (Supplemtary Figs. 9-14).
We next proceeded to label this regenerated apo-ACP-15N with butanoyl-13C4-CoA (Fig. 2c) that contained 13C labels from carbons 1 through 4. Butanoyl-13C4-ACP-15N demonstrated weaker HSQC chemical shift perturbations (Fig. 3c) compared to octanoyl-ACP-15N. Further sample treatment involved one last conversion to the apo- form by AcpH, followed by labeling with octanoyl-8-13C1-CoA containing a single 13C label at carbon 8 (Figs. 2c and 3d). We performed 13C-selective nuclear overhauser effect (NOE) experiments, in which we observe NMR signals for other protons within 5 angstroms from the 13C label, on the butanoyl-13C4-ACP-15N and octanoyl-8-13C1-ACP-15N as a means to gain structural information about substrate-protein interactions. In collecting 13C-edited NOE spectra of the 13C-labeled acyl pantetheines, we observed no NOE signal for butanoyl-13C4-ACP-15N (Supplementary Fig. 15a), whereas octanoyl-8-13C1-ACP produced a notable signal (Supplementary Fig. 15b). This result was likely produced from spatial proximity of an aliphatic proton in an ACP-15N sidechain and the 13CH3 group in the octanoyl-13C1- acyl chain, indicating that the longer acyl chain is immobilized in the protein binding pocket. This finding indicates a lack of dynamic mobility and sequestration of the acyl chain on the NMR time scale. Conversely, the negative result from butanoyl-13C4-ACP-15N indicates that the shorter acyl chain is notably more dynamic in solution. The X-ray crystal structure indicates two states for a tethered butanoyl substrate, one sequestered and one outside the protein24. Our NMR-based finding highlights the differences between solution and crystalline structures, in which the behavior of substrate-tethered ACPs varies substantially in different physical states. We conclude that analysis of ACP-substrate dynamics must necessarily be performed in solution state.
In addition to observing the dynamics of tethered acyl substrates, these NMR studies provide a quantitative evaluation for protein quality after repeated labeling and unlabeling steps. This demonstration offered an ideal testing ground for the reversible labeling method, as we used only one isotope-enriched protein sample for the entire experiment. Any protein degradation or incomplete reactivity would severely compromise the quality of resulting NMR spectra. To provide quality control, we acquired HSQC spectra of purified ACP-15N at each discrete step throughout the process and compared to the original apo-ACP-15N sample (Fig. 3), which revealed predominant protein integrity retention throughout the experiment. Through tracking the ultraviolet absorbance of ACP-15N throughout all conversions (Supplementary Table 1), we observe a final recovery of 27% protein after 5 discrete enzymatic reaction steps. We further evaluated reaction efficiency for all presented reactions (Table 1a,b and Supplementary Figs. 16 and 17), demonstrating that two-step yields above 60% are feasible for most ACP constructs.
Table 1.
Evaluating ACP Sfp/AcpH reaction efficiency
| a | E.coli ACP | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| apo #1 | butanoyl | apo #2 | apo #1 | octanoyl | apo #2 | apo #1 | rhodamine | apo #2 | apo #1 | coumarin | apo #2 | |
| mass (mg) | 1.9 | 1.7 | 1.8† | 1.6 | 1.2 | 1.0 | 1.6 | - | 1.0 | 1.6 | - | 1.4 |
| step yield | - | 92% | 99% | - | 77% | 83% | - | - | 64% | - | - | 85% |
| cycle yield | 98% | 64% | 64% | 85% | ||||||||
| b | MBP-PaACP | GFP-PaACP | Luciferase-ACP | |||||||||
| apo #1 | rhodamine | apo #2 | apo #1 | rhodamine | apo #2 | apo #1 | rhodamine | apo #2 | ||||
| mass (mg) | 1.7 | 1.3 | 0.9 | 2.5 | 2.2 | 1.6 | 1.7 | 1.5 | 1.0 | |||
| step yield | - | 92% | 74% | - | 86% | 75% | - | 82% | 68% | |||
| cycle yield | 57% | 65% | 56% | |||||||||
All free (d) and fusion (e) ACPs were labeled and unlabeled in order to validate reaction yields.
Elevated yield calculation for obtaining apo-ACP from butanoyl-ACP is most likely due to minor protein or buffer contamination for this sample. Reaction completion was monitored by Urea-PAGE (Supplementary Fig. 16) and SDS-PAGE (Supplementary Fig. 17) as appropriate.
This work suggests that AcpH is capable of removing a broad variety of covalently-tethered labels beyond those studied here, in addition to accommodating N and C-terminal ACP fusion partners with ease. Given the multitude of existing opportunities for ACP labeling, particularly in work involving fusion protein applications and natural product biosynthetic studies, we believe that providing a reversible methodology will provide markedly improved flexibility for rapid modification of protein species. Additionally, the cost-saving measure of recovering valuable apo-ACP substrates cannot be overlooked. Due to the wide pantetheine substrate acceptance demonstrated by a combined Sfp and AcpH methodology, various fluorescent and functional tags can be exchanged on a single protein with robustness not offered by previous enzymatic methods.
Online Methods
Determination of Protein Concentration, Protein Gels, Miscellaneous
ACP concentrations were determined by UV absorbance measurements at 280 nm unless otherwise noted. Extinction coefficients were calculated using Expasy (http://www.expasy.ch) “ProtParam” tool: E. coli free ACP = 1490 M-1cm-1, GFP-ACP = 69000 M-1cm-1, MBP-PaACP = 66000 M-1cm-1, Lux-ACP = 85720 M-1cm-1. Non-ACP protein concentrations and fusion-ACPs used in the efficiency analysis (Table 1b) were determined using the Bradford method against a BSA standard. ACP was run on 20% 2M Urea-PAGE to resolve apo/holo/crypto conversions, and 12% SDS-PAGE to evaluate overall purity during NMR workup. ACP fusions MBP-PaACP and GFP-ACP samples were run on 15% acrylamide 2 M Urea-PAGE for fluorescent imaging experiments. Electrophoresis of fluorescent coumarin and rhodamine non-fusion E. coli ACP modifications utilized 10% Tris-Tricine SDS-PAGE.
Gel Imaging
Coomassie stained gels were imaged on a Fluor-S MultiImager (Bio-Rad) using visible light exposure. Coomassie gel images were acquired as “.tiff” and excess white was discarded using the “Auto Levels” feature of Photoshop (Adobe). UV acquisition was also performed on the Fluor-S MultiImager, with short/long wave UV and a 520LP filter, excess black was discarded using the “Auto Levels” feature of Photoshop (Adobe). GFP-fluorescent imaging performed prior to gel fixing was performed on a UVP BioSpectrum (UVP LLC) system with SYBR Green 515nm-570nm emission filter, exciting with trans-illumination from a UVP BioLite with 420BP40 filter. GFP-fluorescent images were collected as “.tiff” and had gray input levels adjusted using Photoshop (Adobe) from “0,1.00,255” to “0,1.00,150” to discard excess black. Rhodamine-labeled protein gels were imaged on a Typhoon (GE Healthcare) gel scanner at 50 μm resolution with a photomultiplier tube (PMT) setting of 450, using 532 nm (green laser) excitation, and 580BP30 emission filter. Typhoon gel images were collected as “.gel” files, converted to “.tiff” in ImageJ (NIH), exported to Photoshop (Adobe) and had gray input levels adjusted from “0,1.00,255” to “80,1.00,255” to discard excess whites collected from the “.gel” file. All gels, with the exception of GFP-ACP containing gels, were fixed with 10% acetic acid, 40% methanol, and 50% water for 1 hour, then rinsed 3 times with water for prior to UV fluorescent imaging and subsequent staining. GFP-ACP images were acquired prior to gel fixing, after which they were fixed and imaged as other gels.
Production of AcpH and Recombinant ACP Constructs
Cloning methods and primers are contained in Supplementary Information (Supplementary Methods & Supplementary Table 2). For expression of E. coli ACP-15N, E. coli BL21 (DE3) cells containing plasmid pET22b encoding C-terminal 6xHis tagged E. coli ACP was cultured in 1 L M9 minimal media supplemented with 1 g/L of N15 enriched ammonium chloride and 100 mg/L ampicillin. Culture was grown to OD600 = 0.6, induced with 1 mM IPTG and incubated 4 hours at 37 °C. E. coli BL-21(DE3) cells containing the MBP-AcpH plasmid were grown in 1L LB, 0.2% D-glucose and 50 μg/mL kanamycin sulfate at 37 °C to OD = 0.6, induced with 1 mM of isopropyl β-D-1-thiogalactopyranoside (IPTG), and grown at 16 °C overnight. Media was centrifuged 30 minutes at 2000 rpm to pellet cells. Cell pellets were stored at -20 °C overnight. AcpH-6xHis construct was grown similarly without glucose. Cells were thawed on ice and suspended in lysis buffer (50 mM TrisCl pH 8, 500 mM NaCl, 10% glycerol) with additional ingredients 0.1 mg/mL lysozyme, 0.1 mM DTT, 5 μg/mL DNase I, 5 μg/mL RNAse A and passed twice through a French pressure device at 1000 psi. Lysate was centrifuged 45 minutes at 10,000 rpm, and supernatant was incubated with amylose resin (New England Biolabs) for MBP-AcpH or Ni-NTA (Novagen) for AcpH-6xHis according to manufacturer protocols. Eluted MBP-AcpH was then concentrated to 10 mg/mL and MBP-AcpH was FPLC-purified with 50 mM TrisCl, 250 mM TrisCl, pH 8.0 buffer to remove contaminating native E. coli MBP. MBP-AcpH was concentrated with 10 kDa Amicon spin filter (Millipore Corp) stored in 40% glycerol at -80 °C after flash freezing aliquots in liquid nitrogen. 6xHis-AcpH was lysed in a similar manner, but purified with Ni-NTA resin (Novagen). Ni-NTA resin with bound protein was washed with 10 mM imidazole and eluted with 300 mM imidazole in lysis buffer. 6xHis-AcpH was desalted to remove imidazole, and flash frozen at -80 °C at 1 mg/mL without further modification. MBP-PaACP and Lux-ACP were expressed in E. coli BL-21 (DE3) in LB with 50 μg/mL kanamycin. GFP-ACP (6xHis-tagged in pCA24N vector)27 was expressed in E. coli K-12 strain AG1 (ASKA library) cells, in LB with 20 μg/mL chloramphenicol. Cells containing fusion ACPs were grown, induced, and purified in an otherwise identical manner to 6xHis-tagged AcpH. MBP- and GFP- fusion ACP 300 mM imidazole elutions were dialyzed into AcpH reaction buffer without Mg2+ and Mn2+ cofactors overnight. Lux-ACP was buffer exchanged using a PD-10 desalting column (GE Healthcare) into AcpH reaction buffer, flash frozen, and stored overnight at -80 °C. Lux-ACP was thawed on ice and AcpH was added to 5 μM, and the reaction incubated at 37 °C for 4 hours. Dialyzed ACP fusions next had appropriate amounts of 1M MgCl2 and MnCl2 added to achieve 15 mM and 1 mM final concentrations, respectively. Free AcpH was added to free ACP and MBP/GFP fusion ACPs at 5 μM final concentration, and the mixture was incubated overnight at 37°C in a rotary wheel. Lux-ACP was reacted for 4 hours at 37 °C. ACP reactions were centrifuged to remove any precipitate, and purified by anion exchange chromatography. Purity evaluation was conducted on MBP-PaACP and GFP-ACP (Supplementary Fig. 3), as well as Lux-ACP (Supplementary Fig. 4) with SDS-PAGE.
Preparation of Coumarin-ACP Standard
DK554 cells were grown, induced, and prepared to generate predominantly apo-ACP. Isopropanol supernatant containing ACP was applied to DEAE resin, and eluted with a sodium chloride gradient. ACP was then labeled using 6xHis-Sfp and coumarin-CoA. Sfp was removed with Ni-NTA resin, and excess coumarin-CoA was removed with size exclusion chromatography on G25 sephadex resin.
Preparation of E. coli DK554 Lysate
50 mL LB media supplemented with 25 μM calcium D-pantothenate, 50 mM D-glucose, and 50 μg/mL kanamycin utilized previously. Media was inoculated with 1 mL of overnight DK554 starter culture and grown to OD = 0.4. IPTG was added at a concentration of 1 mM to the media and was shaken for 5 hours at 37 °C. Media was centrifuged at 4000 rpm at 4°C for 30 minutes to pellet cells. Cell pellets were resuspended in 25 mM TrisCl pH 7.5, 250 mM NaCl, 0.1 mg/mL lysozyme, 10 μM pepstatin, 10 μM leupeptin and passed twice through a French pressure device at 1000 psi.
Removal of coumarin-pantetheine from ACP in 5 mL lysate used 10 μM MBP-AcpH fusion in 600 mL AcpH reaction buffer (50 mM TrisCl, pH 8.0, 100 mM NaCl, 10 % glycerol, 15 mM MgCl2, 1 mM MnCl2) within a 3 kDa MWCO dialysis bag at 37 °C overnight.
Fluorescent Labeling of E. coli DK554 Lysate with Modified Coenzyme A
E. coli DK554 cell lysate with total protein concentration of approximately 2 mg/mL was added to the volume of premade 10X PPTase reaction buffer (500 mM Na-HEPES, 100 mM MgCl2, pH 7.4) that brought the total reaction concentration to 50 mM Na-HEPES pH 7.4, 10 mM MgCl2, 5 μM of coumarin-CoA, and 2 μM Sfp. Samples were incubated at 37°C for 1 hour, followed by centrifugation to remove precipitate, supernatant passage over an equilibrated G50 Sephadex (GE Healthcare) desalting column, and dialysis of the lysate into 50 mM TrisCl pH 8.0, 100 mM NaCl, 10% glycerol to further remove unreacted CoA analog.
AcpH Treatment of Coumarin-ACP in Lysate
Coumarin-labeled DK554 cell lysate supernatant was added to a freshly-prepared 10X AcpH reaction buffer to generate the following reaction concentrations: 50 mM TrisCl pH 8, 150 mM NaCl, 15 mM MgCl2, 1 mM MnCl2. MBP-AcpH was added to 2 μM. Reaction contents were placed in a 3.5 kD MWCO dialysis membrane and dialyzed against 50-fold volume of reaction buffer stirred overnight at 37°C. No remaining coumarin-ACP fluorescence was observed, and a significant amount of MBP-AcpH appeared as precipitate afterwards, as determined by SDS-PAGE analysis (not shown). Post-reaction contents were centrifuged 30 minutes at 4000 rpm at 6°C. Supernatant was dialyzed back into 50 mM TrisCl pH 7.5, 250 mM NaCl in preparation for rhodamine-labeling.
Sfp & AcpH Treatment of Purified Rhodamine-ACP
The demonstrated activity of AcpH on rhodamine-ACP was performed with previously purified 6xHis-tagged apo-ACP. 7 nmol of apo-ACP was treated with 5 μM native Sfp and 24 nmol of rhodamine-CoA in 50 mM TrisCl pH 8, 100 mM NaCl, 10 mM MgCl2 at 37°C for 1 hour. Rhodamine-ACP was re-purified with Ni-NTA resin to remove excess rhodamine-CoA and Sfp, and dialyzed to remove imidazole. Dialyzed rhodamine-ACP was then incubated with and without 7 μM AcpH at 37°C for 2 hours, and the resulting crude reactions were run on SDS-PAGE and imaged to illustrate fluorescent label removal with AcpH.
AcpH Treatment of ACP-15N for NMR Study
An AcpH reaction was conducted to generate each apo-15N-ACP sample prior to labeling and/or analysis. Following NMR acquisition of each sample, ACP was dialyzed into AcpH reaction buffer without cofactors (50 mM TrisCl pH 8.0, 100 mM NaCl). Glycerol was found to be unnecessary for desired AcpH activity and was omitted. Following dialysis, MgCl2 and MnCl2 were added to achieve a final concentration of 15 mM and 1 mM, respectively. Free 6xHis AcpH was added to a concentration of 5-10 μM, and the mixture was incubated at 37°C for 8 hours. Reaction completion was determined by Urea-PAGE analysis. The completed reactions were centrifuged 30 minutes at 4,000 rpm at 6°C to remove precipitate.
Labeling of ACP-15N Using “One-Pot” Sfp Methodology
Apo-ACP-15N was mixed with CoA-A, D, & E, ATP disodium salt, native Sfp, and octanoyl-pantethenamide in a one-pot chemoenzymatic reaction27 to selectively generate octanoyl-ACP-15N in vitro. Additional generation of butanoyl-13C4-ACP-15N and octanoyl-8-13C1-ACP-15N analogs was conducted with the same methodology, except using regenerated apo-ACP-15N with butanoyl-13C-oxypantetheine and oxtanoyl-8-13C1-oxypantetheine. Ni-NTA resin was used to re-purify the ACP-15N after each labeling reaction. Apo/holo/crypto ACP-15N monitoring was conducted with separation on conformationally sensitive Urea-PAGE.
ACP Anion Exchange Purification
All preparative ACP samples were dialyzed into low salt buffer. E. coli ACP and MBP-PaACP ion exchange running buffer was 25 mM L-Histidine pH 6.0. GFP-ACP and Lux-ACP ion exchange buffer was 25 mM bis-Tris, pH 6.0. Supernatants were then applied to a DEAE HiTrap (GE Healthcare) 1 mL or 5 mL column. Free ACP, MBP-ACP, and GFP-ACP were loaded onto columns and washed with 10 mL of 25 mM buffer and 25 mM NaCl and eluted with 5 mL of 25 mM buffer and 500 mM NaCl. LuxACP was loaded onto a 5 mL DEAE HiTrap and washed with a step gradient of 0, 100, 200, 300, and 500 mM NaCl in 25 mM bis-Tris pH 6.0. NMR E. coli ACP-15N samples were then dialyzed into 100 mM sodium phosphate, 1 mM DTT, pH 7.4, and concentrated to 450 μL prior to NMR acquisition. E. coli ACP-15N NMR sample purity was evaluated by SDS-PAGE analysis (Supplementary Fig. 8). Following ion exchange, fusion ACPs were dialyzed or desalted, spin concentrated, and stored at -80°C prior to further use.
Fusion-ACP Rhodamine-CoA Labeling and Label Removal
Purified apo-MBP-PaACP at 200 μM was labeled with 1 mM rhodamine-CoA and 13 μM native Sfp in a reaction volume of 20 μL for 3 hours at 37°C. Purified apo-GFP-ACP at 150 μM was labeled with 1 mM rhodamine-CoA and 7 μM native Sfp in a reaction volume of 35 μL for 3 hours at 37°C. Purified apo-Lux-ACP at 12 μM was labeled with 50 μM rhodamine-CoA and 3 μM native Sfp for 1 hour at 37°C. Fusion ACPs were then re-purified from excess rhodamine-CoA and native Sfp using Ni-NTA resin, and buffer exchanged and concentrated using 10kDa MWCO 0.5 mL Amicon spin filters (Millipore). Label removal of MBP-ACP and GFP-ACP proceeded with 4 μL of the concentrated crypto- fusion ACPs with 10 μM AcpH in 10 μL of AcpH reaction buffer for 3 hours at 37°C. 6 μM crypto-Lux-ACP was reacted with 5 μM AcpH for 3 hours at 37°C. AcpH reaction samples were run immediately afterwards on Urea-PAGE (MBP-ACP and GFP-ACP) or SDS-PAGE (Lux-ACP) with no further purification.
Sfp & AcpH Treatment of E. coli ACP and Fusion-ACPs for Efficiency Analysis
Labeling of butanoyl, octanoyl, and coumarin ACP proceeded via Sfp and CoA A-D-E “one-pot methodology”. Labeling of free ACP and fusion-ACPs with rhodamine proceeded via Sfp and rhodamine-CoA. Free E. coli ACP reactions were conducted overnight at 37°C. MBP-PaACP labeling proceeded via native Sfp; GFP-ACP and luciferase-ACP reactions using Sfp-conjugate proceeded for 6 hours at 37°C. MBP-PaACP was repurified using Ni-NTA and desalted with PD-10 desalting column (GE Healthcare) to remove imidazole. GFP-ACP and luciferase-ACP were separated from solids with a fritted spin column and desalted to remove excess CoA analog. Non-fluorescent E. coli ACPs were quantified using UV spectrometry. Label removal proceeded in AcpH reaction buffer with 10% glycerol via reaction with an AcpH-conjugate for E. coli ACP at 37°C overnight, and room temperature overnight for MBP-PaACP and GFP-ACP. Due to precipitation induced by extended incubation, luciferase ACP was reacted with AcpH at 37°C for 2 hours. Reaction completion was monitored by gel-shifts and/or fluorescence depletion in 20% Urea-PAGE for free ACPs, and 10% SDS-PAGE for the fusion ACPs. Final apo-ACP samples were then desalted into AcpH reaction buffer lacking Mg2+/Mn2+ and subsequently quantified (Table 1a,b).
GFP-ACP: Rhodamine-CoA & Sfp Labeling Monitoring by FRET
Apo-GFP-ACP and rhodamine-CoA were diluted to 100 μM and 200 μM, respectively, in 10 mM TrisCl pH 7.5. 5 μL of this 10X substrate mix was added to 6 wells of a costar 3694 96-well plate (Corning Inc) in triplicate. 10 μL of milliQ water diluent was added to adjust final concentrations. 35 μL of 1.43 μM Sfp in 71.5 mM HEPES pH 7.6, 14.3 mM MgCl2, 1.43 mg/mL BSA (stabilizer) was added to initiate the reaction, with reaction buffer lacking Sfp added to the control. Final 50 μL enzyme reactions contained 10 μM apo-GFP-ACP, 20 μM rhodamine-CoA, 1 μM Sfp, 50 mM HEPES pH 7.6, 10 mM MgCl2. The 96-well plate was centrifuged 2 minutes at 1000 rpm, and fluorescence was monitored at 405 nm excitation and 595 nm emission for 30 minutes at in a Perkin-Elmer HTS 7000 Plus plate reader at room temperature.
Pantetheine Probe Synthesis
All CoA-related probes are depicted in Supplementary Information (Supplementary Fig. 18). Pantetheine probe synthetic methods and chemical spectra are also contained within Supplementary Information (Supplementary Note and Supplementary Figs. 19-42).
Supplementary Material
Acknowledgments
We wish to thank Timothy L. Foley (University of California San Diego, UCSD) for preparing rhodamine-CoA, coumarin-CoA, and coumarin-ACP standard, Stephen Mayfield (UCSD) for providing the luciferase plasmid template, Shiteng Duan (UCSD) for laboratory support, Michael Rothmann (UCSD) for assisting AcpH method development, Roger Tsien (UCSD) for use of GFP-related gel imaging equipment, UCSD Chemistry & Biochemistry Mass Spectrometry Facility, and Jim La Clair (UCSD) for manuscript design input. This research was funded by National Institute of Health grants R21AI090213, R01GM094924, and R01GM095970.
Footnotes
Note: Supplementary information is available on the Nature Methods website (http://www.nature.com/naturemethods/)
Author Contributions:
N.M.K. performed all cloning, subcloning, enzymatic reactions, imaging, and protein purifications (unless otherwise stated). R.W.H. conducted all protein NMR experiments and provided resulting NMR data. R.W.H. prepared the native Sfp, MBP-CoA-A,D,E enzyme stocks utilized for “one pot” chemoenzymatic CoA analog synthesis. A.R.S. synthesized, purified, and characterized all oxopantetheine probes in this work. N.M.K., R.W.H., A.R.S. and M.D.B. wrote the manuscript.
Competing Financial Interests:
The authors declare no competing financial interests.
References
- 1.Luchansky SJ, Argade S, Hayes BK, Bertozzi CR. Biochemistry-US. 2004;43:12358–12366. doi: 10.1021/bi049274f. [DOI] [PubMed] [Google Scholar]
- 2.Stachler MD, Chen I, Ting AY, Bartlett JS. Mol Ther. 2008;16:1467–1473. doi: 10.1038/mt.2008.129. [DOI] [PubMed] [Google Scholar]
- 3.Batra G, et al. Protein expres purif. 2010;74:99–105. doi: 10.1016/j.pep.2010.04.017. [DOI] [PubMed] [Google Scholar]
- 4.Guignet EG, Hovius R, Vogel H. Nat Biotechnol. 2004;22:440–444. doi: 10.1038/nbt954. [DOI] [PubMed] [Google Scholar]
- 5.Gauchet C, Labadie GR, Poulter CD. J Am Chem Soc. 2006;128:9274–9275. doi: 10.1021/ja061131o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rabuka D. Curr Opin Chem Biol. 2010;14:790–796. doi: 10.1016/j.cbpa.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yin J, et al. P Natl Acad Sci USA. 2005;102:15815–15820. doi: 10.1073/pnas.0507705102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mosiewicz KA, Johnsson K, Lutolf MP. J Am Chem Soc. 2010;132:5972–5974. doi: 10.1021/ja9098164. [DOI] [PubMed] [Google Scholar]
- 9.Hinner MJ, Johnsson K. Curr Opin Biotech. 2010;21:766–776. doi: 10.1016/j.copbio.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 10.Foley TL, Young BS, Burkart MD. FEBS J. 2009;276:7134–7145. doi: 10.1111/j.1742-4658.2009.07425.x. [DOI] [PubMed] [Google Scholar]
- 11.Reyes CP, Clair JJL, Burkart MD. Chem Commun. 2010;46:8151–8153. doi: 10.1039/c0cc02876b. [DOI] [PubMed] [Google Scholar]
- 12.Meier JL, et al. ACS Chem Biol. 2009;4:948–957. doi: 10.1021/cb9002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meier JL, Haushalter RW, Burkart MD. Bioorg Med Chem Lett. 2010;20:4936–4939. doi: 10.1016/j.bmcl.2010.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haushalter RW, et al. ACS Chem Biol. 2011;6:413–418. doi: 10.1021/cb200004k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Evans SE, et al. J Mol Biol. 2009;389:511–528. doi: 10.1016/j.jmb.2009.03.072. [DOI] [PubMed] [Google Scholar]
- 16.Meier JL, Burkart MD. Curr Opin Chem Biol. 2011;15:48–56. doi: 10.1016/j.cbpa.2010.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murugan E, Kong R, Sun H, Rao F, Liang Z-X. Protein Expres Purif. 2010;71:132–138. doi: 10.1016/j.pep.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 18.Quadri LE, et al. Biochemistry-US. 1998;37:1585–1595. doi: 10.1021/bi9719861. [DOI] [PubMed] [Google Scholar]
- 19.Lambalot RH, Walsh CT. J Biol Chem. 1995;270:24658–24661. doi: 10.1074/jbc.270.42.24658. [DOI] [PubMed] [Google Scholar]
- 20.Foley TL, Burkart MD. Anal Biochem. 2009;394:39–47. doi: 10.1016/j.ab.2009.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Foley TL, et al. Org Biomol Chem. 2010;8:4601–4606. doi: 10.1039/c0ob00322k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan D, Vogel H. Biochem J. 2010;430:1–19. doi: 10.1042/BJ20100462. [DOI] [PubMed] [Google Scholar]
- 23.Płoskoń E, et al. Chem Biol. 2010;17:776–785. doi: 10.1016/j.chembiol.2010.05.024. [DOI] [PubMed] [Google Scholar]
- 24.Roujeinikova A, et al. Acta Crystallogr D. 2002;58:330–332. doi: 10.1107/s0907444901020091. [DOI] [PubMed] [Google Scholar]
- 25.Roujeinikova A, et al. J Mol Biol. 2007;365:135–145. doi: 10.1016/j.jmb.2006.09.049. [DOI] [PubMed] [Google Scholar]
- 26.Upadhyay SK, et al. J Biol Chem. 2009;284:22390–22400. doi: 10.1074/jbc.M109.014829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masanari K, et al. DNA Res. 2005;12:291–299. [Google Scholar]
- 28.Worthington AS, Burkart MD. Org Biomol Chem. 2006;4:44–46. doi: 10.1039/b512735a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.