

Significance
Gut microbes are increasingly recognized as influential components of animal biology. Genomic, mechanistic, and evolutionary aspects of gut symbiont specialization remain understudied, however, largely due to the complexity of gut communities, especially in vertebrate systems. We show that the simple microbiota of eusocial bees exhibits host specificity and that coresident species in the bee gut possess complementary capabilities for energy metabolism, implying their occupancy in distinct ecological niches. In addition, coresidence in the gut of a host species results in horizontal exchange of genes between unrelated symbionts. Strains in different hosts have diverged, and honey bee symbionts are evolutionarily and functionally distinct from their bumble bee counterparts, indicating that gut symbionts may be critical elements in biological differences among bee species.
Keywords: bacterial genomics, strain variation, symbiosis
Abstract
Gilliamella apicola and Snodgrassella alvi are dominant members of the honey bee (Apis spp.) and bumble bee (Bombus spp.) gut microbiota. We generated complete genomes of the type strains G. apicola wkB1T and S. alvi wkB2T (isolated from Apis), as well as draft genomes for four other strains from Bombus. G. apicola and S. alvi were found to occupy very different metabolic niches: The former is a saccharolytic fermenter, whereas the latter is an oxidizer of carboxylic acids. Together, they may form a syntrophic network for partitioning of metabolic resources. Both species possessed numerous genes [type 6 secretion systems, repeats in toxin (RTX) toxins, RHS proteins, adhesins, and type IV pili] that likely mediate cell–cell interactions and gut colonization. Variation in these genes could account for the host fidelity of strains observed in previous phylogenetic studies. Here, we also show the first experimental evidence, to our knowledge, for this specificity in vivo: Strains of S. alvi were able to colonize their native bee host but not bees of another genus. Consistent with specific, long-term host association, comparative genomic analysis revealed a deep divergence and little or no gene flow between Apis and Bombus gut symbionts. However, within a host type (Apis or Bombus), we detected signs of horizontal gene transfer between G. apicola and S. alvi, demonstrating the importance of the broader gut community in shaping the evolution of any one member. Our results show that host specificity is likely driven by multiple factors, including direct host–microbe interactions, microbe–microbe interactions, and social transmission.
Host specialization is a key evolutionary process in many symbionts. For bacteria, closely related strains of the same species may carry unique gene assemblages favoring the colonization of certain host species over others. The genetic basis of host specificity has been of considerable interest in the study of pathogens, but only a few studies of this process are available for mutualistic symbionts, including the normal gut microbiota (1–3). Despite tremendous advances in this field, most comparative studies of gut microbes rely on metagenomic and 16S rRNA gene surveys that typically give very coarse-grained information about evolutionary processes at the subspecies level. Modeling the differentiation of individual strains and discerning the factors that drive their specialization require both complete genome sequences and tractable systems with which to test hypotheses.
In vertebrates, a well-recognized example of strain-level diversification is with Lactobacillus reuteri, a highly host-specific gut microbe found in diverse mammals and birds, for which multiple genome sequences and mouse-model approaches are available (4, 5). L. reuteri strains not only carry unique genes that restrict host colonization but also exhibit different patterns of genomic evolution according to their preferred host (6). Congruence between host phylogeny and bacterial community composition in many animals (7–9) suggests that strain-level host specificity in gut bacteria is commonplace. Recently, such specificity has also been proposed to occur in bees (10, 11), where 16S rRNA gene sequence analysis revealed gut symbiont phylogeny correlating with host species rather than geographic origin (12).
The bee gut microbiota is simple compared with that of most vertebrates. Eight species (defined as strains sharing >97% 16S rRNA identity) comprise over 95% of bacteria in the adult workers of the honey bee Apis mellifera (13). These species include the betaproteobacterium Snodgrassella alvi (family Neisseriaceae) and the gammaproteobacterium Gilliamella apicola (family Orbaceae), the dominant Gram-negative members of the gut community, with each comprising up to 30–39% of the microbiota (13). These species appear to be unique to the eusocial honey bees (Apis spp.) and their cousins, the bumble bees (Bombus spp.) (10). There is accumulating evidence that G. apicola and S. alvi are mutualistic symbionts with roles in both pathogen defense (14) and nutrition (15). Their highly restricted distribution and phylogenetic correlation with their hosts are suggestive of a lengthy coevolutionary history with bees and with each other, yet very little is known about their functional capabilities and symbiotic interactions (11, 15, 16).
In this paper, we examine the genomic characteristics of G. apicola and S. alvi from A. mellifera and Bombus spp. and assess possible mechanisms responsible for the observed host specificity. We also uncover the metabolic capabilities of these symbionts and describe factors that may underlie the gut colonization process. As widespread, economically important insects with a relatively simple and well-defined microbiota, bees hold great promise as an alternative, invertebrate model for understanding specialized host-associated microbial communities.
Results and Discussion
Genome Characteristics.
The genomes of three S. alvi and three G. apicola, previously isolated (11), were sequenced (Fig. 1). Strains wkB1T and wkB2T were isolated from A. mellifera, whereas strains wkB11 and wkB12 were from Bombus bimaculatus and strains wkB29 and wkB30 were from Bombus vagans. Genomes of the type strains, G. apicola wkB1T and S. alvi wkB2T, were closed and assembled to finished status. The S. alvi wkB2T genome consists of a single circular chromosome of 2,527,978 bp encoding 2,299 proteins, 59 tRNAs, and 4 rRNA operons. S. alvi strains wkB12 and wkB29 were assembled to 34 and 86 scaffolds, respectively, and both of these genomes are slightly smaller than that of wkB2T, have a greater number of mobile elements, but carry a similar number of protein-coding genes. Unlike other closely related Neisseriaceae, no highly repeated sequences for DNA uptake are found in S. alvi (17).
Fig. 1.

Schematics and statistics of sequenced G. apicola and S. alvi genomes. Protein-coding regions of wkB1T and wkB2T are plotted clockwise in dark red (+ strand) and light red (− strand). The locations of orthologous proteins in the Bombus-derived strains are shown in green and blue. G+C, guanine and cytosine.
G. apicola wkB1T possesses a single 3,139,412-bp chromosome encoding 2,809 proteins, 51 tRNAs, and 4 rRNA operons. The genomes of strains wkB11 and wkB30 were assembled to 61 and 140 scaffolds, respectively. Assembly of strain wkB30 was hindered by a large number of transposable elements that mostly fall into three types. The origins of these particular elements are not clear, because they are absent from the other strains, but a degenerated clustered regularly interspaced short palindromic repeat (CRISPR)-Cas system in wkB30 may explain their prevalence (SI Results). The most striking difference between wkB1T and the two Bombus-derived strains is in genome size: Both wkB11 and wkB30 are over 800 kb smaller and have a reduced complement of protein-coding genes (Figs. 1 and 2A).
Fig. 2.

(A) Numbers of shared orthologs and unique genes in G. apicola and S. alvi genomes. (B) Gene content as categorized by the Kyoto Encyclopedia of Genes and Genomes (KEGG) subsystem. Within genomes, ∼50% of genes were not assigned to a category and ∼28–38% of predicted genes were annotated as hypothetical or putative; these are not shown here. (C) Pairwise dS values of all orthologs, plotted against each other (e.g., the ∼1:1 relationship of red points shows that genes are equally divergent between the Apis symbiont and either of the Bombus symbionts). dS values extend from 0 to 4 on all axes.
For genes that could be functionally categorized, the greatest discrepancy between wkB1T and the Bombus-derived G. apicola is in those encoding carbohydrate metabolism (Fig. 2B). Strain wkB1T possesses a substantially larger number of these genes than do wkB11 and wkB30, as well as Orbus hercynius CN3T, an outgroup to G. apicola (18). This finding is congruent with our previous analysis showing that the Apis gut metagenome is enriched in carbohydrate metabolism genes (15), suggesting a lineage-specific expansion of carbohydrate processing capabilities in honey bee G. apicola. These gene sets may result from a history of horizontal transfer: The sugar-importing phosphotransferase system genes of Orbaceae genomes often have closest identity to those in Firmicutes (Table S1), and other genes, such as those for pectin degradation, have also been proposed to be horizontally acquired (15). It is possible that G. apicola has, like certain Lactobacillus spp. (19, 20), an extensive pan-genome that facilitates the gain and loss of metabolic capabilities among strains as suited to their environment. The relative paucity of carbohydrate metabolism genes in wkB11 and wkB30 could reflect our limited sampling of G. apicola diversity or might represent a genuine consequence of genome evolution brought about by the lifestyle of their Bombus hosts. In contrast to Apis, Bombus colonies are small (∼50–400 workers) and are founded anew annually by a single queen (21). Thus, Bombus microbes likely experience genetic bottlenecks that would reduce access to the G. apicola pan-genome and lead to genome reduction. Host ecology has similarly been suggested to influence lineage-specific evolution in the vertebrate gut symbiont L. reuteri (6). Both Apis and Bombus have similar diets of pollen and nectar; however, the greater reliance of Apis on processed food stores (beebread and honey), differences in foraging preferences (22), and the effect of domestication should not be ruled out as factors affecting gene repertoires in G. apicola.
S. alvi strains, in contrast, have less gene content variation than G. apicola (Fig. 2B). Strains wkB12 and wkB29 are very closely related, with high average nucleotide identity (97.5% in orthologs) and low silent site divergence (dS; Fig. 2C). All S. alvi strains share a large core gene set (Fig. 2A) and encode very similar metabolic networks (Fig. S1). Of the 634 unique wkB2T genes, only 65 possess a KEGG-annotated function. These 65 include genes for nonribosomal peptide synthesis of siderophores, type 6 secretion systems (T6SS), type I and type III restriction modification, phage-related proteins, and a 14-kb region involved in putative lipid/fatty acid metabolism. The accessory nature of these genes suggests that compared with G. apicola, S. alvi has a more conserved core metabolism and a smaller pan-genome. This conservation may result from a less promiscuous lifestyle and greater niche specialization. Indeed, S. alvi appears to have a tighter association with its host, as evidenced by both phylogenetic results implying largely vertical inheritance (12) and microscopy showing S. alvi localizing close to the gut intima (16) (Fig. 3A).
Fig. 3.

(A) Location of the gut microbial community. A bee with its gut removed shows the ileum and rectum, where most bacteria reside. An ileum cross-section shows the position of S. alvi (blue hatching) adjacent to the gut wall and G. apicola (red) toward the lumen (illustration based on ref. 16). (B) Metabolic pathways and interactions predicted from G. apicola and S. alvi genomes. Missing pathways are dashed, whereas pathways inferred to be complete are solid. Green lines denote possible interspecies interactions. Strains vary in the presence or absence of certain features, as indicated by circular icons. cyt. bo, cytochrome bo complex; FDH, formate dehydrogenase; GlcNAc, N-acetylglucosamine; NDH-1, NADH dehydrogenase I; T1SS, type 1 secretion system; YadA, Yersinia adhesin. *Only in wkB2T. †Not all may be synthesized. A more detailed analysis of symbiont metabolism is included in SI Results and Figs. S1–S3.
Metabolic Reconstruction.
Both G. apicola and S. alvi have relatively small genomes with reduced functional capabilities, which is consistent with their being specialized gut symbionts. S. alvi has lost the ability to use carbohydrates for carbon or energy: The glycolysis (Embden–Meyerhof–Parnas), pentose phosphate, and Entner–Doudoroff pathways needed to convert sugars to pyruvate are all missing key enzymes, and thus are predicted to be nonfunctional (Fig. 3B and Fig. S1). This is surprising, considering that the bee diet consists mainly of carbohydrates. Instead, S. alvi possesses transporters for uptake of carboxylates, such as citrate, malate, α-ketoglutarate, and lactate. These can be used directly in the tricarboxylic acid cycle (TCA) cycle or, in the case of lactate, can be converted to pyruvate via lactate dehydrogenase. S. alvi is an obligate aerobe possessing NADH dehydrogenase and cytochrome bo and bd oxidases, but it lacks the TCA cycle enzyme succinyl-CoA synthetase, which catalyzes the interconversion of succinyl-CoA and succinate. Although genes encoding enzymes to bypass this step of the cycle are not detected (Fig. S2), we predict that a noncanonical, yet functional, TCA cycle is present because S. alvi possesses no other major routes for energy production and carbon metabolism. Even though S. alvi cannot uptake and catabolize carbohydrates, carbohydrates can still be synthesized from TCA intermediates via a complete gluconeogenesis pathway; this is necessary because carbohydrates are precursors of peptidoglycan, LPS, and nucleic acids. Other basic biosynthetic pathways are present, including those for purine and pyrimidine nucleotides and all 21 proteinogenic amino acids except selenocysteine. Vitamins B2 (riboflavin) and B6 (pantothenate) can be synthesized de novo, and strain wkB2T can also synthesize vitamin B3 and its derivatives (NAD+ and NADP+) from aspartate.
G. apicola’s central metabolism sharply contrasts with and, in some ways, complements that of S. alvi. G. apicola is a facultative anaerobe that lacks many genes of the TCA cycle and electron transport chain (Fig. S3). Although the hypoxia-induced cytochrome bd oxidase is present, cytochrome bo and most of the subunits of NADH dehydrogenase are not. Hence, its main mode of energy production is not aerobic respiration but anaerobic fermentation of carbohydrates (23). Glycolysis and pentose phosphate pathways, as well as (in strains wkB1T and wkB30) the Entner–Doudoroff pathway, are present, allowing G. apicola to generate ATP and biosynthetic precursors directly from carbohydrate catabolism. The dominant role of carbohydrate uptake and utilization in G. apicola obviates the need for gluconeogenesis, and, accordingly, two key enzymes for producing sugars from pyruvate are missing. G. apicola may be unable to synthesize selenocysteine, cysteine, methionine, glutamate, glutamine, and proline due to incomplete sulfur assimilation and TCA pathways (SI Results). It also lacks enzymes for synthesizing the pyrimidine precursor orotate and compensates by possessing salvage pathways for exogenous nucleosides.
That G. apicola may be acquiring amino acids and pyrimidines from S. alvi or that they contribute vitamins to each other is an intriguing possibility. Another case for symbiont metabolic complementarity is found in the partitioning of nutritional niches. G. apicola almost exclusively catabolizes carbohydrates, whereas S. alvi has lost such ability. Sugar fermentation produces formate and lactate that are excreted and can subsequently be used to drive oxidative phosphorylation (both symbionts possesses formate dehydrogenase) or can be assimilated for carbon and energy by S. alvi (which has a lactate permease and several lactate dehydrogenases). However, S. alvi is not dependent on G. apicola for survival; sufficient nutrition is supplied via the host diet or the host itself such that S. alvi can thrive in the absence of any other gut bacteria (Fig. 4). Other members of the bee gut microbiota, such as Lactobacillus and Bifidobacterium (24, 25), are also fermentative, and hence may have similar syntrophic interactions with S. alvi. Resolving the community metabolic dynamics will require further elucidation of the environmental conditions in the gut, expression and regulation of metabolic pathways, and symbiont modulation by the host.
Fig. 4.

Host specificity of S. alvi strains, as shown by colonization efficiency. Gut bacterial load (CFU) was counted 5 d after oral inoculation. The sample size for each treatment is ≥12. Means, 95% confidence interval of means, and the results of Mann–Whitney U tests (*P < 0.0001) are shown. In competition treatments, strain wkB2T was coinoculated with strain wkB12 or wkB29 (1:10 ratio in A. mellifera, 10:1 ratio in B. impatiens). Percentage indicates average wkB2T prevalence in competitions.
Host Colonization and Specialization.
The host-specific distribution of G. apicola and S. alvi strains found by 16S rRNA surveys (10, 12) might be explained by specialized, coevolved host–microbe interactions. The genomic basis of this specificity could lie in the presence or absence of certain metabolic features or in functional differences within host–interaction factors. Examples include the symbiont Xenorhabdus, in which certain loci enable colonization of specific nematode hosts (26), and the mammalian gut symbiont L. reuteri, where only strains residing in native hosts can up-regulate secretion and adhesion genes to trigger biofilm formation (27). Likewise, strains of pathogens are often restricted in their host ranges by their virulence factors: These include plasmids and pathogenicity islands in Salmonella enterica (28) and type III secretion systems in Pseudomonas syringae (29).
The bee symbionts carry various genes associated with bacterial colonization functions, including adherence, immune evasion, virulence, and cell–cell communication factors, that are candidates for affecting host specificity. In G. apicola, these include type IVa pili, T6SS in wkB1T and wkB11, and flp pili and capsular polysaccharides in wkB1T and wkB30 (Fig. 3B and Fig. S3). Type IVa pili were also found in S. alvi, as were type I secretion system RTX proteins and YadA-like adhesins. These features are best known for their roles in pathogenesis, but in gut symbionts, they may instead function in mutualistic interactions (30–32). Capsular polysaccharides, RTX proteins, type IV pili, T6SS, and adhesins can take part in biofilm synthesis and modulation (33–38), reinforcing the idea that biofilm formation is a key process in bee gut colonization (15, 16). Biofilms may help determine host specificity (27) and provide a suitable substrate for facilitating other cross-species interactions.
To demonstrate that host specificity has a functional basis and is not merely a correlation due to transmission history (12), we fed cultured S. alvi to laboratory-reared honey bees (A. mellifera) and bumble bees (Bombus impatiens) and recorded colonization success in the gut. Unlike G. apicola (14), experimentally introduced S. alvi was able to persist in newly emerged, germ-free bees reliably and in the absence of other microbiota members. Bees were given a one-time inoculum of a sucrose solution with no bacteria (negative control) or with ∼106 cfu of strain wkB2T, wkB12, or wkB29. After 5 d, strains in their native host (Apis for wkB2T and Bombus for wkB12 and wkB29) reached counts of ∼107 cfu, whereas strains in nonnative hosts did not proliferate beyond the initial 106 cfu of the inoculum and were mostly cleared from the gut (Fig. 4). We also coinoculated native and nonnative strains in a ratio of 1:10 to test if native strains can colonize despite a numerical disadvantage. In natural conditions, gut symbionts have many opportunities to be horizontally transferred between hosts and may be forced to coexist or compete with other strains. All coinoculated samples reached colony-forming unit levels comparable to monoinoculated samples, and almost all recovered colony-forming units were of the native strain (Fig. 4). Thus, native strains can overcome a large numerical disadvantage and completely outcompete nonnative strains. These results strongly suggest a physiological basis to the observed host specificity and support the idea of specialized coevolved interactions between bees and their associated symbiont strains.
Interestingly, strains wkB12 and wkB29 were not isolated from B. impatiens, but rather B. bimaculatus and B. vagans, respectively. Although we show that S. alvi strains appear to be adapted to their host genera (Apis or Bombus), the ability of wkB12 and wkB29 to colonize B. impatiens raises the question of whether strain specialization extends to the level of host species. Bombus species have distinctive gut community compositions (39), but many are sympatric and can possibly exchange symbionts at foraging sites.
Genome Evolution.
For both G. apicola and S. alvi, measures of dS confirmed earlier gene phylogenies that showed strains from Bombus to be more closely related to each other (lower dS) than to strains from Apis (Fig. 2C and Table S2). Measures of dS are largely independent from effects of selection, and hence are a proxy for the relative length of time two strains have diverged. The high dS values between the strains from Apis and Bombus (∼2 for G. apicola and ∼1 for S. alvi) suggest long divergence times, plausibly since the separation of Apis and Bombus ∼80 Mya. The lower dS between Bombus-derived strains indicates recent divergence or ongoing gene transfer and recombination. These lower dS values were associated with elevated dN/dS (ratio of nonsynonymous to synonymous substitutions per site), both genome-wide and among individual genes (Fig. S4A and Table S2). This is consistent with the idea that purifying selection has had insufficient time to eliminate deleterious nonsynonymous polymorphisms in cases of recent gene divergence (40).
In the two Bombus strains of S. alvi (wkB12 and wkB29), all genes were nearly identical, as indicated by their consistently low dS (Fig. 2C). However, between the Bombus strains of G. apicola (wkB11 and wkB30), dS values were widely dispersed, with many divergent genes but also with genes with high identity. These two groups can clearly be distinguished in Fig. S4A, which shows a large set of genes with dS < 1.2 that is not found for the comparison between Apis- and Bombus-derived strains (e.g., wkB1 vs. wkB11). Thus, a fraction of the core gene set is recombining between strains within Bombus but not between strains from different host genera. In cases where orthologs are not recombining, dS values are often as high between Bombus-derived strains as between strains from Apis and Bombus, indicating that deeply branching symbiont lineages may coexist within a host clade. The recombining genes (dS < 1.2) in wkB11 and wkB30 are not highly clustered in the genomes (Fig. S4B), suggesting they are not part of recently acquired genomic islands.
Symbiont Horizontal Gene Transfer and Gut Community Evolution.
Long-standing associations in host–microbe symbioses are mostly considered in terms of direct interactions between a host and a single microbe. However, bacteria, particularly those in the gut, often live in complex communities comprising many species and strains within species. Here, we established that bee gut symbionts have dynamic genomes and appear to be specialized in their ability to colonize different hosts. Within host lineages, it is plausible that members of the microbiota may have a reciprocal impact on each other’s evolutionary trajectory by sharing genes that are adaptive in their environment. To test this, we looked for evidence of horizontal gene transfer (HGT) between G. apicola and S. alvi.
Using a conservative criterion for detecting recent HGT, we found 87 S. alvi genes that had high identity to genes in G. apicola (Table S3). The distribution of predicted transfers (Fig. 5) indicates that most HGT occurs within the Apis gut community or within the Bombus community but not between Apis and Bombus microbes. This supports the hypothesis of a coevolved microbiota. The types of genes shared provide insight into the selective forces that may be shaping these gut communities. A large proportion of shared genes between Bombus-derived S. alvi and G. apicola were part of restriction modification systems, whereas Apis symbionts shared many RHS-domain proteins (Fig. 5 and Fig. S5). Restriction modification systems degrade foreign DNA and are used in defense against bacteriophages, suggesting that members of the Bombus microbiota face common viral threats. CRISPR elements, another widespread system of phage defense, are abundant in G. apicola genomes (SI Results and Fig. S6) and may act synergistically with restriction modification systems (41). RHS-domain proteins can be involved in intercellular competition through a toxin/antitoxin mechanism and may be secreted by T6SSs (42). Indeed, our strains carrying RHS proteins also possess T6SSs, and RHS and T6SS loci flank each other in wkB2T (Fig. S5). Strains that possess a version of RHS protein that is incompatible with the rest of the microbiota might be killed or otherwise excluded from that community (42). These results suggest that bee gut microbes are coevolving to interact with each other and against exogenous agents. Thus, in addition to host-specific physiological barriers to gut colonization, the gut bacterial community may also play a role in restricting the ability of strains to establish in new hosts.
Fig. 5.
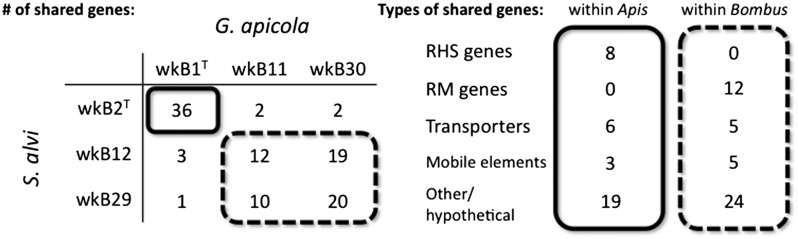
HGT in the bee gut microbiota. (Left) Number of shared genes of high identity between G. apicola and S. alvi strains is shown. Strains within the same host share more genes. (Right) Among transferred genes, several classes are overrepresented, such as RHS and restriction modification (RM) genes.
An evolutionarily dynamic, yet stably inherited, gut community could be beneficial to both bacterial symbionts and their host. The ability of the microbiota to share genes permits rapid adaptation of the entire community to changing conditions, such as exposure to antibiotics used in apiculture (43). In Bombus, gut communities from different colonies differ in their ability to confer protection against strains of a protozoan parasite, suggesting that gut communities can recognize and defend against a diverse set of invaders (44).
Conclusions
Bees are an emerging system for the study of gut microbial communities, having the advantages of a small, well-defined microbiota, as well as possessing the stability and social transmission routes of more complex (e.g., mammalian) systems. Its relative simplicity and the ability to cultivate all major members in vitro (45) make the bee gut a powerful model for investigating fundamental questions in microbial ecology, such as the origins, maintenance, and functions of strain-level variation and the dynamics of heterogeneous gut community assembly. In addition, as shown here, this system is amenable to the study of host specialization and coevolution, among other aspects of symbiosis. How symbiotic partners evolve specificity to each other is of great interest, given that symbiosis is widespread in organisms of health, economic, and environmental consequence. Considering the importance of bees as agricultural pollinators and producers, understanding the biology of their bacteria will likely have future implications in the realm of applied sciences, such as in the engineering and application of probiotics.
Materials and Methods
Genome Sequencing and Annotation.
Strains were cultured on blood heart infusion agar (11) and DNA was extracted using phenol-chloroform and sequenced on Illumina HiSeq2000 or PacBio RS platforms. Genome assembly, gap closing, gene annotation, and metabolic pathway reconstruction were done by a combination of automated and manual methods. Details are described in SI Materials and Methods.
Genomes generated in this study are deposited in the GenBank as follows: wkB1T (accession no. CP007445), wkB2T (accession no. CP007446), wkB11 (accession no. JFON00000000), wkB12 (accession no. JFZW00000000), wkB29 (accession no. JFZV00000000), and wkB30 (accession no. JFZX00000000).
Gut Colonization Experiments.
S. alvi strains wkB2T, wkB12, and wkB29 were grown for 24 h in Insectagro DS2 media (Corning, Inc.) at 35 °C and 10% (vol/vol) CO2. Pupae of A. mellifera and B. impatiens were aseptically removed from their cells and kept in a 34 °C incubation chamber. Newly eclosed bees between 0–48 h of age were starved for 3–5 h and then fed with 5 μL of 1:1 wt/wt sucrose-water containing ∼106 cells of S. alvi or a blank control. Fed bees were caged together (A. mellifera in groups of 10) or individually (B. impatiens), provisioned with 1:1 wt/wt sucrose-water and sterile (irradiated) bee pollen, and returned to incubation at 34 °C. After 5 d, bee midguts and hindguts were aseptically removed, homogenized, and diluted in Insectagro DS2 media. Dilutions were plated on heart infusion agar (Difco) supplemented with 5% sheep’s blood and incubated at 35 °C and 10% CO2 for 48 h. Colony-forming units were counted to estimate the number of cells in each gut (detection limit at 5.56 × 104 cfu). To differentiate between strains in the competition assays, colonies were restreaked to plates containing 25 μg/mL oxytetracycline; strain wkB2T is resistant to this antibiotic, whereas wkB12 and wkB29 are not (11). wkB2T prevalence was calculated from 2,272 restreaked colonies across 52 samples. Detailed protocols and statistical analyses are discussed in SI Methods and Materials.
Genome Analyses.
Orthologous genes were identified with OrthoMCL (46) using the RAST-predicted gene set for all genomes and BLAST cutoffs of e−5 and 70% match length. The dS and dN analysis was done with PAML 4 codeml (47) using maximum likelihood and the F81 model. Genome-wide dS and dN values (Table S2) were obtained from concatenated, codon-aligned orthologs; this procedure was also repeated to obtain values for each ortholog pair (Fig. 2C and Fig. S4A). To detect horizontally transferred genes, a BLASTn search of all G. apicola genes against all S. alvi genes (and the reciprocal) was performed using a cutoff of e−50. Hits from both species were then queried against the GenBank nr database and only those with closest identity to each other were retained.
Supplementary Material
Acknowledgments
We thank Eli Powell, Kelsey Bartlett, and Amanda Mancenido for technical assistance; Kim Hammond for management of honey bee hives; and Guilin Wang for PacBio sequencing support. The genome of O. hercynius CN3T was generously provided by Gottfried Wilharm and the Wellcome Trust Sanger Institute. This work was funded by Yale University and the Canadian Natural Sciences and Engineering Research Council Postgraduate Scholarship (to W.K.K.), Swiss National Science Foundation (NSF) Postdoctoral Fellowships 140157 and 147881 (to H.K.) and 135986 (to P.E.), European Molecular Biology Organization Fellowship ALTF 1317-2011 (to P.E.), and US NSF Dimensions of Biodiversity Awards 1046153 and 1415604 (to N.A.M.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. CP007445, CP007446, JFON00000000, JFZW00000000, JFZV00000000, and JFZX00000000).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1405838111/-/DCSupplemental.
References
- 1.Maltz M, Graf J. The type II secretion system is essential for erythrocyte lysis and gut colonization by the leech digestive tract symbiont Aeromonas veronii. Appl Environ Microbiol. 2011;77(2):597–603. doi: 10.1128/AEM.01621-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maltz MA, Weiss BL, O’Neill M, Wu Y, Aksoy S. OmpA-mediated biofilm formation is essential for the commensal bacterium Sodalis glossinidius to colonize the tsetse fly gut. Appl Environ Microbiol. 2012;78(21):7760–7768. doi: 10.1128/AEM.01858-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McFall-Ngai M. Divining the essence of symbiosis: Insights from the squid-vibrio model. PLoS Biol. 2014;12(2):e1001783. doi: 10.1371/journal.pbio.1001783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oh PL, et al. Diversification of the gut symbiont Lactobacillus reuteri as a result of host-driven evolution. ISME J. 2010;4(3):377–387. doi: 10.1038/ismej.2009.123. [DOI] [PubMed] [Google Scholar]
- 5.Walter J, Britton RA, Roos S. Host-microbial symbiosis in the vertebrate gastrointestinal tract and the Lactobacillus reuteri paradigm. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4645–4652. doi: 10.1073/pnas.1000099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frese SA, et al. The evolution of host specialization in the vertebrate gut symbiont Lactobacillus reuteri. PLoS Genet. 2011;7(2):e1001314. doi: 10.1371/journal.pgen.1001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ley RE, et al. Evolution of mammals and their gut microbes. Science. 2008;320(5883):1647–1651. doi: 10.1126/science.1155725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ochman H, et al. Evolutionary relationships of wild hominids recapitulated by gut microbial communities. PLoS Biol. 2010;8(11):e1000546. doi: 10.1371/journal.pbio.1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanders JG, et al. Stability and phylogenetic correlation in gut microbiota: Lessons from ants and apes. Mol Ecol. 2014;23(6):1268–1283. doi: 10.1111/mec.12611. [DOI] [PubMed] [Google Scholar]
- 10.Martinson VG, et al. A simple and distinctive microbiota associated with honey bees and bumble bees. Mol Ecol. 2011;20(3):619–628. doi: 10.1111/j.1365-294X.2010.04959.x. [DOI] [PubMed] [Google Scholar]
- 11.Kwong WK, Moran NA. Cultivation and characterization of the gut symbionts of honey bees and bumble bees: Description of Snodgrassella alvi gen. nov., sp. nov., a member of the family Neisseriaceae of the Betaproteobacteria, and Gilliamella apicola gen. nov., sp. nov., a member of Orbaceae fam. nov., Orbales ord. nov., a sister taxon to the order ‘Enterobacteriales’ of the Gammaproteobacteria. Int J Syst Evol Microbiol. 2013;63(Pt 6):2008–2018. doi: 10.1099/ijs.0.044875-0. [DOI] [PubMed] [Google Scholar]
- 12.Koch H, Abrol DP, Li J, Schmid-Hempel P. Diversity and evolutionary patterns of bacterial gut associates of corbiculate bees. Mol Ecol. 2013;22(7):2028–2044. doi: 10.1111/mec.12209. [DOI] [PubMed] [Google Scholar]
- 13.Moran NA, Hansen AK, Powell JE, Sabree ZL. Distinctive gut microbiota of honey bees assessed using deep sampling from individual worker bees. PLoS ONE. 2012;7(4):e36393. doi: 10.1371/journal.pone.0036393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koch H, Schmid-Hempel P. Socially transmitted gut microbiota protect bumble bees against an intestinal parasite. Proc Natl Acad Sci USA. 2011;108(48):19288–19292. doi: 10.1073/pnas.1110474108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Engel P, Martinson VG, Moran NA. Functional diversity within the simple gut microbiota of the honey bee. Proc Natl Acad Sci USA. 2012;109(27):11002–11007. doi: 10.1073/pnas.1202970109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinson VG, Moy J, Moran NA. Establishment of characteristic gut bacteria during development of the honeybee worker. Appl Environ Microbiol. 2012;78(8):2830–2840. doi: 10.1128/AEM.07810-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frye SA, Nilsen M, Tønjum T, Ambur OH. Dialects of the DNA uptake sequence in Neisseriaceae. PLoS Genet. 2013;9(4):e1003458. doi: 10.1371/journal.pgen.1003458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Volkmann M, et al. Orbus hercynius gen. nov., sp. nov., isolated from faeces of wild boar, is most closely related to members of the orders ‘Enterobacteriales’ and Pasteurellales. Int J Syst Evol Microbiol. 2010;60(Pt 11):2601–2605. doi: 10.1099/ijs.0.019026-0. [DOI] [PubMed] [Google Scholar]
- 19.Smokvina T, et al. Lactobacillus paracasei comparative genomics: Towards species pan-genome definition and exploitation of diversity. PLoS ONE. 2013;8(7):e68731. doi: 10.1371/journal.pone.0068731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Douillard FP, et al. Comparative genomic and functional analysis of 100 Lactobacillus rhamnosus strains and their comparison with strain GG. PLoS Genet. 2013;9(8):e1003683. doi: 10.1371/journal.pgen.1003683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michener CD. The Social Behavior of the Bees: A Comparative Study. Cambridge, MA: Harvard Univ Press; 1974. [Google Scholar]
- 22.Leonhardt SD, Blüthgen N. The same, but different: Pollen foraging in honeybee and bumblebee colonies. Apidologie (Celle) 2012;43:449–464. [Google Scholar]
- 23.Engel P, Kwong WK, Moran NA. Frischella perrara gen. nov., sp. nov., a gammaproteobacterium isolated from the gut of the honeybee, Apis mellifera. Int J Syst Evol Microbiol. 2013;63(Pt 10):3646–3651. doi: 10.1099/ijs.0.049569-0. [DOI] [PubMed] [Google Scholar]
- 24.Olofsson TC, Vásquez A. Detection and identification of a novel lactic acid bacterial flora within the honey stomach of the honeybee Apis mellifera. Curr Microbiol. 2008;57(4):356–363. doi: 10.1007/s00284-008-9202-0. [DOI] [PubMed] [Google Scholar]
- 25.Bottacini F, et al. Bifidobacterium asteroides PRL2011 genome analysis reveals clues for colonization of the insect gut. PLoS ONE. 2012;7(9):e44229. doi: 10.1371/journal.pone.0044229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cowles CE, Goodrich-Blair H. The Xenorhabdus nematophila nilABC genes confer the ability of Xenorhabdus spp. to colonize Steinernema carpocapsae nematodes. J Bacteriol. 2008;190(12):4121–4128. doi: 10.1128/JB.00123-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frese SA, et al. Molecular characterization of host-specific biofilm formation in a vertebrate gut symbiont. PLoS Genet. 2013;9(12):e1004057. doi: 10.1371/journal.pgen.1004057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sabbagh SC, Forest CG, Lepage C, Leclerc JM, Daigle F. So similar, yet so different: Uncovering distinctive features in the genomes of Salmonella enterica serovars Typhimurium and Typhi. FEMS Microbiol Lett. 2010;305(1):1–13. doi: 10.1111/j.1574-6968.2010.01904.x. [DOI] [PubMed] [Google Scholar]
- 29.Sarkar SF, Gordon JS, Martin GB, Guttman DS. Comparative genomics of host-specific virulence in Pseudomonas syringae. Genetics. 2006;174(2):1041–1056. doi: 10.1534/genetics.106.060996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu CH, Lee SM, Vanlare JM, Kasper DL, Mazmanian SK. Regulation of surface architecture by symbiotic bacteria mediates host colonization. Proc Natl Acad Sci USA. 2008;105(10):3951–3956. doi: 10.1073/pnas.0709266105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chow J, Mazmanian SK. A pathobiont of the microbiota balances host colonization and intestinal inflammation. Cell Host Microbe. 2010;7(4):265–276. doi: 10.1016/j.chom.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dulla GF, Go RA, Stahl DA, Davidson SK. Verminephrobacter eiseniae type IV pili and flagella are required to colonize earthworm nephridia. ISME J. 2012;6(6):1166–1175. doi: 10.1038/ismej.2011.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Valle J, et al. Broad-spectrum biofilm inhibition by a secreted bacterial polysaccharide. Proc Natl Acad Sci USA. 2006;103(33):12558–12563. doi: 10.1073/pnas.0605399103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Satchell KJ. Structure and function of MARTX toxins and other large repetitive RTX proteins. Annu Rev Microbiol. 2011;65:71–90. doi: 10.1146/annurev-micro-090110-102943. [DOI] [PubMed] [Google Scholar]
- 35.Burrows LL. Pseudomonas aeruginosa twitching motility: Type IV pili in action. Annu Rev Microbiol. 2012;66:493–520. doi: 10.1146/annurev-micro-092611-150055. [DOI] [PubMed] [Google Scholar]
- 36.Russell AB, Peterson SB, Mougous JD. Type VI secretion system effectors: Poisons with a purpose. Nat Rev Microbiol. 2014;12(2):137–148. doi: 10.1038/nrmicro3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aschtgen MS, Bernard CS, De Bentzmann S, Lloubès R, Cascales E. SciN is an outer membrane lipoprotein required for type VI secretion in enteroaggregative Escherichia coli. J Bacteriol. 2008;190(22):7523–7531. doi: 10.1128/JB.00945-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Linke D, Riess T, Autenrieth IB, Lupas A, Kempf VA. Trimeric autotransporter adhesins: Variable structure, common function. Trends Microbiol. 2006;14(6):264–270. doi: 10.1016/j.tim.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 39.Cariveau DP, Elijah Powell J, Koch H, Winfree R, Moran NA. Variation in gut microbial communities and its association with pathogen infection in wild bumble bees (Bombus) ISME J. 2014 doi: 10.1038/ismej.2014.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Castillo-Ramírez S, et al. The impact of recombination on dN/dS within recently emerged bacterial clones. PLoS Pathog. 2011;7(7):e1002129. doi: 10.1371/journal.ppat.1002129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dupuis MÈ, Villion M, Magadán AH, Moineau S. CRISPR-Cas and restriction-modification systems are compatible and increase phage resistance. Nat Commun. 2013;4:2087. doi: 10.1038/ncomms3087. [DOI] [PubMed] [Google Scholar]
- 42.Koskiniemi S, et al. Rhs proteins from diverse bacteria mediate intercellular competition. Proc Natl Acad Sci USA. 2013;110(17):7032–7037. doi: 10.1073/pnas.1300627110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tian B, Fadhil NH, Powell JE, Kwong WK, Moran NA. Long-term exposure to antibiotics has caused accumulation of resistance determinants in the gut microbiota of honeybees. MBio. 2012;3(6):e00377–e12. doi: 10.1128/mBio.00377-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koch H, Schmid-Hempel P. Gut microbiota instead of host genotype drive the specificity in the interaction of a natural host-parasite system. Ecol Lett. 2012;15(10):1095–1103. doi: 10.1111/j.1461-0248.2012.01831.x. [DOI] [PubMed] [Google Scholar]
- 45.Engel P, et al. Standard methods for research on Apis mellifera gut symbionts. Journal of Apicultural Research. 2013;52(4) Available at http://dx.doi.org/10.3896/IBRA.1.52.4.07. [Google Scholar]
- 46.Li L, Stoeckert CJ, Jr, Roos DS. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13(9):2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]