

Significance
The activated B-cell–like (ABC) subtype of diffuse large B-cell lymphoma (DLBCL) is an aggressive cancer that can only be cured in roughly 40% of cases. These malignant cells rely on the NF-κB signaling pathway for survival. Here, we report that genetic or pharmacologic interference with bromodomain and extraterminal domain (BET) chromatin proteins reduces NF-κB activity and ABC DLBCL viability. Unexpectedly, the mechanism involves inhibition of IκB kinase, the key cytoplasmic enzyme that activates the NF-κB pathway. The NF-κB pathway in ABC DLBCL is activated by B-cell receptor signaling, which can be blocked by the BTK kinase inhibitor ibrutinib. BET inhibitors synergized with ibrutinib to decrease growth of ABC DLBCL tumors in mouse models. BET inhibitors should be evaluated in ABC DLBCL clinical trials.
Keywords: cancer therapy, drug synergism
Abstract
In the activated B-cell–like (ABC) subtype of diffuse large B-cell lymphoma (DLBCL), NF-κB activity is essential for viability of the malignant cells and is sustained by constitutive activity of IκB kinase (IKK) in the cytoplasm. Here, we report an unexpected role for the bromodomain and extraterminal domain (BET) proteins BRD2 and BRD4 in maintaining oncogenic IKK activity in ABC DLBCL. IKK activity was reduced by small molecules targeting BET proteins as well as by genetic knockdown of BRD2 and BRD4 expression, thereby inhibiting downstream NF-κB–driven transcriptional programs and killing ABC DLBCL cells. Using a high-throughput platform to screen for drug–drug synergy, we observed that the BET inhibitor JQ1 combined favorably with multiple drugs targeting B-cell receptor signaling, one pathway that activates IKK in ABC DLBCL. The BTK kinase inhibitor ibrutinib, which is in clinical development for the treatment of ABC DLBCL, synergized strongly with BET inhibitors in killing ABC DLBCL cells in vitro and in a xenograft mouse model. These findings provide a mechanistic basis for the clinical development of BET protein inhibitors in ABC DLBCL, particularly in combination with other modulators of oncogenic IKK signaling.
The activated B-cell like subtype (ABC) of diffuse large B-cell lymphoma (DLBCL) has an aggressive clinical course compared with other DLBCL subtypes, with an overall survival of only 40% with current multidrug chemotherapies (1, 2). In recent years, detailed genetic and functional genomic analyses unveiled the key oncogenic mechanisms that sustain the aggressiveness of this subtype. Notably, all ABC DLBCLs rely on constitutive NF-κB activation for survival (3). Various oncogenic events converge on NF-κB to promote lymphomagenesis. About 10% of ABC DLBCL tumors have activating mutations affecting CARD11, a scaffolding protein required for the assembly of the CARD11–BCL10–MALT1 (CBM) complex. Mutant CARD11 proteins spontaneously generate cytoplasmic CBM aggregates that drive constitutive NF-κB activity (4). ABC DLBCL tumors with wild-type CARD11 use other mechanisms to activate NF-κB. In 20% of cases, signals emanating from the B-cell receptor (BCR) are augmented by somatically acquired mutations targeting the BCR subunits CD79A and CD79B (5). In 39% of ABC DLBCLs, NF-κB is activated by somatic mutations targeting MyD88, an adaptor protein in the Toll-like receptor (TLR) pathway (6). In normal B cells, stimulus-dependent engagement of the BCR and MyD88 pathways activates IκB kinase (IKK), which phosphorylates ΙκBα, thereby promoting its degradation and allowing NF-κB transcription factors to enter the nucleus and activate a distinctive set of target genes. By contrast, ABC DLBCL cells become addicted to constitutive activity of IKK such that its inhibition is lethal (7). Recent therapeutic efforts to target oncogenic signaling in ABC DLBCL have focused on ibrutinib, a selective inhibitor of the kinase BTK that transmits signals from the BCR to the NF-κB pathway (5).
Bromodomain and extraterminal domain (BET) proteins are a family of epigenetic adaptors that bind to acetylated chromatin and promote RNA Pol2-dependent transcription (8). Proposed mechanisms underlying BET protein transcriptional activation include recruitment of the transcriptional elongation complex pTEFb, chromatin remodeling, and histone chaperone activity (9, 10). Recently, different small-molecule inhibitors of BET proteins have been developed that competitively interfere with BET protein binding to acetylated lysine residues of histones (11, 12). These molecules are highly toxic to various cancer cell lines, including models of Burkitt lymphoma, multiple myeloma, acute myelogenous leukemia (AML), and MLL-rearranged leukemia (13–16). In multiple myeloma, AML, and Burkitt lymphoma, the toxicity of the BET inhibitor JQ1 appears to stem from its ability to regulate the oncogene MYC. Our interest in this class of small molecules was piqued by the demonstration that the BET inhibitor I-BET could down-regulate expression of NF-κB target genes in macrophages (11).
Recent studies correlated the ability of BET inhibitors to down-regulate gene expression to the presence of nearby superenhancers (SEs), which are large clusters of regulatory genomic regions that are enriched for the binding of BET proteins, mediator complex, and master regulators (17). A recent study used BET inhibition to identify SEs in DLBCL and suggested that down-modulation of lineage-specific factors was the reason behind the toxicity of these compounds (18). This study did not focus on potential differences in the mechanism of JQ1 toxicity between ABC and germinal center B-cell (GCB) DLBCL, which rely on distinct regulatory pathways for survival (2). In the present study, we investigated the ability of BET inhibitors to inhibit oncogenic NF-κB signaling in ABC DLBCL. Mechanistic studies unexpectedly uncovered a profound influence of BET proteins on cytoplasmic signaling through IKK activity in ABC DLBCL.
Results
BET Proteins Sustain ABC DLBCL Cell Survival.
We treated a panel of nine ABC DLBCL lines with the BET inhibitor JQ1 and observed a strong, dose-dependent toxicity in all lines: the average 50% inhibition concentration (IC50) ranged from 56 to 243 nM, which was on a par with the 101 nM IC50 observed with the multiple myeloma cell line LP1, which is known to be sensitive to JQ1 (Fig. 1A) (13). In addition to ABC DLBCL, JQ1 was toxic to cell line models of Burkitt lymphoma and GCB DLBCL with comparable IC50 values (Fig. S1A). Three structurally distinct BET inhibitors—JQ1, CPI203, and IBET-151—yielded similar toxicity profiles, suggesting on-target inhibition of BET proteins (Fig. S1B). JQ1 treatment of ABC DLBCL cells invoked a time- and dose-dependent increase in the apoptotic cell fraction, as measured by flow cytometry for active Caspase 3 and cleaved Parp1, suggesting that reduced cell survival is a prominent factor in JQ1 toxicity for these cells (Fig. 1B and Fig. S1C).
Fig. 1.

JQ1 toxicity in ABC DLBCL. (A) Viability of ABC DLBCL cell lines after JQ1 treatment, as assessed by MTS assay at day 4 posttreatment. The multiple myeloma line LP1 is shown as positive control for toxicity. (B) Apoptotic cells (percentage of total), as assessed by a dual intracellular-flow assay for active Caspase 3 and cleaved Parp-1, after JQ1 treatment for the indicated times. See Fig. S1C for a representative staining. (C) Shown is the fraction of live, shRNA-expressing (GFP+) cells over time after shRNA induction, compared with the day 0 preinduction values. Error bars represent SEM of triplicates.
To investigate which BET proteins are essential for ABC DLBCL survival, we evaluated the toxicity of short hairpin RNAs (shRNAs) targeting BRD2 and BRD4, the two BET family members most highly expressed in DLBCL by gene expression profiling (Fig. S1 D and E). Inducible expression of BRD2 and BRD4 shRNA constructs coexpressing GFP in ABC DLBCL cells produced a time-dependent depletion of GFP+, shRNA-expressing cells (Fig. 1C and Fig. S1F). Moreover, knockdown of BRD2 and BRD4 cooperated with JQ1 in the killing of ABC DLBCL cells, supporting the conclusion that both BRD2 and BRD4 are required to maintain ABC DLBCL viability. Ectopic expression of BRD2 and BRD4 was able to rescue ABC DLBCL cells from the toxicity of their respective shRNAs, demonstrating the specificity of the shRNAs (Fig. S1G).
Mechanism of JQ1 Toxicity in ABC DLBCLs.
To further define the molecular basis of JQ1 toxicity in ABC DLBCL, we performed ChIP-seq to identify BRD4 and RNA polymerase II (Pol2) genomic binding sites in the presence or absence of JQ1. We focused on regions in the vicinity of protein-coding genes (window from −15 kb relative to the transcriptional start site and including the entire gene body; Materials and Methods). In HBL1 ABC DLBCL cells and in LP1 multiple myeloma cells, we observed BRD4 binding near the majority of genes (HBL1: n = 13,976; LP1: n = 13,640 of 23,505 RefSeq genes; Fig. S2A). JQ1 treatment globally impaired BRD4 binding, as expected (Fig. 2A). For each cell line, we defined a set of 500 genes that had the most significant decrease in BRD4 binding following JQ1 treatment (promoter, upstream, and gene body locations combined) and performed gene set enrichment analysis using a database of gene expression signatures that reflect signaling and regulatory processes in normal and malignant B cells (19). The top enriched signatures in HBL1 cells included a set of genes highly expressed in ABC DLBCL, whereas LP1-enriched signatures reflected key transcriptional programs of multiple myeloma biology (Fig. S2B and Table S1). ChIP-seq confirmed MYC as a BRD4 target in both cell types, even though the degree of BRD4 binding to MYC and the decrease in elongating RNA Pol2 following JQ1 treatment were clearly more pronounced in the myeloma line LP1 (Fig. S2C). To extend these findings, we performed a time course analysis of gene expression changes following JQ1 treatment of HBL1 and LP1 cells. Hierarchical clustering analysis revealed that a significant fraction of the JQ1–down-regulated genes were affected in an ABC DLBCL-specific manner (Fig. 2B and Fig. S2D). Consistent with ChIP-seq results, a signature of MYC target genes was the top enriched signature among genes down-regulated in the multiple myeloma line LP1, whereas a smaller enrichment was seen in the ABC DLBCL line HBL1 (Fig. S2E). Of note, shRNA-mediated knockdown of MYC did not synergize with JQ1 in the killing of ABC DLBCL (Fig. S2F), and ectopic expression of MYC failed to rescue ABC DLBCL from the toxicity of BRD2 and BRD4 shRNAs (Fig. S2G). These findings suggest that the toxicity induced by BET protein inhibition in ABC DLBCL is likely to be multifactorial. Indeed, the enriched signatures in the HBL1 ABC DLBCL included different gene sets that define the transcriptional output of critical ABC DLBCL signaling pathways, reflecting oncogenic MYD88 signaling (MYD88 signatures), chronic active BCR signaling (BCR signatures), and constitutive expression of NF-κB target genes (NFkB signatures) (Fig. 2C; see Table S2 for details). Genes specifically down-regulated by JQ1 in ABC DLBCL included IL6 and IL10, two bona fide NF-κB targets that promote malignancy via autocrine JAK/STAT signaling (20).
Fig. 2.

Mechanism of JQ1 toxicity in ABC DLBCLs. (A) Heat maps of BRD4 ChIP-seq in HBL1 and LP1 cells, after 3-h treatment with either DMSO or 500 nM JQ1. For each cell line, heat maps were ranked based on BRD4 occupancy in the DMSO sample and peaks were centered on a ±2-kb window from their apex. See Materials and Methods for details. (B) Genes down-regulated by JQ1 in the HBL1 ABC DLBCL line were selected (less than −1 log2 fold change, at least two out of four time points), and heat maps generated to compare the effect of JQ1 treatment among HBL1 and LP1 cells. (C) Top enriched signatures among genes down-regulated by JQ1 in the HBL1 cells were selected. Enrichment ratios are shown for both HBL1 and LP1 cells. See Table S2 for a detailed signature definition. (D) Relative activity of an NF-κB–dependent luciferase reporter in the indicated ABC DLBCL lines treated overnight (16 h) with either DMSO, JQ1, or the IKKβ inhibitor MLN120b. (E) Relative activity of an NF-κB–dependent luciferase reporter in HBL1 and TMD8 cells, after induction of either control shRNA or BRD4 shRNA for the indicated time points. (F) Top enriched signatures among genes down-regulated after both BRD2 and BRD4 knockdown were selected. Enrichment ratios are shown for shBRD2, shBRD4, and shBDR2+shBRD4 combined analyses. See Table S3 for details. Error bars represent SEM of triplicates. For signature enrichment analyses, error bars represent an estimate of the SE. See Materials and Methods for details.
JQ1 treatment resulted in dose-dependent decrease of NF-κB activity in four different ABC DLBCLs lines, as assessed by an NF-κB–dependent luciferase reporter (Fig. 2D). The magnitude of NF-κB inhibition was similar to that produced by treatment with MLN120b, a specific IKKβ inhibitor (7). Similarly, induction of a BRD4 shRNA resulted in time-dependent decrease in NF-κB–dependent transcription (Fig. 2E). Finally, gene expression profiling performed after BRD2 or BRD4 knockdown confirmed strong inhibition of MYD88, BCR, and NF-κB-related gene signatures, whereas only a modest enrichment for MYC target genes was observed (Fig. 2F and Table S3).
BET Protein Inhibition Attenuates IKKβ Signaling in ABC DLBCL.
In our effort to dissect the effect of JQ1 on the NF-κB pathway, we observed a strong decrease in phosphorylated IKKβ (p-IKK) following JQ1 treatment in four different ABC DLBCL lines, indicating inhibition of IKK activity (Fig. 3A). As expected, no detectable p-IKK was observed in GCB DLBCL lines that do not rely on constitutive NF-κB for survival. Consistent with decreased IKK signaling, accumulation of IκBα was observed in all of the ABC DLBCL lines treated with JQ1, whereas no effects on IκBα protein levels were observed in GCB DLBCL lines. Two structurally distinct BET inhibitors, JQ1 and IBET-151, stabilized IκBα in ABC DLBCL lines to a similar extent, suggesting that this effect was due to on-target inhibition of BET protein function (Fig. 3B).
Fig. 3.
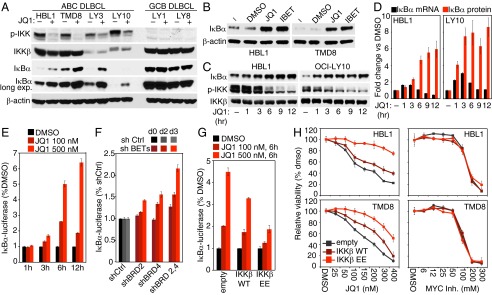
JQ1 affects NF-κB signaling in ABC DLBCLs. (A) ABC DLBCL and GCB DLBCL cell lines were treated for 16 h with either DMSO or JQ1 (500 nM). Whole-cell lysates were analyzed by Western blot. (B) ABC DLBCL cell lines were either left untreated or treated for 16 h with DMSO, 500 nM JQ1, or 500 nM I-BET 151. Whole-cell lysates were analyzed by Western blot. (C) ABC DLBCL cell lines were treated with 500 nM JQ1 for the indicated times. Whole-cell lysates were analyzed by Western blot. (D) ABC DLBCL cell lines were treated with 500 nM JQ1 for the indicated times. IκBα mRNA levels where analyzed by TaqMan Q-PCR. IκBα protein levels were analyzed in parallel by Western blot. (E) Relative IκBα-luciferase activity in TMD8 cells, after treatment with DMSO, 100 nM JQ1, or 500 nM JQ1 for the indicated times. (F) Relative IκBα-luciferase activity in TMD8 cells. Cells were infected with the indicated shRNAs, selected, and then induced for the indicated times before measuring the IκBα-luciferase activity. (G) Relative IκBα-luciferase activity in TMD8 cells. Cells were infected with the indicated expression vectors, selected, and then induced for 2 d before a 6-h JQ1 treatment. (H) HBL1 and TMD8 ABC DLBCL cell lines were infected with either empty, IKKβ WT, or IKKβ EE expression vectors, selected, and preinduced for 2 d before seeding for MTS assay. Shown is the relative cell viability at day 4 after drug treatment. Error bars represent SEM of triplicates, except for D, OCI-LY10 cells, n = 2.
JQ1 treatment caused a time-dependent accumulation of total IκBα beginning after 1 h (Fig. 3 C and D). Quantitative PCR for IκBα mRNA revealed an approximately twofold to threefold induction beginning after 1 h of JQ1 treatment, but this effect waned by 6 h (Fig. 3D). Notably, IκBα protein levels increased more dramatically (approximately fivefold to eightfold) following JQ1 treatment and were maintained at later time points (9–12 h) when IκBα mRNA levels had returned to baseline. During the same time course, p-IKK levels began to decrease detectably at 3 h and continued to drop at later time points. Thus, although the induction of IκBα mRNA may contribute to the rise of IκBα protein at early time points, we surmised that the continued rise in IκBα protein at later times could be due to a decrease in IKKβ activity.
To measure IKKβ activity quantitatively, we used a reporter system in which IκBα is fused in frame to firefly luciferase (7). Inhibition of IKKβ activity causes an increase in luciferase activity, due to increased stability of the fusion reporter. JQ1 treatment induced a time- and dose-dependent stabilization of the IκBα-luciferase (Fig. 3E), as did knockdown of BRD2, BRD4, or both BET proteins together (Fig. 3F). To investigate further the role of IKK inhibition in JQ1-induced toxicity, we examined the effects of ectopic expression of wild-type IKKβ or of a dominant-active IKKβ isoform (IKKβ EE). As expected, dominant-active IKKβ decreased IκBα-luciferase levels in ABC DLBCL cells (Fig. S3 A and B). When these cells were treated with JQ1, the dominant-active IKKβ EE isoform blunted the rise in IκBα-luciferase levels, whereas wild-type IKKβ had only a modest effect (Fig. 3G). Consistent with these findings, the dominant-active IKKβ EE isoform was able to partially rescue ABC DLBCL lines from JQ1 toxicity (Fig. 3H, Left). This effect was not due to a general prosurvival effect of dominant active IKKβ because this isoform was unable to rescue ABC DLBCL cells from the toxicity of the MYC/MAX dimerization inhibitor 10058-F4 (21) (Fig. 3H, Right). Together, these data suggest that a major aspect of JQ1 toxicity in ABC DLBCL is its ability to attenuate IKKβ and NF-κB activity.
JQ1 Synergizes with Ibrutinib in Killing ABC DLBCL Lines.
Given the ability of JQ1 to inhibit IKKβ, we explored whether it would synergize with other drugs to kill ABC DLBCL cells. To this end, we used a high-throughput drug screening platform to test pairs of compounds at various doses to identify synergistic combinations (22). We interrogated a library of 466 targeted agents that are either approved or are in early stages of development for cancer therapy. In the initial screen, we combined in a matrix format six serial dilutions of JQ1 with six serial dilutions of each library compound, and measured the effect on viability of the TMD8 ABC DLBCL line after 48 h. JQ1–drug interactions were ranked according to the “excess over the highest single agent” (HSA) method, a standard method used to discover synergistic combinations (23) (Fig. S4A, full screen results are available at https://tripod.nih.gov/matrix-client/rest/matrix/blocks/222/table). The highest scoring JQ1 interacting agent was SPC-839 (NCGC00161703), a quinazoline IKKβ inhibitor (24). Other high-ranking compounds included the BTK kinase inhibitor ibrutinib (#4) as well as multiple inhibitors of the PI (3) kinase/mTOR pathway. A secondary 10 × 10 matrix screen confirmed strong synergistic toxicity of JQ1 plus SPC-839 (Fig. 4A) or ibrutinib (Fig. 4B), as evidenced by isobologram analysis of viability data. Moreover, these drug combinations synergized in the induction of apoptosis, as assessed by a luminescence-based caspase 3/7 activation assay (Fig. S4 B and C).
Fig. 4.

JQ1 synergize with ibrutinib in killing ABC DLBCLs. (A) Combination responses for the BET inhibitor JQ1 and the IKKβ inhibitor SPC-839 as judged by a 10 × 10 viability matrix. TMD8 cells viability was measured at 48 h posttreatment by CellTiterGlo assay (Left). An isobologram analysis of viability data are shown on the Right. (B) Same as in A, but the combination between JQ1 and the BTK inhibitor ibrutinib was analyzed. (C) The indicated cell lines were treated with a large concentration range of BTK inhibitor ibrutinib, in combination with either DMSO or three different concentration of JQ1. Shown is the relative viability of each combination subset, as assessed by MTS assay at day 4 posttreatment. (D) Relative IκBα-luciferase activity in the ABC DLBCL TMD8 cell line. Cells were treated overnight (16 h) with indicated concentration of ibrutinib, in combination with either DMSO or three different concentration of JQ1. Error bars represent SEM of triplicates.
We expanded the analysis of JQ1/ibrutinib synergy to a panel of ABC DLBCL, GCB DLBCL, and multiple myeloma lines, all of which were killed by JQ1 treatment alone (Fig. S4D). To illustrate the synergistic effects of this combination, we normalized data from 96-h viability assays relative to the effect of JQ1 as a single agent (Fig. 4C). In these plots, a shift of the JQ1 dose-response curves toward a lower IC50 compared with the ibrutinib-only curves indicates greater toxicity of the combination than either agent alone. We observed synergistic toxicity of the JQ1/ibrutinib combination in three ABC DLBCL lines but not in the GCB DLBCL or myeloma lines (Fig. 4C). Moreover, JQ1 and ibrutinib synergized in blocking IKK activity, as indicated by stabilization of the IκBα-luciferase reporter (Fig. 4D).
Efficacy of BET Inhibition Plus Ibrutinib in an ABC DLBCL Xenograft Model.
We next investigated the effect of BET protein inhibition in a xenograft model of ABC DLBCL, using the BET inhibitor CPI203, which is structurally related to JQ1 and has a similar toxicity spectrum (Fig. S1B), but has a better bioavailability profile in mice (25). Like JQ1, CPI203 synergized with ibrutinib in the killing of the ABC DLBCL lines in vitro (Fig. S5A). As a single agent, CPI203 produced significant, but not complete, inhibition of DLBCL tumor growth when mice were injected i.p. twice daily (5 mg/kg) (Fig. 5A). To explore the potential synergy between CPI203 and ibrutinib in this model, we used ibrutinib at a dose (2 mg/kg) that caused only minimal tumor inhibition (Fig. 5B, blue line). In combination with 5 mg/kg CPI203, this dose of ibrutinib produced full inhibition of tumor growth, illustrating the potency of this combination (Fig. 5B, green line). BET protein inhibition by CPI203 was well tolerated by the mice, causing only a modest weight loss, and the combination with ibrutinib did not cause additional toxicity (Fig. S5B). Notably, the observed in vivo toxicity of CPI203 and ibrutinib was associated with inhibition of NF-κB activation, as evidenced by decreased mRNA expression levels of two NF-κB target genes, IL6 and IL10, and the drug combination reduced the expression of these cytokines to almost undetectable levels in all tumors (Fig. 5C).
Fig. 5.

Combination therapy in a xenograft model of ABC DLBCL. (A) Human TMD8 ABC DLBCL cells were established as a s.c. tumor in NOD/SCID mice and treated by i.p. injection with either vehicle or BET Inhibitor CPI203 (5 mg/kg). Mice were treated for 21 d, and tumor growth was measured as a function of tumor volume. (B) Human TMD8 ABC DLBCL cells were established as a s.c. tumor in CB17 SCID mice and treated by i.p. injection with vehicle, BET inhibitor CPI203 (5 mg/kg), BTK inhibitor ibrutinib (2 mg/kg), or with a combination of CPI203 and ibrutinib. Mice were treated for 12 d, and tumor growth was measured as a function of tumor volume. (C) Total RNA was extracted from xenograft biopsies. Human IL6, IL10, and MYC mRNA levels were measured by TaqMan RT Q-PCR. For each gene, the relative abundance was calculated vs. B2M values, arbitrarily set to 1,000. Error bars represent SEM. (A) n = 3 mice per group. (B) n = 9 mice per group. (C) n = 7 for vehicle group, n = 8 for combination group, and n = 9 for single-arm groups.
Discussion
Our mechanistic analysis of BET inhibitor toxicity in ABC DLBCL revealed a role for BET proteins in promoting oncogenic kinase activity of IKK, thereby sustaining ABC DLBCL viability. These observations were reinforced by our high-throughput combinatorial drug screen, which revealed strong synergy between the BET inhibitor JQ1 and multiple signal transduction inhibitors that target signaling pathways leading to IKK activation in ABC DLBCL. Most notable was the combination of JQ1 with the BCR pathway inhibitor ibrutinib, which synergized with BET inhibitors to kill ABC DLBCL cells and to prevent ABC DLBCL xenograft growth.
Our initial hypothesis was that BET inhibitors would be toxic to ABC DLBCL cells due to a direct negative effect on the transcription of NF-κB target genes, based on previous work suggesting this mechanism of action in macrophages (11). However, our analysis unexpectedly uncovered an influence of nuclear BET proteins on cytoplasmic IKK signaling. JQ1 down-regulated several gene expression signatures in ABC DLBCL that are induced by oncogenic signaling pathways that activate IKK. The effect of JQ1 on gene expression signatures of MYC activity, which is a major reason for its toxicity in myeloma cells, was much less pronounced in the HBL1 ABC DLBCL lines than in the LP1 myeloma line. JQ1 also inhibited the expression of many other genes in ABC DLBCL, for example, the lymphoid-restricted coactivator OCA-B (POU2AF1), as previously noted (18). Notably, ectopic expression of a dominant-active isoform of IKKβ partially rescued ABC DLBCL cells from JQ1 toxicity, whereas ectopic provision of MYC did not. Thus, although JQ1 has pleiotropic effects on gene expression in ABC DLBCL, its effect on IKK activity and NF-κB–dependent transcription contributes significantly to its toxicity.
Several non-mutually exclusive mechanisms may account for this observation. One model would suggest that BET inhibitors reduced IKK activity in ABC DLBCL by decreasing the transcription of genes encoding signaling proteins that function upstream of IKK. The BET proteins are highly enriched in transcriptional regulatory domains known as SEs, which control expression of key tissue-specifying genes (26). Indeed, SEs have been identified in DLBCL cells near genes encoding lymphoid-restricted transcription factors (18). ABC DLBCLs rely on constitutive signaling emanating from the BCR or from MYD88, and each pathway uses a cascade of kinases and signaling adapters to activate IKK. By gene expression profiling, the expression of genes encoding signaling proteins in the BCR or MyD88 pathways declined modestly or not at all following JQ-1 treatment (Fig. S6 A and B). Nonetheless, the combined transcriptional down-regulation of these genes might still result in the observed potent inhibition of IKK activity. A second possibility is that BET proteins might act in the cytoplasm and modulate signaling by binding to acetylated proteins in the pathways upstream of IKK in ABC DLBCL. This possibility seems remote because a careful fractionation of nuclear and cytoplasmic proteins from ABC DLBCL cells revealed no more BRD4 in the cytoplasm than could be accounted for by contamination with nuclear components, as assessed by histone H3 immunoblotting (Fig. S7 A and B). A final possibility is that BET proteins may mediate “inside-out” signaling from the nucleus to the cytoplasm that modulates IKK activity. Precedent for this type of signaling comes from analysis of DNA damage-induced activation of IKK, which relies on ATM-dependent phosphorylation of nuclear IKKγ (NEMO) (27). However, an inhibitor of ATM kinase that blocks DNA damage-induced IKK activation had no effect on IKK activity or the viability of ABC DLBCL cells, demonstrating that any potential inside-out signaling in these cells is mechanistically distinct (Fig. S7 C and D).
Irrespective of mechanism, our studies have important implications for the clinical development of BET inhibitors in ABC DLBCL. Most importantly, the ability of BET inhibitors to inhibit oncogenic NF-κB activity in ABC DLBCL makes their therapeutic development for the treatment of this disease highly rational, and suggests a measureable endpoint to gauge their pharmacodynamic efficacy. BET inhibitors strongly decreased expression of the NF-κB target genes IL6 and IL10 in ABC DLBCL xenografts, raising the possibility that levels of these cytokines in the blood could be used to monitor BET inhibitor activity. Our work further argues for early evaluation of combinations involving BET inhibitors and BCR pathway inhibitors such as ibrutinib. Given the pleiotropic role of BET proteins in transcriptional regulation, BET inhibitors could have on-target side effects that might limit the doses that can be achieved clinically. By pairing a BET inhibitor with ibrutinib, lower doses might yield equivalent antitumor responses while minimizing general toxicity. On the flip side, ibrutinib has shown efficacy in ABC DLBCL, but responses are not durable in most patients, highlighting the need to develop drug combinations to improve ibrutinib efficacy (reviewed in ref. 28). Importantly, combined treatment of mice with the BET inhibitor CPI203 and ibrutinib did not cause any additional toxicity beyond the mild effect of CPI203 on animal weight, suggesting that this drug combination may be tolerated clinically.
Materials and Methods
For shRNA toxicity screen, cell lines were engineered to express an ecotropic retroviral receptor and the bacterial tetracycline repressor (6). For the high-throughput drug combination screen, viability was assessed by CellTiterGlo (Promega) (22). All animal experiments were carried out in accordance with the National Cancer Institute Animal Care and Use Committee guidelines. For a detailed explanation of all of the methods, please see SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Kathleen Meyer for help with the Gene Expression Omnibus (GEO) database submission. This research was supported by the Intramural Research Programs of the National Institutes of Health, National Cancer Institute, Center for Cancer Research, and the National Human Genome Research Institute; the Frederick National Laboratory for Cancer Research, National Institutes of Health, including Contract HHSN261200800001E and Grant U54CA143930; the Division of Preclinical Innovation, National Center for Advancing Translational Sciences; and the Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research.
Footnotes
The authors declare no conflict of interest.
Data deposition: The gene expression data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE58791). The ChIP-seq data reported in this paper have been deposited in Short Read Archive (SRA), www.ncbi.nlm.nih.gov/sra (accession no. SRP043524).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1411701111/-/DCSupplemental.
References
- 1.Alizadeh AA, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769):503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 2.Shaffer AL, 3rd, Young RM, Staudt LM. Pathogenesis of human B cell lymphomas. Annu Rev Immunol. 2012;30:565–610. doi: 10.1146/annurev-immunol-020711-075027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med. 2001;194(12):1861–1874. doi: 10.1084/jem.194.12.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lenz G, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science. 2008;319(5870):1676–1679. doi: 10.1126/science.1153629. [DOI] [PubMed] [Google Scholar]
- 5.Davis RE, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463(7277):88–92. doi: 10.1038/nature08638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ngo VN, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470(7332):115–119. doi: 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lam LT, et al. Small molecule inhibitors of IkappaB kinase are selectively toxic for subgroups of diffuse large B-cell lymphoma defined by gene expression profiling. Clin Cancer Res. 2005;11(1):28–40. [PubMed] [Google Scholar]
- 8.Florence B, Faller DV. You bet-cha: A novel family of transcriptional regulators. Front Biosci. 2001;6:D1008–D1018. doi: 10.2741/florence. [DOI] [PubMed] [Google Scholar]
- 9.Jang MK, et al. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19(4):523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 10.LeRoy G, Rickards B, Flint SJ. The double bromodomain proteins Brd2 and Brd3 couple histone acetylation to transcription. Mol Cell. 2008;30(1):51–60. doi: 10.1016/j.molcel.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nicodeme E, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468(7327):1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Filippakopoulos P, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delmore JE, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mertz JA, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci USA. 2011;108(40):16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dawson MA, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478(7370):529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zuber J, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478(7370):524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lovén J, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chapuy B, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013;24(6):777–790. doi: 10.1016/j.ccr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaffer AL, et al. A library of gene expression signatures to illuminate normal and pathological lymphoid biology. Immunol Rev. 2006;210:67–85. doi: 10.1111/j.0105-2896.2006.00373.x. [DOI] [PubMed] [Google Scholar]
- 20.Lam LT, et al. Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-kappaB pathways in subtypes of diffuse large B-cell lymphoma. Blood. 2008;111(7):3701–3713. doi: 10.1182/blood-2007-09-111948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yin X, Giap C, Lazo JS, Prochownik EV. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene. 2003;22(40):6151–6159. doi: 10.1038/sj.onc.1206641. [DOI] [PubMed] [Google Scholar]
- 22.Mathews L, et al. High-throughput combinatorial screening identifies drugs that cooperate with ibrutinib to kill ABC diffuse large B-cell lymphoma cells. Proc Natl Acad Sci USA. 2014;111(6):2349–2354. doi: 10.1073/pnas.1311846111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borisy AA, et al. Systematic discovery of multicomponent therapeutics. Proc Natl Acad Sci USA. 2003;100(13):7977–7982. doi: 10.1073/pnas.1337088100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palanki MS, et al. Structure-activity relationship studies of ethyl 2-[(3-methyl-2,5-dioxo(3-pyrrolinyl))amino]-4-(trifluoromethyl)pyrimidine-5-carboxylate: An inhibitor of AP-1 and NF-kappaB mediated gene expression. Bioorg Med Chem Lett. 2002;12(18):2573–2577. doi: 10.1016/s0960-894x(02)00517-6. [DOI] [PubMed] [Google Scholar]
- 25.King B, et al. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell. 2013;153(7):1552–1566. doi: 10.1016/j.cell.2013.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whyte WA, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153(2):307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCool KW, Miyamoto S. DNA damage-dependent NF-κB activation: NEMO turns nuclear signaling inside out. Immunol Rev. 2012;246(1):311–326. doi: 10.1111/j.1600-065X.2012.01101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Young RM, Staudt LM. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat Rev Drug Discov. 2013;12(3):229–243. doi: 10.1038/nrd3937. [DOI] [PMC free article] [PubMed] [Google Scholar]