

Significance
During proliferation, eukaryotic cells go through a defined series of phases. The primary regulators of this process are kinases paired with protein partners called cyclins. Exactly how these specific cyclin–kinase pairs orchestrate the myriad cellular events during each cell phase has been difficult to define largely because it has proven challenging to identify their substrates. Here, we describe a method to identify cyclin- and phase-specific substrates with high confidence and also to pinpoint their sites of modification. The method enabled us to identify the phosphatase Cdc14 as a substrate of a cyclin–kinase pair that acts during DNA synthesis in budding yeast and to uncover a new means by which this major antagonist of the cyclin–kinase pair is itself controlled.
Keywords: kinase substrate identification, NESKA
Abstract
In eukaryotes, cell cycle progression is controlled by cyclin/cyclin-dependent kinase (CDK) pairs. To better understand the details of this process, it is necessary to dissect the CDK’s substrate pool in a cyclin- and cell cycle stage-specific way. Here, we report a mass spectrometry-based method that couples rapid isolation of native kinase–substrate complexes to on-bead phosphorylation with heavy-labeled ATP (ATP-γ-18O4). This combined in vivo/in vitro method was developed for identifying cyclin/CDK substrates together with their sites of phosphorylation. We used the method to identify Clb5 (S-cyclin)/Cdc28 and Cln2 (G1/S-cyclin)/Cdc28 substrates during S phase in Saccharomyces cerevisiae (Cdc28 is the master CDK in budding yeast). During the work, we discovered that Clb5/Cdc28 specifically phosphorylates S429 in the disordered tail of Cdc14, an essential phosphatase antagonist of Cdc28. This phosphorylation severely decreases the activity of Cdc14, providing a means for modulating the balance of CDK and phosphatase activity.
During proliferation, a eukaryotic cell must faithfully replicate its genome and segregate the resulting genetic material equally into two progeny cells. A host of cellular events during this process must happen in a precisely ordered manner. Although it has been well established that cyclin-dependent kinases (CDKs) constitute the primary driving force that order and coordinate these cell cycle events (1–3), the detailed picture of how CDK orchestrates the myriad cellular events during cell cycle progression remains to be fully elucidated. At different phases of the cell cycle, the CDKs associate with specific cyclins. These particular cyclin/CDK pairs preferentially phosphorylate sets of largely distinct substrates (4–7). To investigate this intricate relationship among cell cycle phase, cyclin/CDKs, and their substrates, it is necessary to dissect the CDK phosphorylated substrates in a cyclin-specific and cell cycle phase-specific manner. We report here a mass spectrometry-based native enzyme–substrate complex kinase assay (NESKA) that was developed to address this challenge. NESKA combines rapid isolation of native kinase–substrate complexes with on-bead phosphorylation using heavy labeled ATP within these complexes, providing site-specific in vivo and in vitro phosphorylation information. It allows us to screen for specific cyclin-CDK substrates in a hypothesis-free manner within a chosen time window during cell cycle progression.
We applied NESKA to investigate phosphorylation substrates of Clb5/Cdc28 during S-phase in budding yeast (Cdc28 encodes the main CDK in budding yeast; Clb5 is the major S-phase cyclin that specializes in the regulation of DNA synthesis; refs. 8–10). It has been shown that CDK phosphorylation of two proteins, Sld2 and Sld3, initiates DNA replication (11–14). Here, we find that NESKA correctly identified a crucial phosphorylation site on Sld2 with Clb5/Cdc28. In addition, we discovered that Cdc14 is specifically phosphorylated by Clb5-Cdc28 during S phase. Cdc14 is an essential phosphatase that antagonizes CDK activity during mitotic exit (15–19). We demonstrate that this phosphorylation site severely inhibits Cdc14, providing a way for modulating the balance of CDK and phosphatase activity.
Results
Development of NESKA and Its Application to Clb5/Cdc28.
We developed the present mass spectrometry-based NESKA (Fig. 1A) based on affinity purification techniques optimized for studying protein–protein interactions (20–24). Importantly, at a given stage in the cell cycle, synchronized cells are rapidly injected into liquid nitrogen to preserve their state. These frozen cells are cryogenically ground into a fine powder to aid in rapid extraction and isolation of cyclin/CDK–substrate complexes. Next, we perform affinity isolation of the tagged cyclin/CDKs sufficiently rapidly and gently to preserve at least a portion of the substrate interactions. In this study, we coupled this affinity isolation procedure with a subsequent on-bead kinase assay for screening the phosphorylated substrates of a targeted kinase. To distinguish this in vitro on-bead phosphorylation from in vivo phosphorylation, we added stable isotope-labeled ATP-γ-18O4 as a cofactor. During this reaction, the affinity-captured kinase transfers the “heavy” phosphate group from ATP-γ-18O4 to interacting protein substrates; this heavy phosphate group can be readily distinguished from preexisting “light” in vivo phosphate groups by mass spectrometry. We consider such in vitro phosphorylated proteins as putative substrates of the targeted kinase because they meet two criteria: (i) they physically interact with the affinity purified kinase and (ii) they are phosphorylated by the on-bead kinase activity, which is dominated by the captured kinase. Often the same site is phosphorylated both in vitro (heavy) and in vivo (light) (Table 1 and SI Appendix, Table S1). This redundancy provides an additional layer of confidence that such sites are bona fide phosphorylation targets of the captured kinase. However, it is worth noting that the lack of a corresponding in vivo phosphorylation does not necessarily indicate that the in vitro version is an artifact because in vivo phosphorylation levels are generally lower than the corresponding in vitro levels and can fall below MS detection limits and/or are not picked up for tandem MS sequencing. Indeed, taking advantage of high resolution extracted ion chromatograms (XIC) (25), we identified several coeluting light in vivo phosphopeptides that were only sequenced by tandem MS in their heavy in vitro form (SI Appendix, Fig. S1 and Table 1).
Fig. 1.
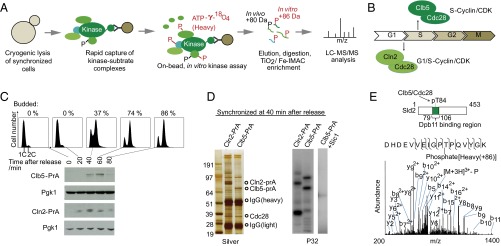
Mass spectrometric identification of cyclin-specific CDK substrates at a defined stage of the cell cycle. Scheme of the NESKA methodology (A); roles of Clb5/Cdc28 and Cln2/Cdc28 in cell cycle regulation (B); and synchronous cell cycle progression after release from G1/S arrest was monitored by FACS analysis, bud index, and cyclin expression (C). Pgk1 was used as a loading control for Western blotting. The 40-min time point after release was determined to be S phase. (D) Affinity isolation of Clb5/Cdc28 and Cln2/Cdc28 complexes with binding substrates. Cln2, Clb5, and Cdc28 were the major bands except for the contaminant IgG bands on the silver-stained gel. Autoradiography with ATP-γ-32P showed that cyclin-CDK retained activity on the beads. Recombinant Sic1 abolished the kinase activity of Clb5/Cdc28 beads. (E) Sld2 was phosphorylated at T84, within the Dpb11 binding region, by Clb5/Cdc28 during S phase, as determined by NESKA.
Table 1.
Identified substrates of Clb5/Cdc28 and Cln2/Cdc28 during S phase
| Protein | Major cellular process | Phospho-site | In vitro | In vivo |
| Clb5/Cdc28 | ||||
| Sic1 | CDK inhibition | T [5] | + | + |
| T [173] | + | + | ||
| S [191] | + | + | ||
| YHR097C | ND | T [76] | + | — |
| Cdc14 | Mitotic exit | S [429] | + | — |
| Boi2 | Polar growth | S [652] | + | — |
| Sld2 | DNA replication | T [84] | + | — |
| Far1 | CDK inhibition | S [114] | + | +(MS) |
| S [174] | — | + | ||
| Bik1 | Microtubule association | S [117] | + | — |
| Net1 | Mitotic exit | S [252] | + | +(MS) |
| T [676] | + | +(MS) | ||
| Sgn1 | mRNA metabolism | T [42] | + | — |
| Tif4632 | Translation initiation | S [474] | + | +(MS) |
| Ubp3 | Protein deubiquitination | S [695] | + | — |
| Def1 | RNAPII degradation | T [249] | + | — |
| S [260] | + | + | ||
| S [646] | +(MS) | + | ||
| Mbf1 | Transcription regulation | S [143] | + | — |
| Cln2/Cdc28 | ||||
| Whi5 | Repressor of G1 transcription | S [154], S [156] | + | +(MS) |
| Srl3 | Whi5 paralog | T [5], T [14] | + | — |
| T [14] | + | +(MS) | ||
| T [40] | + | +(MS) | ||
| S [212] | + | +(MS) | ||
| Grr1 | G1/S transition | S [199] | + | + |
| S [1101] | + | + | ||
| Ace2 | G1 transcription | S [80] | + | + |
| S [392], T [384] | + | + | ||
| T [422] | + | — | ||
| S [709], S [714] | — | + | ||
| Gyp1 | Vesicle transportation | S [107] | + | — |
| YPL014W | ND | S [345] | + | + |
| Gic2 | Cell polarity | S [334], S [337] | + | — |
| Def1 | RNAPII degradation | T [249] | + | — |
| S [260] | — | + | ||
| S [646] | +(MS) | + | ||
Substrates with codon adaptation index <0.3 (∼20,000 copies per cell) are shown. An extended Table S1 is included in SI Appendix. +, identified by tandem MS; +(MS), identified through coeluted heavy/light peak pair in high resolution extracted ion chromatogram.
There are several other technical features that also contribute to the capacity of the NESKA development for identifying kinase substrates and their sites of phosphorylation: (i) Reduced phosphatase levels on beads and potent phosphatase inhibitors enable higher levels of in vitro phosphorylation; and (ii) phosphopeptide enrichment significantly reduces the background and facilitates MS detection (26).
Here, we applied NESKA to specifically targeted Clb5/Cdc28 substrates during S phase (Fig. 1B), providing both cyclin specificity and cell cycle phase specificity. To achieve such specificity, we arrested yeast cell cycle progression at the G1/S boundary with α-factor then removed the factor to allow synchronous progression. FACS analysis and bud index counting showed that the 40-min time point represents the S phase because DNA is observed to be replicating from one to two copies and ∼40% of the yeast cells have budded (Fig. 1C). This finding is also consistent with the expression profile of Clb5, which begins to be expressed in S phase (Fig. 1C).
Cln2 was chosen as a control for studying cyclin specificity because it plays a different role than Clb5 in cell cycle control (3). Although Cln2 is also present during S phase, it appears slightly earlier (Fig. 1C). To test whether our affinity-captured Clb5/Cdc28 retains kinase activity, we carried out 32P autoradiography experiments on the affinity beads (Fig. 1D). We observed that Clb5/Cdc28 was active toward coprecipitated interacting proteins and introduced a different phosphorylation pattern compared with Cln2/Cdc28 control (Fig. 1D). Importantly, after incubation with recombinant CDK inhibitor Sic1, the on-bead kinase activity was abolished (Fig. 1D). Because Sic1 is highly specific toward Clb/Cdc28 (27), this result demonstrates that the observed on-bead kinase activity can be fully attributed to Clb5/Cdc28. This result further implies that heavy phosphopeptides generated on bead in vitro are produced by Clb5/Cdc28.
As evidence of the specificity of NESKA, we identified Sld2 as a substrate of Clb5/Cdc28 but not of the Cln2/Cdc28 control (Fig. 1E and Table 1). It has also been reported that Sld2 is an essential substrate of Clb5/Cdc28 and that its phosphorylation regulates DNA replication during S phase (11, 12). In particular, the presently identified Thr84 site has been demonstrated to play a significant role in this phosphorylation-dependent regulation by promoting Dpb11 binding (12). Because Sld2 is of low abundance [codon adaptation index = 0.132 (28); experimental copy number 700 per cell (29)], its identification by NESKA demonstrates the specificity and high sensitivity of this method.
Additional evidence for the specificity of NESKA was provided by our observation of a doubly phosphorylated Whi5 peptide isolated with Cln2/Cdc28 (SI Appendix, Fig. S2) but not with Clb5/Cdc28. Whi5 is an inhibitor of the transcription factor SBF that activates G1/S transition genes (30, 31). Upon phosphorylation, it is excluded from the nucleus and SBF is relieved from inhibition (30, 31). Our identification of phosphorylated Whi5 is in good agreement with the known role of Cln2/Cdc28 in the G1/S transition (32). Furthermore, these phosphorylated sites lie within in the nuclear export signal region of Whi5 and have been shown to regulate its cellular localization by Cln/Cdc28 (33). In addition, Srl3, a Whi5 paralog, was also identified as a Cln2-Cdc28 substrate (Table 1).
By comparing the substrate list of Clb5/Cdc28 and Cln2/Cdc28 (Table 1), we found that different substrates prefer different cyclins, even during the same stage of the cell cycle (here S phase), supporting the idea that cyclin specificity is an important part of cell cycle regulation (3). Also, the diverse cellular processes in which these substrates are involved point to the broad regulatory function of cyclin-CDKs (3). For example, we identified Gic2, an effector of Cdc42, as a substrate for Cln2/Cdc28. This observation is consistent with Cln2’s role in promoting budding (34). Meanwhile, we identified a microtubule association protein Bik1 as a substrate for Clb5/Cdc28, which could provide a linkage from Clb5 to mitotic spindle formation (3).
Cdc14 Is Phosphorylated by Clb5/Cdc28 During S Phase at Ser429.
Among the identified Clb5/Cdc28 substrates (Table 1), we became particularly interested in Cdc14 because it is an essential phosphatase that antagonizes CDK activity during mitotic exit and resets the phosphorylation state of CDK substrates (15–19). During S phase, the majority of Cdc14 is sequestered in the nucleolus by interaction with Net1 (35, 36) (Net1 was also among the identified substrates of Clb5-Cdc28; Table 1). It has been reported that mislocalization of Cdc14 during S phase interferes with DNA synthesis (37).
Our identified in vitro phosphorylation site, S429, is in the middle of Cdc14’s disordered tail and distant from the catalytic PTP motif in its primary sequence (Fig. 2A). Because no in vivo phosphorylation was detected for S429 in our NESKA assay, we infer that the in vivo phosphopeptide is present at levels below the detection limit of the mass spectrometer. To investigate this inference, we genomically tagged and affinity purified Cdc14 from synchronized S-phase yeast culture. The coprecipitation of nucleolus components (Net1, Rpa190, and Rpa135) suggests that Cdc14 is properly localized in the nucleolus in S phase (Fig. 2B). In vivo phosphorylated S429 was identified from this enriched sample and the tandem MS pattern of the corresponding phosphopeptide closely resembled the in vitro derived version (Fig. 2B). The intensity of in vivo phosphorylated peptide is ∼7% of its unmodified counterpart (SI Appendix, Fig. S3). Moreover, time-resolved quantitative MS analysis indicates that this phosphorylation emerges in S phase, persists through M phase, and is gradually removed thereafter (Fig. 2C and SI Appendix, Fig. S4) (mutation of a nearby Clb5 substrate recognition “RXL” site doesn’t appear to change the pS429 level). Taken together, these results suggest that S429 of Cdc14 is phosphorylated by Clb5/Cdc28 during S phase.
Fig. 2.
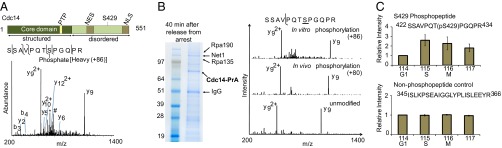
Cdc14 is phosphorylated at S429 by Clb5/Cdc28 during S phase. (A) Cdc14-S429 was phosphorylated by Clb5/Cdc28 in the NESKA assay. S429 is located in the disordered tail of Cdc14. *, neutral loss of phosphate and H2O molecules. #, neutral loss of two H2O molecules. (B) S429 of Cdc14 was confirmed to be phosphorylated in vivo during S phase. Cdc14 was enriched by affinity purification. The tandem MS patterns of unmodified, in vivo phosphorylated, and in vitro phosphorylated peptides are similar. (C) Cell-cycle regulated S429 phorsphorylation was quantified by iTRAQ (66). The error bars represent SEM of quantified peptides from three iTRAQ experiments. S429 is phosphorylated during S phase and gradually dephosphorylated.
Phosphorylation of Cdc14 on Ser429 Inhibits Its Phosphatase Activity.
To investigate the functional role that this phosphorylation site may play, we introduced a phosphomimetic mutation in which S429 is replaced by two glutamic acids. It has been reported that two glutamic acids can provide a good mimic of the phosphate group because they also present two negative charges at physiological pH (38) (Fig. 3A). Because S429 is located in the disordered tail, we posit that one extra residue should cause little structural distortion. In addition, phosphorylation sites in disordered regions have often been associated with local charge effects (39).
Fig. 3.
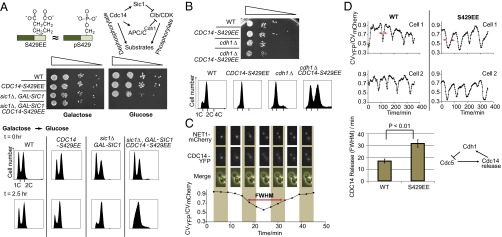
Phosphomimetic mutant Cdc14-S429EE shows phenotypes consistent with inhibited phosphatase activity. (A) Cdc14-S429EE mutation is synthetically lethal with sic1∆. Expression of Sic1 under GAL promoter was turned off by switching from galactose to glucose medium. The double mutant was arrested during S phase, as FACS analysis showed. (B) Cdc14-S429EE has a slowed growth rate when combined with cdh1∆. FACS analysis showed increased 4C population and indicated defective cytokinesis. (C) Quantification of Cdc14 release from the nucleolus during mitotic exit by time-course microscopy (43). Nucleolus was visualized by its structural component Net1 fused with mCherry. Cdc14 was fused with YFP. The coefficient of variation ratio of YFP and mCherry signals correlates with Cdc14 release. FWHM, full width at half maximum. (D) Cdc14-S429EE has prolonged Cdc14 release compared with wild-type control. The mean FWHM of Cdc14 release is 17 min for wild type and 32 min for S429EE mutant. Errors bar represent SEM of measurements from a total of 12 cell cycles from five different cells. P value was calculated with Student t test.
This S429EE mutation by itself does not cause obvious effects on growth, but it induces a significant decrease of the 1C population as measured by FACS (Fig. 3B, Lower). When combined with sic1 deletion, the double mutant is no longer viable (Fig. 3A). In addition, we determined that the S429EE mutation has no effect on Cdc14 abundance or its binding pattern to other proteins (SI Appendix, Fig. S8). These findings suggest that the phosphomimetic S429EE mutation inhibits Cdc14 activity. Such inhibited Cdc14 activity would make the CDK inhibitor Sic1 essential for counteracting CDK activity. FACS analysis showed that the double mutant is arrested during S phase (Fig. 3A, Lower). Sic1 and Cdc14 both promote replication origin relicensing during mitotic exit by antagonizing Clb-Cdk activity, which may partially account for the arrest of the CDC14-S429EE sic1∆ double mutant in S phase (40, 41). These results suggest that S429 phosphorylation of Cdc14 inhibits its activity. However, we did not observe a clear phenotype associated with the nonphosphorylatable Cdc14 S429A mutation (SI Appendix, Fig. S5), perhaps because there are redundant mechanisms that are sufficient to control Cdc14. Net1 inhibits Cdc14 through nucleolar sequestration, and Clb5/Cdc28 phosphorylation of Cdc14 could conceivably be an additional safety mechanism in case of failure of nucleolar sequestration. To test this hypothesis, we combined the Cdc14 nonphosphorylatable mutation with net1 deletion. The low percentage at which we obtained double mutants (two of nine spores carrying net1∆) tentatively suggests a combinatorial effect. However, the severely compromised fitness of these double mutants, and that of the net1∆ parent strain, prevented detailed phenotypic characterization (SI Appendix, Fig. S5, Lower).
We also investigated the combination of Cdc14 S429EE mutation and cdh1 deletion. Cdh1 promotes mitotic cyclin degradation and inhibits CDK activity during late mitosis (3). This double mutant showed a slower growth rate (Fig. 3B), and defects in finishing cytokinesis correctly, indicated by the accumulation of cells with four copies of DNA (Fig. 3B). These results are consistent with the idea that S429EE inhibits Cdc14 activity, thereby making other CDK inhibiting mechanisms stand out. Consistent phenotypes were observed when S429EE is combined with cdc15 temperature-sensitive mutation (SI Appendix, Fig. S6). Cdc15 is part of the mitotic exit network and promotes the release of Cdc14 (42). It is lethal to combine S429EE mutation with a cdc15 mutation that compromises Cdc15 activity.
Next, we studied how the phosphomimetic mutation affects the cellular localization of Cdc14. During most of the cell cycle, Cdc14 is sequestered in the nucleolus by Net1 (35, 36) and only released upon mitotic exit to counteract CDK activity. We monitored this dynamic release of Cdc14 through a previously established quantitative fluorescence microscopic assay (43) (Fig. 3C). In this assay, the release of Cdc14 is assessed through the colocalization of Cdc14-YFP and Net1-mCherry, a nucleolus marker. We observed prolonged Cdc14-S429EE release compared with wild type (Fig. 3D)—this finding agrees well with a model wherein Cdc14 regulates its oscillated release autonomously through a negative feedback loop (43–45).
Finally, we developed an MS-based phosphatase assay with stable isotope-labeled phosphopeptide substrates (Fig. 4A). These substrates are derived from a known Cdc14 target Cdh1-pS239 (46). This assay enabled us to directly compare the phosphatase activity of the phosphomimetic S429EE mutant with wild type. To determine whether inserting an additional residue will distort the Cdc14 structure, thus impact its activity, we also included a S429AA mutant as a control (Fig. 4A). Using purified bacterially expressed phosphatases (SI Appendix, Fig. S7), we found that the S429EE mutant has severely inhibited phosphatase activity, whereas the S429AA control has comparable activity to wild type (Fig. 4B). Furthermore, we determined that in vitro phosphorylation of S429 by recombinant Clb5-Cdc28 also inhibits Cdc14 activity (SI Appendix, Fig. S9). The modest inhibitory effect (∼15%) is consistent with the in vitro phosphorylation level (∼25%) of Cdc14 and its self-dephosphorylation (SI Appendix, Fig. S9). [The self-dephosphorylation may suggest a positive feedback mechanism. A similar phosphoregulation has been reported on fission yeast Cdc14/Clp1 during late mitosis (47).] In conclusion, both our genetic and biochemical studies demonstrate that phosphorylation of S429 on Cdc14 inhibits its enzymatic activity.
Fig. 4.

Recombinant Cdc14-S429EE shows severely inhibited activity in an MS-based phosphatase assay. (A) The Cdc14-S429EE mutant dephosphorylates the Cdh1 phosphopeptide at a much slower rate compared with wild-type and S429AA controls. (B) The MS peak heights of the phosphopeptides were used for quantification. Error bars represent SD of three experiments.
Discussion
Kinases are central to the regulation of many cellular pathways. A variety of methods have been successfully coupled with MS-based proteomics to identify substrates of targeted kinases (48–50). These methods include, for example, marking substrates with ATP analogs that are used by a target mutant kinase but not by the endogenous kinases (51, 52); purification of substrates by affinity isolation with the kinase of interest as bait (53–55); alterations in phosphorylation patterns induced by specific kinase inhibition or stimulation (39, 56–61); and in vitro kinase assays (57, 62, 63). Here, we present a method for identifying substrates of cyclin/CDKs that combines rapid affinity isolation with an on-bead kinase assay, providing both in vitro and in vivo phosphorylation-site information with high specificity and sensitivity.
Our affinity purification-based approach (NESKA) requires that putative substrates physically interact with the kinase, which greatly improves the chance of identifying direct substrates compared with the specific kinase inhibition/stimulation approach. Furthermore, carrying out the in vitro kinase assay within native kinase–substrate complexes minimizes artifacts associated with in vitro kinase assays on cell lysates and with the use of excess recombinant kinase. Because NESKA uses heavy ATP, it can identify both in vitro and in vivo phosphorylation of the same site, which significantly increases the confidence of the identification.
Our NESKA assay is suited to dissect the substrate pool of CDK, whose function depends on the cell cycle and properties of the cyclin components. We identified the substrate pool of Clb5/Cdc28 during S phase and compared them with the substrates of Cln2/Cdc28 during the same S phase. In agreement with the literature (11, 12), we discovered a phosphorylation site on Sld2 with Clb5/Cdc28. This site is located in a region that is known to promote Dpb11 interaction upon phosphorylation. In contrast, we did not detect this site with Cln2/Cdc28 but found Whi5 phosphorylation sites within its NES region. These results demonstrate that NESKA is capable of obtaining cyclin-specific information at a defined phase of the cell cycle. Although NESKA identifies a variety of specific substrates (Table 1 and SI Appendix, Table S1) and a majority (∼70%; 19 of 26) of them are consistent with previous proteomic scale studies (39, 53, 64, 65) (SI Appendix, Table S2), other authentic substrates are likely missing because the affinity purification step biases our assay toward the higher-affinity targets. We envisage that in vivo cross-linking may further extend our coverage of substrates to those that associate with lower affinities. Nevertheless, the low overlap between the putative substrates of Clb5/Cdc28 versus Cln2/Cdc28 shown in Table 1 (just Def1), clearly demonstrates differential substrate preference of these cyclin/CDKs even within the same time within S phase (3).
One previously unknown Clb5/Cdc28 substrate within our list, Cdc14, drew our particular interest because of its central role as an essential cell cycle phosphatase. We also identified its binding partner Net1 as a Clb5/Cdc28 substrate. In budding yeast, Cdc14 is sequestered in the nucleolus through binding with Net1 during most of the cell cycle (35, 36). It is briefly released during early anaphase and mitotic exit to antagonize CDK activity before being sequestered again. Because a Cdc14 mutation that prevents its sequestration is synthetically lethal with clb5 deletion (37), it was of interest to investigate the function of our identified Cdc14-S429 phosphorylation by Clb5/Cdc28 during S phase.
The phosphomimetic S429EE mutation showed clear phenotypes, including synthetic lethality with sic1 deletion and cytokinesis defects with cdh1 deletion—consistent with inhibition of the Cdc14 phosphatase activity. These observations reinforce the role of Cdc14 in antagonizing CDK activity in budding yeast. In addition, S429EE shows prolonged Cdc14 release, supporting the idea that Cdc14 activity negatively regulates its own release (43–45). Our MS-based phosphatase assay compared the S429EE mutant to the wild-type enzyme, clearly demonstrating that the mutant has severely inhibited activity.
In summary, we have described an MS-based assay (NESKA) that should be readily implementable in many different organisms to screen substrates for a given kinase. The assay provides combined in vitro and in vivo phosphorylation site information in a subunit-specific and time-resolved manner. With ever-increasing MS sensitivity, steadily improving phosphopeptide enrichment techniques and ongoing efforts to stabilize enzyme–substrate complex in vivo, even more complete phosphorylation information should be obtainable by NESKA in the future.
Methods
The MATa bar1∆ strains with PrA-tagged cyclins in their native loci were arrested with α-factor. Arrested yeast cells were washed and resuspended in fresh YPD to resume synchronous cell cycle progression. The 40-min time point after the release was determined to be the S phase by FACS analysis and bud index. Cells from this time point were harvested and frozen in liquid nitrogen as droplets. These frozen droplets were cryogenically ground into fine powder with a planetary ball mill (PM-100; Retsch) to thoroughly lyse the cells while preserving labile protein complexes. The cell powder (typically 2–4 g) was resuspended in ice-cold solubilization buffer with brief sonication. The resulting cell lysate solution was cleared by a short 2-min centrifugation at 27,000 × g. Supernatant was further filtered through 1.6-µm GF/A filter (Whatman). The cleared lysate was incubated with IgG magnetic beads (1 mL/1 mg beads) for 30 min at 4 °C on a rotator. After incubation, the beads were washed and resuspended in kinase reaction buffer [3 µL/1 mg beads; 20 mM Hepes, 10 mM sodium chloride, 1 mM magnesium chloride, and 1× phosphatase inhibitor mixture (Thermo) at pH 7.4] supplemented with 1 mM ATP-γ-18O4 (heavy ATP; Cambridge Isotope Laboratories) (for P32 autoradiography, 0.5 µCi ATP-γ-32P was added). After 30-min incubation at room temperature, the reaction solution was removed and saved. The bound proteins were eluted, combined with the saved reaction solution, and digested either in gel or in solution. To enrich phosphopetides, either a TiO2 or Fe-IMAC column was used.
More experimental details are available in SI Appendix, SI Methods.
Supplementary Material
Acknowledgments
We thank Kresti Pecani, Erica Jacobs, Lu Bai, Kelly Molloy, Beatrix Ueberheide, and Wenzhu Zhang for assistance and suggestions. This work was supported by the National Institutes of Health Grants 2P41GM103314 and GM103362 (to B.T.C.) and GM47238 and GM78153 (to F.R.C.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1409666111/-/DCSupplemental.
References
- 1.Morgan DO. Cyclin-dependent kinases: Engines, clocks, and microprocessors. Annu Rev Cell Dev Biol. 1997;13:261–291. doi: 10.1146/annurev.cellbio.13.1.261. [DOI] [PubMed] [Google Scholar]
- 2.Murray AW. Recycling the cell cycle: Cyclins revisited. Cell. 2004;116(2):221–234. doi: 10.1016/s0092-8674(03)01080-8. [DOI] [PubMed] [Google Scholar]
- 3.Bloom J, Cross FR. Multiple levels of cyclin specificity in cell-cycle control. Nat Rev Mol Cell Biol. 2007;8(2):149–160. doi: 10.1038/nrm2105. [DOI] [PubMed] [Google Scholar]
- 4.Cross FR, Yuste-Rojas M, Gray S, Jacobson MD. Specialization and targeting of B-type cyclins. Mol Cell. 1999;4(1):11–19. doi: 10.1016/s1097-2765(00)80183-5. [DOI] [PubMed] [Google Scholar]
- 5.Ikui AE, Archambault V, Drapkin BJ, Campbell V, Cross FR. Cyclin and cyclin-dependent kinase substrate requirements for preventing rereplication reveal the need for concomitant activation and inhibition. Genetics. 2007;175(3):1011–1022. doi: 10.1534/genetics.106.068213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loog M, Morgan DO. Cyclin specificity in the phosphorylation of cyclin-dependent kinase substrates. Nature. 2005;434(7029):104–108. doi: 10.1038/nature03329. [DOI] [PubMed] [Google Scholar]
- 7.Kõivomägi M, et al. Dynamics of Cdk1 substrate specificity during the cell cycle. Mol Cell. 2011;42(5):610–623. doi: 10.1016/j.molcel.2011.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Epstein CB, Cross FR. CLB5: A novel B cyclin from budding yeast with a role in S phase. Genes Dev. 1992;6(9):1695–1706. doi: 10.1101/gad.6.9.1695. [DOI] [PubMed] [Google Scholar]
- 9.Schwob E, Nasmyth K. CLB5 and CLB6, a new pair of B cyclins involved in DNA replication in Saccharomyces cerevisiae. Genes Dev. 1993;7(7A):1160–1175. doi: 10.1101/gad.7.7a.1160. [DOI] [PubMed] [Google Scholar]
- 10.Donaldson AD. The yeast mitotic cyclin Clb2 cannot substitute for S phase cyclins in replication origin firing. EMBO Rep. 2000;1(6):507–512. doi: 10.1093/embo-reports/kvd108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masumoto H, Muramatsu S, Kamimura Y, Araki H. S-Cdk-dependent phosphorylation of Sld2 essential for chromosomal DNA replication in budding yeast. Nature. 2002;415(6872):651–655. doi: 10.1038/nature713. [DOI] [PubMed] [Google Scholar]
- 12.Tak YS, Tanaka Y, Endo S, Kamimura Y, Araki H. A CDK-catalysed regulatory phosphorylation for formation of the DNA replication complex Sld2-Dpb11. EMBO J. 2006;25(9):1987–1996. doi: 10.1038/sj.emboj.7601075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanaka S, et al. CDK-dependent phosphorylation of Sld2 and Sld3 initiates DNA replication in budding yeast. Nature. 2007;445(7125):328–332. doi: 10.1038/nature05465. [DOI] [PubMed] [Google Scholar]
- 14.Zegerman P, Diffley JF. Phosphorylation of Sld2 and Sld3 by cyclin-dependent kinases promotes DNA replication in budding yeast. Nature. 2007;445(7125):281–285. doi: 10.1038/nature05432. [DOI] [PubMed] [Google Scholar]
- 15.Jaspersen SL, Charles JF, Tinker-Kulberg RL, Morgan DO. A late mitotic regulatory network controlling cyclin destruction in Saccharomyces cerevisiae. Mol Biol Cell. 1998;9(10):2803–2817. doi: 10.1091/mbc.9.10.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Visintin R, et al. The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol Cell. 1998;2(6):709–718. doi: 10.1016/s1097-2765(00)80286-5. [DOI] [PubMed] [Google Scholar]
- 17.Zachariae W, Schwab M, Nasmyth K, Seufert W. Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science. 1998;282(5394):1721–1724. doi: 10.1126/science.282.5394.1721. [DOI] [PubMed] [Google Scholar]
- 18.Bloom J, et al. Global analysis of Cdc14 phosphatase reveals diverse roles in mitotic processes. J Biol Chem. 2011;286(7):5434–5445. doi: 10.1074/jbc.M110.205054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwab M, Lutum AS, Seufert W. Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell. 1997;90(4):683–693. doi: 10.1016/s0092-8674(00)80529-2. [DOI] [PubMed] [Google Scholar]
- 20.Cristea IM, Williams R, Chait BT, Rout MP. Fluorescent proteins as proteomic probes. Mol Cell Proteomics. 2005;4(12):1933–1941. doi: 10.1074/mcp.M500227-MCP200. [DOI] [PubMed] [Google Scholar]
- 21.Fernandez-Martinez J, et al. Structure-function mapping of a heptameric module in the nuclear pore complex. J Cell Biol. 2012;196(4):419–434. doi: 10.1083/jcb.201109008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, et al. Escherichia coli condensin MukB stimulates topoisomerase IV activity by a direct physical interaction. Proc Natl Acad Sci USA. 2010;107(44):18832–18837. doi: 10.1073/pnas.1008678107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oeffinger M, et al. Comprehensive analysis of diverse ribonucleoprotein complexes. Nat Methods. 2007;4(11):951–956. doi: 10.1038/nmeth1101. [DOI] [PubMed] [Google Scholar]
- 24.Zhong Y, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11(4):468–476. doi: 10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hites R, Biemann K. Computer evaluation of continuously scanned mass spectra of gas chromatographic effluents. Anal Chem. 1970;42:855–860. [Google Scholar]
- 26.Dunn JD, Reid GE, Bruening ML. Techniques for phosphopeptide enrichment prior to analysis by mass spectrometry. Mass Spectrom Rev. 2010;29(1):29–54. doi: 10.1002/mas.20219. [DOI] [PubMed] [Google Scholar]
- 27.Schwob E, Böhm T, Mendenhall MD, Nasmyth K. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell. 1994;79(2):233–244. doi: 10.1016/0092-8674(94)90193-7. [DOI] [PubMed] [Google Scholar]
- 28.Sharp PM, Li WH. The codon Adaptation Index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15(3):1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghaemmaghami S, et al. Global analysis of protein expression in yeast. Nature. 2003;425(6959):737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 30.Costanzo M, et al. CDK activity antagonizes Whi5, an inhibitor of G1/S transcription in yeast. Cell. 2004;117(7):899–913. doi: 10.1016/j.cell.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 31.de Bruin RA, McDonald WH, Kalashnikova TI, Yates J, 3rd, Wittenberg C. Cln3 activates G1-specific transcription via phosphorylation of the SBF bound repressor Whi5. Cell. 2004;117(7):887–898. doi: 10.1016/j.cell.2004.05.025. [DOI] [PubMed] [Google Scholar]
- 32.Skotheim JM, Di Talia S, Siggia ED, Cross FR. Positive feedback of G1 cyclins ensures coherent cell cycle entry. Nature. 2008;454(7202):291–296. doi: 10.1038/nature07118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taberner FJ, Quilis I, Igual JC. Spatial regulation of the start repressor Whi5. Cell Cycle. 2009;8(18):3010–3018. [PubMed] [Google Scholar]
- 34.Yoshida S, Pellman D. Plugging the GAP between cell polarity and cell cycle. EMBO Rep. 2008;9(1):39–41. doi: 10.1038/sj.embor.7401142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shou W, et al. Exit from mitosis is triggered by Tem1-dependent release of the protein phosphatase Cdc14 from nucleolar RENT complex. Cell. 1999;97(2):233–244. doi: 10.1016/s0092-8674(00)80733-3. [DOI] [PubMed] [Google Scholar]
- 36.Visintin R, Hwang ES, Amon A. Cfi1 prevents premature exit from mitosis by anchoring Cdc14 phosphatase in the nucleolus. Nature. 1999;398(6730):818–823. doi: 10.1038/19775. [DOI] [PubMed] [Google Scholar]
- 37.Bloom J, Cross FR. Novel role for Cdc14 sequestration: Cdc14 dephosphorylates factors that promote DNA replication. Mol Cell Biol. 2007;27(3):842–853. doi: 10.1128/MCB.01069-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Strickfaden SC, et al. A mechanism for cell-cycle regulation of MAP kinase signaling in a yeast differentiation pathway. Cell. 2007;128(3):519–531. doi: 10.1016/j.cell.2006.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holt LJ, et al. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science. 2009;325(5948):1682–1686. doi: 10.1126/science.1172867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noton E, Diffley JF. CDK inactivation is the only essential function of the APC/C and the mitotic exit network proteins for origin resetting during mitosis. Mol Cell. 2000;5(1):85–95. doi: 10.1016/s1097-2765(00)80405-0. [DOI] [PubMed] [Google Scholar]
- 41.Lengronne A, Schwob E. The yeast CDK inhibitor Sic1 prevents genomic instability by promoting replication origin licensing in late G(1) Mol Cell. 2002;9(5):1067–1078. doi: 10.1016/s1097-2765(02)00513-0. [DOI] [PubMed] [Google Scholar]
- 42.Bardin AJ, Amon A. Men and sin: What’s the difference? Nat Rev Mol Cell Biol. 2001;2(11):815–826. doi: 10.1038/35099020. [DOI] [PubMed] [Google Scholar]
- 43.Lu Y, Cross FR. Periodic cyclin-Cdk activity entrains an autonomous Cdc14 release oscillator. Cell. 2010;141(2):268–279. doi: 10.1016/j.cell.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Visintin C, et al. APC/C-Cdh1-mediated degradation of the Polo kinase Cdc5 promotes the return of Cdc14 into the nucleolus. Genes Dev. 2008;22(1):79–90. doi: 10.1101/gad.1601308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Manzoni R, et al. Oscillations in Cdc14 release and sequestration reveal a circuit underlying mitotic exit. J Cell Biol. 2010;190(2):209–222. doi: 10.1083/jcb.201002026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bremmer SC, et al. Cdc14 phosphatases preferentially dephosphorylate a subset of cyclin-dependent kinase (Cdk) sites containing phosphoserine. J Biol Chem. 2012;287(3):1662–1669. doi: 10.1074/jbc.M111.281105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wolfe BA, McDonald WH, Yates JR, 3rd, Gould KL. Phospho-regulation of the Cdc14/Clp1 phosphatase delays late mitotic events in S. pombe. Dev Cell. 2006;11(3):423–430. doi: 10.1016/j.devcel.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 48.Guo M, Huang BX. Integration of phosphoproteomic, chemical, and biological strategies for the functional analysis of targeted protein phosphorylation. Proteomics. 2013;13(3-4):424–437. doi: 10.1002/pmic.201200274. [DOI] [PubMed] [Google Scholar]
- 49.Johnson SA, Hunter T. Kinomics: Methods for deciphering the kinome. Nat Methods. 2005;2(1):17–25. doi: 10.1038/nmeth731. [DOI] [PubMed] [Google Scholar]
- 50.Hattori S, Iida N, Kosako H. Identification of protein kinase substrates by proteomic approaches. Expert Rev Proteomics. 2008;5(3):497–505. doi: 10.1586/14789450.5.3.497. [DOI] [PubMed] [Google Scholar]
- 51.Blethrow JD, Glavy JS, Morgan DO, Shokat KM. Covalent capture of kinase-specific phosphopeptides reveals Cdk1-cyclin B substrates. Proc Natl Acad Sci USA. 2008;105(5):1442–1447. doi: 10.1073/pnas.0708966105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morandell S, et al. QIKS—Quantitative identification of kinase substrates. Proteomics. 2010;10(10):2015–2025. doi: 10.1002/pmic.200900749. [DOI] [PubMed] [Google Scholar]
- 53.Archambault V, et al. Targeted proteomic study of the cyclin-Cdk module. Mol Cell. 2004;14(6):699–711. doi: 10.1016/j.molcel.2004.05.025. [DOI] [PubMed] [Google Scholar]
- 54.Pagliuca FW, et al. Quantitative proteomics reveals the basis for the biochemical specificity of the cell-cycle machinery. Mol Cell. 2011;43(3):406–417. doi: 10.1016/j.molcel.2011.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Breitkreutz A, et al. A global protein kinase and phosphatase interaction network in yeast. Science. 2010;328(5981):1043–1046. doi: 10.1126/science.1176495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kettenbach AN, et al. Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci Signal. 2011;4(179):rs5. doi: 10.1126/scisignal.2001497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xue L, et al. Sensitive kinase assay linked with phosphoproteomics for identifying direct kinase substrates. Proc Natl Acad Sci USA. 2012;109(15):5615–5620. doi: 10.1073/pnas.1119418109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smolka MB, Albuquerque CP, Chen SH, Zhou H. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc Natl Acad Sci USA. 2007;104(25):10364–10369. doi: 10.1073/pnas.0701622104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matsuoka S, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316(5828):1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 60.Kosako H, et al. Phosphoproteomics reveals new ERK MAP kinase targets and links ERK to nucleoporin-mediated nuclear transport. Nat Struct Mol Biol. 2009;16(10):1026–1035. doi: 10.1038/nsmb.1656. [DOI] [PubMed] [Google Scholar]
- 61.Blagoev B, Ong SE, Kratchmarova I, Mann M. Temporal analysis of phosphotyrosine-dependent signaling networks by quantitative proteomics. Nat Biotechnol. 2004;22(9):1139–1145. doi: 10.1038/nbt1005. [DOI] [PubMed] [Google Scholar]
- 62.Troiani S, et al. Searching for biomarkers of Aurora-A kinase activity: Identification of in vitro substrates through a modified KESTREL approach. J Proteome Res. 2005;4(4):1296–1303. doi: 10.1021/pr050018e. [DOI] [PubMed] [Google Scholar]
- 63.Huang SY, Tsai ML, Chen GY, Wu CJ, Chen SH. A systematic MS-based approach for identifying in vitro substrates of PKA and PKG in rat uteri. J Proteome Res. 2007;6(7):2674–2684. doi: 10.1021/pr070134c. [DOI] [PubMed] [Google Scholar]
- 64.Ubersax JA, et al. Targets of the cyclin-dependent kinase Cdk1. Nature. 2003;425(6960):859–864. doi: 10.1038/nature02062. [DOI] [PubMed] [Google Scholar]
- 65.Ptacek J, et al. Global analysis of protein phosphorylation in yeast. Nature. 2005;438(7068):679–684. doi: 10.1038/nature04187. [DOI] [PubMed] [Google Scholar]
- 66.Ross PL, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3(12):1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]