

Significance
Neurodegenerative diseases are debilitating conditions that result from degeneration of the nervous system causing symptoms including ataxia and/or dementia. Calmodulin-binding transcription activator 1 (CAMTA1) is a transcription factor enriched in the brain with the highest levels of expression in the cerebellar granular layer and Purkinje cells, midbrain, pons, and hippocampus. When CAMTA1 is deleted from the nervous system of mutant mice, we observe a clear loss of Purkinje cells and reduced cerebellar size. Thus, we conclude that CAMTA1 plays a predominant role in the maturation and survival of cerebellar neurons rather than in the initial development of these cells. The neurological abnormalities associated with CAMTA1-mutant mice allow mechanistic analysis of these abnormalities and disease modeling.
Keywords: CAMTA2, dimerization, palindromic DNA, neural genes
Abstract
Members of the calmodulin-binding transcription activator (CAMTA) family of proteins function as calcium-sensitive regulators of gene expression in multicellular organisms ranging from plants to humans. Here, we show that global or nervous system deletion of CAMTA1 in mice causes severe ataxia with Purkinje cell degeneration and cerebellar atrophy, partially resembling the consequences of haploinsufficiency of the human CAMTA1 locus. Gene-expression analysis identified a large collection of neuronal genes that were dysregulated in the brains of CAMTA1-mutant mice, and elucidation of a consensus sequence for binding of CAMTA proteins to DNA revealed the association of CAMTA-binding sites with many of these genes. We conclude that CAMTA1 plays an essential role in the control of Purkinje cell function and survival. CAMTA1-mutant mice provide a model to study the molecular mechanisms of neurodegenerative diseases and for screening potential therapeutic interventions for such disorders.
First identified in plants as regulators of stress and cold-sensitive changes in gene expression, calmodulin-binding transcription activator (CAMTA) transcription factors subsequently have been implicated in signal-dependent gene expression in metazoans ranging from fruit flies to humans (1–7). CAMTA proteins share a series of functional motifs, including a CG-1 DNA-binding motif, a TIG domain implicated in DNA binding and dimerization of transcription factors, calmodulin-binding IQ motifs, and ankyrin repeats that mediate oligomerization (1).
There are two mammalian CAMTA genes, CAMTA1 and CAMTA2, which share extensive homology and partially overlapping expression patterns. CAMTA1 and 2 are highly expressed in the brain with lower levels of expression in the heart (7). Previously, we reported that CAMTA2 plays an important role in pathological cardiac remodeling, which occurs in response to aberrant calcium signaling (7). CAMTA2-KO mice are viable and do not show obvious phenotypes but are resistant to cardiac stress (7). The functions of CAMTA1 have not been analyzed in mice. However, haploinsufficiency of CAMTA1, caused by mutations and genomic rearrangements, has been shown to cause sporadic and familial nonsyndromic intellectual deficiency and childhood ataxia in humans (8, 9). The mechanistic basis of these abnormalities has not been defined. CAMTA1 also has been implicated as a tumor suppressor in neuroblastoma and glioblastoma (10, 11).
To explore the functions of CAMTA1 in vivo, we generated mice with a conditional loss-of-function allele. Global deletion or deletion in the nervous system of CAMTA1 resulted in severe ataxia, accompanied by Purkinje cell degeneration, decreased cerebellar size, and disruption of neuronal gene expression. Through an unbiased determination of the optimal CAMTA DNA-binding site, we found CAMTA-binding sites to be associated with a high percentage of dysregulated neuronal genes in the cerebellum of CAMTA1-mutant mice. We conclude that CAMTA1 is an essential regulator of Purkinje cell function and survival. CAMTA1-mutant mice provide a model for understanding the mechanistic basis of ataxias and cerebellar function.
Results
Generation of CAMTA1-KO Mice.
The mouse CAMTA1 gene (OTTMUSG00000010309 in Vega Genome Browser), located on chromosome 4, contains at least 20 exons with multiple splice variants, spanning more than 800 kb of DNA. To investigate the functions of CAMTA1 in vivo, we generated a conditional mutant allele of the gene by inserting a loxP site in the intron upstream of exon 9 and a FLP recognition target (FRT)-neo-FRT-LoxP cassette immediately downstream of exon 9 through homologous recombination in ES cells (Fig. S1A). Cre-mediated recombination of this allele introduced a frame-shift mutation after amino acid 284 of CAMTA1, eliminating the majority of the functional domains and creating a loss-of-function allele. Based on mutagenesis studies of conserved domains in CAMTA2 (7), the CG-1 domain contained within the residual portion of the mutated CAMTA1 gene would not be expected to function as a dominant-negative mutant. Targeting of CAMTA1 in ES cells and mice was confirmed by Southern blot analysis and PCR (Fig. S1 B and C).
Male mice heterozygous for the conditional CAMTA1fl/+ allele were bred to females carrying a CAG-Cre transgene, which mediates gene excision at the zygote stage (12), generating CAMTA1+/− offspring. Mice heterozygous for the CAMTA1+/− allele appeared phenotypically normal, although we did not subject these mice to detailed phenotypic analysis. CAMTA1+/− mice were intercrossed to generate CAMTA1−/− (CAMTA1-KO) mice. Homozygosity of the CAMTA1-KO allele resulted in postnatal death before age 4 wk (Table S1). CAMTA1-KO mice were severely runted and displayed ataxia (Fig. S2A and Movie S1). Although CAMTA1 has been suggested to function as a tumor suppressor within neurons and glial cells (10, 11), we never observed any tumors in CAMTA1-KO mice before death.
Neuronal Expression of CAMTA1.
Previous studies reported strong expression of CAMTA1 in the cerebellum, as well as in the hippocampi and olfactory bulbs of embryonic and neonatal mice (9). Consistent with these findings, in situ hybridization of adult brain sections with CAMTA1-specific probes showed that CAMTA1 transcripts were highly enriched in the cerebellum (Fig. 1A). Within the cerebellum, CAMTA1 expression was enriched in Purkinje cells, and the granule layer and was absent in KO mice (Fig. 1B).
Fig. 1.

Expression of CAMTA1 in mouse cerebellum. In situ hybridization analysis of sagittal section of adult mouse brain (2 mo old) of (A) WT and (B) CAMTA1-nKO shows expression pattern of CAMTA1 in the cerebellum. The boxed region in the upper panels is enlarged in the lower panels. Signal shows expression of CAMTA1 in the WT Purkinje cells. Minimal expression of CAMTA1 is seen in the Purkinje cell layer of CAMTA1-nKO mice. Red arrows indicate the localization of Purkinje cells. (Scale bars: Upper, 600 μm; Lower, 100 μm.)
Motor Abnormalities of CAMTA1fl/fl, Nestin-Cre Mice.
To determine the cellular basis of the neurological phenotype of mice with global deletion of CAMTA1, we generated CAMTA1 deletions by crossing CAMTA1fl/fl mice with mice bearing a Nestin-Cre transgene, which is expressed throughout the nervous system (13). RT-PCR analysis of total RNA from cerebella of CAMTA1fl/fl, Nestin-Cre mice (referred to as “CAMTA1-nKO mice”) and in situ hybridization of CAMTA1-nKO brain sections revealed that CAMTA1 was deleted successfully (Fig. 1B and Fig. S1D).
Consistent with the ataxia observed in mice with global deletion of CAMTA1, specific deletion of CAMTA1 in the nervous system with Nestin-Cre resulted in severe ataxia as assessed by multiple motor tasks and general observation (Fig. 2 and Fig. S2 B–D). The ataxic phenotype was observed in every CAMTA1-nKO mouse over the age of 3 mo. Nestin-Cre transgenic mice containing WT CAMTA1 alleles did not display ataxia (13). CAMTA1-nKO mice were able to breed at young ages but did so at a reduced frequency and did not show reduced survival relative to CAMTA1fl/fl (control) littermates up to age 1 y. The severe runting observed upon global deletion of CAMTA1 was less pronounced and was seen in only a subset of nKO mice, suggesting that CAMTA1 has additional functions beyond the cell types in which Nestin-Cre is active.
Fig. 2.
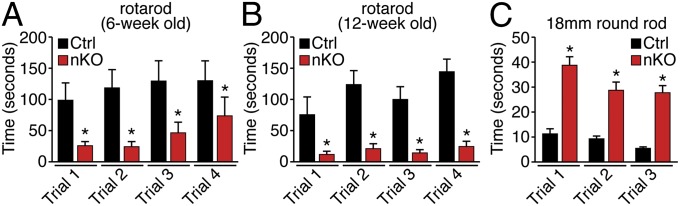
Motor deficits of CAMTA1-nKO mice. (A and B) Motor coordination performance on a rotarod with slow acceleration from 5–25 rpm over 5 min was assessed in CAMTA1fl/fl control and CAMTA1-nKO mice at age 6 (A) and 12 (B) wk. n = 6 control mice, n = 5 CAMTA1-nKO mice; *P < 0.05. Results from four independent trials are shown. (C) CAMTA1-nKO and CAMTA1fl/fl control mice at age 6 wk were evaluated for motor deficits using the beam walk test. The time needed to traverse an 18-mm round wooden rod was recorded in multiple trials. n = 6 control mice, n = 4 CAMTA1-nKO mice; *P < 0.001. Results from three independent trials are shown.
To assess motor coordination quantitatively, nKO mice and CAMTA1fl/fl littermates were subjected to rotarod testing at 6 and 12 wk of age. In multiple trials, CAMTA1-nKO mice displayed a significant reduction in time spent on the rotarod (Fig. 2 A and B). By age 6 wk CAMTA1-nKO mice also experienced highly significant delays in the time needed to cross balance beams of multiple sizes on several successive attempts (Fig. 2C and Fig. S2 B and C). The loss of motor coordination was evident in all CAMTA1-nKO mice and increased in a rostro-caudal gradient as depicted in Movie S2.
We also deleted the conditional CAMTA1 allele using a Pax3-Cre knockin allele, which is expressed in embryonic somites and the dorsal neural tube, where precursors of the central nervous system originate (14). These mice also developed severe ataxia by 6 wk of age. Therefore we focused our subsequent studies on the nKO mice.
Progressive Cerebellar Atrophy and Degeneration of Purkinje Cells in CAMTA1-nKO Mice.
To delineate the cellular basis of the ataxic phenotype of CAMTA1-nKO mice, adult brains from CAMTA1fl/fl and CAMTA1-nKO mice were analyzed. The behavioral results of the accelerating rotarod tests strongly suggested impairments in motor coordination; however, the severity of the phenotype may mask striatal/dopamine-dependent motor skill learning. No gross changes in neural architecture were observed in the forebrain or midbrain of nKO mice (Fig. 3A). Therefore, we focused our attention on cerebellar structure. Deletion of CAMTA1 did not alter the normal cerebellar architecture at 2–3 wk of age but caused a dramatic loss of Purkinje cells and a decrease in cerebellar size at age 3 mo, concomitant with the loss of motor coordination (Fig. 3 A and B). The loss of Purkinje cells, readily visualized by immunostaining for Calbindin (Fig. 3 C and D), also correlated with a significant decrease in the width of the molecular and granular layers within the cerebellum of CAMTA1-nKO mice (Fig. 3 E–H).
Fig. 3.

Degeneration of Purkinje cells and cerebellar atrophy in CAMTA1-nKO mice. (A) Gross morphology of brains from adult WT and CAMTA1-nKO mice. (B) H&E stain of sagittal sections of brain from 3-wk-old (Upper) and adult (Lower) WT and CAMTA1-nKO mice. (Scale bar, 400 μm.) (C) Calbindin-D28K immunohistochemical staining (red) of Purkinje cells in the cerebellum of 6-mo-old CAMTA1-nKO and littermate control mice. Purkinje cells are uniformly organized in the cerebellum of the control mouse but are reduced and disarrayed in the cerebellum of the CAMTA1-nKO mouse. Nuclei are stained with DAPI (blue). gl, granular layer. (Scale bar, 100 μm.) (D) Quantification of the size of calbindin-positive Purkinje cells in the cerebellum (n = 3). (E and F) H&E staining of sagittal sections of the brain from CAMTA1-nKO and littermate control mice at age 2–3 wk (E) and 3 mo (F). White arrows point to Purkinje cells. (Scale bar, 400 μm Upper, 40 μm Lower.) n = 6; *P < 0.05. gl, granular layer; ml, molecular layer. (G and H) Quantification of the Purkinje cells of CAMTA1-nKO and littermate control mice at age 2–3 wk (G) and 3 mo (H). Purkinje cell number is not reduced in CAMTA1-nKO mice at age 2–3 wk but is decreased at age 3 mo along with reductions in the width of the molecular layer and granular layer. Data are presented as mean ± SD.
Identification of a DNA Recognition Motif for CAMTA Proteins.
CAMTA proteins from fruit flies and plants bind to a CG-rich DNA sequence via the CG-1 motif near their N termini (4, 6). To investigate the DNA-binding properties of mammalian CAMTA proteins, we fused various regions of mouse CAMTA1 and CAMTA2 to the maltose-binding protein (MBP). Among various CAMTA–MBP fusions, we were able to obtain stable protein only by using a cDNA fragment coding for the first 630 amino acids of mouse CAMTA2, which encompasses the CG motif, transcription activation domain, and TIG motif (Fig. S3). The CG-1 and TIG motifs of CAMTA1 and 2 are highly homologous (Fig. S3).
The bacterially expressed MBP–CAMTA2 fusion protein was used in iterative gel mobility shift assays with random oligonucleotide pools. Following four rounds of selection, we identified a consensus sequence for CAMTA-DNA binding of PyGCANTGCG (Fig. 4 A and B and Fig. S4A). Here “Py” represents T or C, and “N” represents A, T, or G. Gel mobility shift assays showed that the mobility of an oligonucleotide probe containing the CAMTA consensus sequence was retarded by MBP–CAMTA2, but not by MBP alone (Fig. S4B). Mutation of the core sequence CGCATTGCG to CGgtTcaaG completely abolished the interaction of MBP–CAMTA2 and DNA, whereas a mutation in the sequence adjacent to the core consensus did not abolish the interaction between MBP–CAMTA2 and DNA (Fig. S4B). However, the latter mutation reduced DNA binding by MBP–CAMTA2, indicating that the adjacent sequences influence interaction with CAMTA protein.
Fig. 4.

Identification of the CAMTA DNA-binding site and adjacent A/T-rich sequence. (A) Strategy for selecting CAMTA-binding DNA sequences using a DNA oligonucleotide library containing random nucleotides at 18 variable positions flanked by primers A and B. 32P-labeled double-stranded oligo-pools were generated by primers A and B and incubated with MBP or MBP-FLAG-CAMTA2 (amino acids 1–625). The DNA–CAMTA complex was resolved on a native polyacrylamide gel (Gel shift). DNA was recovered from the gel and labeled for the next round of selection. (B) Gel mobility shift of the DNA–CAMTA complex on a 4% native polyacrylamide gel. DNA recovered from the gel was used for the next round of selection. Four cycles of selection were performed. C1, the first cycle of selection; C2, the second cycle of selection; C4, the fourth cycle of selection. Anti-FLAG was used for supershift. (C) Sequences of the CAMTA consensus site (WT) and mutations (M1–M15) used for gel mobility shift assays. The relative binding of CAMTA to the different oligos is summarized. (D) Gel mobility shift assays to detect interactions between CAMTA2 and the oligos shown in C. (E) CAMTA consensus site with the adjacent A/T-rich sequence mutations (green) used for gel mobility shift assays. The relative binding of CAMTA to the different oligos is summarized. (F) Gel mobility shift assays to detect interactions between CAMTA2 and the oligos listed in E. Full-length CAMTA2 was translated in vitro using pcDNA (Life Technologies)-CAMTA2 as template. Anti-FLAG antibody was used for supershift. pcDNA empty vector was used as template in lysate controls.
To characterize the consensus DNA-binding sequence of CAMTA proteins further, we generated 15 mutations of the core consensus sequence (Fig. 4C). Gel-shift assays using full-length CAMTA2 translated in vitro showed that four mutations—M1, M6, M11, and M12—did not abolish the interaction of CAMTA proteins with the DNA, whereas three mutations—M2, M8, and M10—greatly decreased the interaction (Fig. 4 C and D). All other mutations (M3–M5, M7, M9, and M13-14) abolish interaction of CAMTA proteins with DNA (Fig. 4 C and D). These results suggested that the consensus DNA-binding sequence of CAMTA could be (T/C)GCANTGCG, where “N” stands for any nucleotide.
Adjacent to the consensus DNA-binding sequence of CAMTA, we noticed A/T-rich sequences that are known to increase DNA flexibility and to be crucial for target site recognition of GC box-binding proteins (15). To test the relevance of the A/T-rich sequences in the CAMTA-binding site, we generated nine mutations (f1 to f9) with reduced length of A/T-rich sequence (Fig. 4E). Four mutations, f6–f9, greatly decreased the interaction of CAMTA protein and the DNA probe (Fig. 4F), suggesting that the adjacent A/T-rich sequences also are crucial for the core sequence binding by CAMTA.
To examine the ability of CAMTA proteins to activate transcription through the identified consensus sequence, we constructed a reporter (1×CAMTA-luc), consisting of one copy of the CAMTA consensus sequence CGCATTGCG linked to a TATA box of the E1b virus promoter. In transfected COS cells, both CAMTA1 and CAMTA2 were able to activate reporter gene expression, with CAMTA1 conferring higher activation than CAMTA2 (Fig. S4C). Changing the core CGCATTGCG to CGgtTcaaG abolished the transcriptional activity of CAMTA proteins on the reporter (Fig. S4C), indicating that the consensus binding site CGCATTGCG is required for transcriptional activation by CAMTA proteins.
Replacement of the conserved lysine K108 in the CG-1 domain of CAMTA2 with alanine or of the corresponding lysine K141 in CAMTA1 with glutamic acid completely disrupted the DNA-binding activity of CAMTA proteins (Fig. S4 D–F). These mutant proteins also were unable to activate the 1×CAMTA-luc reporter in COS cells (Fig. S4G).
Dimerization of CAMTA on the Palindromic Core Sequence.
The CAMTA consensus binding site shows a symmetry surrounding the central variable N position, suggesting a bipartite mode of DNA binding (Fig. 5A). Gel mobility shift assays were performed with FLAG-tagged N terminus (amino acids 1–625) of CAMTA2 (termed MBP-FLAG-CAMTA) incubated with DNA probe containing the CAMTA-binding site to generate two bands on the gel (Fig. 5B). Both bands showed a supershift when incubated with anti-FLAG antibody. The ratio of the intensity of the upper band to that of the lower band was decreased when less CAMTA protein was added to the same amount of DNA probe. Only the lower band was detectable when the least amount of CAMTA protein was added. These results suggest that the higher band is a complex of a CAMTA homodimer and the DNA probe, whereas the lower band is a complex of a CAMTA monomer and the DNA probe.
Fig. 5.

Dimerization of CAMTA on the palindromic core sequence. (A) Schematic of CAMTA dimers on the palindromic core sequence. (B) Gel mobility shift assay consisting of recombinant protein of the N terminus (amino acids 1–625) of CAMTA (MBP-FLAG-CAMTA) incubated with its DNA-binding site generates two bands on the gel. Both bands were supershifted by anti-FLAG antibody. When decreasing amounts of CAMTA protein are added (designated by the black triangle), the ratio of the intensity of the upper band to the intensity of the lower band is decreased. The higher band is a complex of a CAMTA homodimer and DNA probe. The lower band is a complex of a CAMTA monomer and DNA probe. (C) Schematic of the in vitro pull-down assay of dimerization of CAMTA. (D) Pull-down assay showing 35S-labeled, myc-tagged CAMTA incubated with FLAG-CAMTA and the DNA core sequence could be pulled down with anti-FLAG beads, whereas 35S-labeled, myc-tagged CAMTA alone or with DNA was not pulled down by anti-FLAG beads.
To confirm dimerization of CAMTA on DNA, we performed an in vitro pull-down assay using in vitro-translated FLAG-CAMTA as pull-down “bait” and in vitro-translated 35S-labeled myc-CAMTA as “prey” to detect interactions. Equal amounts of FLAG-CAMTA and 35S-myc-CAMTA were mixed with DNA and anti-FLAG beads (Fig. 5C). If CAMTA forms a dimer when binding to DNA, 35S-myc-CAMTA should be pulled down with anti-FLAG beads (Fig. 5C, Left). If CAMTA forms a monomer when binding DNA, anti-FLAG beads cannot capture 35S-myc-CAMTA (Fig. 5C, Right). The results show that 35S-myc-CAMTA can be pulled down with anti-FLAG beads, but not with IgG beads, in the presence of FLAG-CAMTA. In the absence of FLAG-CAMTA, 35S-myc CAMTA was not pulled down by anti-FLAG beads. Without the DNA probe, the interaction between 35S-myc CAMTA and FLAG-CAMTA was decreased greatly (Fig. 5D). Taken together, these data support the conclusion that CAMTA binds to its palindromic DNA core sequence as a dimer.
Changes in Neuronal Gene Expression in CAMTA1-nKO Mice.
To begin to define the molecular basis of CAMTA1 function in the nervous system, we compared the gene-expression profiles of cerebella of control and nKO mice by microarray analysis. These studies identified 203 genes that were dysregulated by at least 1.5-fold in CAMTA1-nKO cerebella (Fig. 6). Of these 203 genes, 69 were down-regulated and 134 were up-regulated in CAMTA1-nKO mice. Gene Ontology analysis revealed that 84 of the 203 genes were involved in neuronal functions or in the protection of neurons from apoptosis (Fig. 6). Selected genes containing putative CAMTA consensus sites within 5 kb upstream of the transcriptional start site that are down- and up-regulated in CAMTA1-nKO cerebellum are listed in Table S2.
Fig. 6.

Identification of dysregulated genes in CAMTA1-nKO cerebellum. Microarray analysis of RNA isolated from cerebella of control and CAMTA1-nKO mice identified 203 genes that were up- or down-regulated by at least 1.5-fold in CAMTA1-nKO mice.
Analysis of DNA sequences associated with genes down-regulated in the CAMTA1-nKO cerebella revealed that at least 11 genes are associated with CAMTA consensus sites (T/C)GCANTGCG (Table S2). Interestingly, Snhg11 (NM_175692.3 Riken cDNA A930034L06), the most down-regulated gene in CAMTA1-nKO cerebella, is highly expressed in Purkinje cells within the cerebellar granular layer, corresponding to the expression pattern of CAMTA1. Another gene strongly down-regulated in nKO cerebella, Gtl2, has been shown to function as a tumor suppressor (16). Numerous other CAMTA1-sensitive genes that we found to be associated with consensus CAMTA-binding sites are involved in stress responsiveness, signal transduction, and protein metabolism. Among genes up-regulated in CAMTA1-nKO cerebellum, transthyretin (TTR) was up-regulated 51-fold in nKO mice (Table S2). TTR has been implicated in neuroprotection (17, 18). The CAMTA consensus site does not exist within 5 kb upstream of the TTR gene; therefore up-regulation of TTR may be a secondary response to neurodegeneration in CAMTA1-nKO brain. Somatostatin (Sst), a multifunctional peptide, also was up-regulated in nKO brain. Increased expression of Sst in neurons is associated with motor coordination disabilities in Huntington’s chorea (19). Sst accumulation increases in response to β-amyloid–induced neurotoxicity in cortical neurons (20). Up-regulation of genes in response to neuronal cell death partially suggests neurodegeneration in CAMTA1-nKO mice. Thus, deletion of CAMTA1 in the nervous system dysregulates the expression of a broad collection of genes required for survival and homeostasis of neuronal cells, including Purkinje cells and granule neurons.
Discussion
The results of this study demonstrate an essential role of the CAMTA1 transcription factor in maintaining cerebellar function in mice and suggest that this calcium-sensitive transcription factor is required for coordination of gene expression and survival of Purkinje cells. The absence of CAMTA1 in mice results in severe ataxia and neuronal atrophy. Through the identification of the consensus DNA-binding site for mammalian CAMTA proteins and the analysis of dysregulated cerebellar genes in CAMTA1-KO mice, we identified a large collection of genes that likely mediate the essential functions of CAMTA1 in cerebellar development and function. Given the many genes that are apparently regulated by CAMTA1 in the cerebellum, it seems likely that the ataxic abnormalities of CAMTA1-nKO mice reflect the combined functions of many of these genes rather than that of a single downstream target gene.
CAMTA1 is highly enriched in the brain, with the highest levels of expression in the cerebellar granular layer and Purkinje cells, midbrain, pons, and hippocampus. During the early stages of postnatal development, the cerebellum undergoes a dramatic increase in size and developmental changes that culminate in maturation of the adult cerebellum (21). The morphological changes occurring in the cerebellum include maturation of Purkinje cells with an increase in dendritic arborization, concomitant with an increase in the molecular layer and granular layer within the cerebellum. Based on histological analysis, the anatomy and size of the cerebellum appeared normal in 2- to 3-wk-old CAMTA1-nKO mice. However, we observed a clear loss of Purkinje cells and reduced cerebellar size in mutant mice by age 3 mo. Thus, we conclude that CAMTA1 plays a predominant role in the maturation and survival of cerebellar neurons rather than in the initial development of these cells.
CAMTA1 is one of many genes located within the 1p36 chromosomal locus, which corresponds to a common chromosomal breakpoint associated with a broad range of clinical features, including neurological defects, in humans (22). In this regard, multiple SNPs that correlate to cognitive abnormalities have been identified in the CAMTA1 gene (8). The importance of CAMTA1 for normal neurological function was highlighted further by the identification of human subjects with heterozygous chromosomal rearrangements in the CAMTA1 locus who present with congenital cerebellar ataxia, in accord with our findings in mice (9). It should be emphasized, however, that these human phenotypes result from heterozygous CAMTA1 mutations, whereas the abnormalities observed in mice result from homozygous gene deletion. There also are some distinctions between the neurologic phenotypes seen in our CAMTA1-nKO mice and humans with CAMTA1 haploinsufficiency. In particular, the ataxia seen in humans does not appear to be progressive and has not been shown to be accompanied by neuronal degeneration as seen in our homozygous mutant mice.
Although CAMTA1 is expressed predominantly in the brain, it also is expressed in the heart (7), and it has been reported that CAMTA1 is required for differentiation of adult stem cells into myocardial cells (23). We did not detect any overt cardiac abnormalities in mice with global deletion of CAMTA1. Interestingly, however, a polymorphism in CAMTA1 revealed an association in patients with cardiovascular disease who had poor cognitive performance, suggesting the potential involvement of CAMTA1 in both cardiovascular and neural function (24).
Our previous studies demonstrated regulation of CAMTA proteins by association with class II histone deacetylases (HDACs) (7). In this regard, HDAC4 associates with ataxin-1 and myocyte enhancer transcription factor 2 (MEF2) to regulate neuronal survival (25). The MEF2/HDAC5/calcium/calmodulin-dependent protein kinase II (CaMKII)-signaling pathway also regulates survival of cerebellar granule cells (26), and our results show that deletion of CAMTA1 leads to a decrease in the granule cell layer in adult mice. Thus, we speculate that the influence of CAMTA on neuronal survival is subject to signal-dependent regulation by the MEF2/HDAC5/CaMKII pathway.
In plants, CAMTA proteins activate stress-responsive genes by binding to a CG-rich motif, CG(C/T)G (2, 3, 5, 6). We found that mammalian CAMTA proteins also bind to a CG-rich motif, (T/C)GCANTGCG, although this sequence is distinct from that of plant CAMTA binding. In the absence of CAMTA1, numerous genes are dysregulated in the cerebellum. Among these genes, Eif4a2, Fbxl3a, and Magee1 are involved in protein synthesis and degradation. Neurodegenerative disorders often are associated with the accumulation of misfolded disease-specific proteins, causing repression of protein translation (27, 28), which has been associated with synaptic failure and neuronal loss (27). It will be interesting to investigate whether loss of protein quality is a major trigger to degeneration of Purkinje cells and other neurons in the absence of CAMTA1. The functions of numerous other CAMTA1-dependent genes, such as Snhg11, Snurf, and Snrpn, are currently unknown. How these genes affect the functions of cerebellar neurons and other cells of the nervous system in which CAMTA1 is expressed remains to be determined.
The neurological abnormalities associated with CAMTA1-mutant mice allow mechanistic analysis of these abnormalities and disease modeling. Further studies of the CAMTA-signaling pathway in the nervous system may contribute to the identification of therapeutic targets for intervention in neurodegenerative diseases. Understanding how the derangements in neuronal gene expression associated with CAMTA1 deletion relate to those associated with other transcription factor mutations, such as those in TATA box-binding protein (29, 30), also will be of interest.
Materials and Methods
Generation of CAMTA1 Conditional-KO Mice.
CAMTA1 conditional-KO mice were generated by inserting sites for Cre-mediated recombination upstream and downstream of exon 9 through homologous recombination, as described in SI Materials and Methods. Southern blot analysis was performed as previously described (7). All animal procedures were approved by the University of Texas Southwestern Medical Center Institutional Animal Care and Use Committee.
Histological and Immunohistochemical Analysis.
Brains were harvested from CAMTA1 control and nKO mice. Histology and immunohistochemistry were performed as described previously (31). Frozen sections were stained with anti–calbindin-D28K antibody (1:500) (C9848; Sigma). The size of calbindin-positive Purkinje cells was assessed using Stereo Investigator (MBF Bioscience).
In Situ Hybridization.
In situ hybridization was performed as described (32). Briefly, an 807-bp cDNA fragment located in exon 9 of the mouse CAMTA1 gene was isolated by PCR and subcloned into a pGEM-T Easy vector (forward primer: TGTCCGAGGTCACTAACGAG; reverse primer: GAGGTGCAAGGAGGAAGTAGA). Digoxin-labeled sense or antisense riboprobes were generated by in vitro transcription with SP6 or T7 RNA polymerase (Roche). In situ hybridization then was performed on sagittal cryostat sections of 16 µm.
Beam Walk and Rotarod Performance Test.
Neurobehavioral tests were performed at the University of Texas Southwestern Rodent Behavior Core Facility, as described in SI Materials and Methods.
CAMTA Site Selection and Transfection Assays.
DNA-binding site selection using CAMTA2-MBP fusion proteins and transfection assays were performed as described in SI Materials and Methods.
RNA and Microarray Analysis.
RNA was isolated from mouse brain using TRIzol reagent and was used for microarray analysis performed by the University of Texas Southwestern Microarray Core Facility using the MouseWG-6 v2.0 BeadChips (Illumina) or was used for quantitative PCR as described in SI Materials and Methods.
Reporter Activity Assays.
Plasmid constructs and transfection assays for luciferase reporter assays are described in SI Materials and Methods.
Statistics.
Values are presented as the mean ± SE. Student t test was performed for paired analysis. Repeated-measure two-way ANOVA with Tukey’s multiple comparisons post hoc test was used to analyze behavior studies. P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Jose Cabrera for graphics and Shari Birnbaum at the University of Texas Southwestern Rodent Behavior Core Facility for mouse behavior analysis. This work was supported in part by grants from National Institutes of Health and by Grant I-0025 from the Robert A. Welch Foundation (to E.N.O.). C.E.G. was supported by Fellowship 7-09-CVD-04 from the American Diabetes Association.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1411251111/-/DCSupplemental.
References
- 1.Bouché N, Scharlat A, Snedden W, Bouchez D, Fromm H. A novel family of calmodulin-binding transcription activators in multicellular organisms. J Biol Chem. 2002;277(24):21851–21861. doi: 10.1074/jbc.M200268200. [DOI] [PubMed] [Google Scholar]
- 2.Doherty CJ, Van Buskirk HA, Myers SJ, Thomashow MF. Roles for Arabidopsis CAMTA transcription factors in cold-regulated gene expression and freezing tolerance. Plant Cell. 2009;21(3):972–984. doi: 10.1105/tpc.108.063958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galon Y, et al. Calmodulin-binding transcription activator (CAMTA) 3 mediates biotic defense responses in Arabidopsis. FEBS Lett. 2008;582(6):943–948. doi: 10.1016/j.febslet.2008.02.037. [DOI] [PubMed] [Google Scholar]
- 4.Han J, et al. The fly CAMTA transcription factor potentiates deactivation of rhodopsin, a G protein-coupled light receptor. Cell. 2006;127(4):847–858. doi: 10.1016/j.cell.2006.09.030. [DOI] [PubMed] [Google Scholar]
- 5.Kim Y, Park S, Gilmour SJ, Thomashow MF. Roles of CAMTA transcription factors and salicylic acid in configuring the low-temperature transcriptome and freezing tolerance of Arabidopsis. Plant J. 2013;75(3):364–376. doi: 10.1111/tpj.12205. [DOI] [PubMed] [Google Scholar]
- 6.Pandey N, et al. CAMTA 1 regulates drought responses in Arabidopsis thaliana. BMC Genomics. 2013;14:216. doi: 10.1186/1471-2164-14-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song K, et al. The transcriptional coactivator CAMTA2 stimulates cardiac growth by opposing class II histone deacetylases. Cell. 2006;125(3):453–466. doi: 10.1016/j.cell.2006.02.048. [DOI] [PubMed] [Google Scholar]
- 8.Huentelman MJ, et al. Calmodulin-binding transcription activator 1 (CAMTA1) alleles predispose human episodic memory performance. Hum Mol Genet. 2007;16(12):1469–1477. doi: 10.1093/hmg/ddm097. [DOI] [PubMed] [Google Scholar]
- 9.Thevenon J, et al. Intragenic CAMTA1 rearrangements cause non-progressive congenital ataxia with or without intellectual disability. J Med Genet. 2012;49(6):400–408. doi: 10.1136/jmedgenet-2012-100856. [DOI] [PubMed] [Google Scholar]
- 10.Henrich KO, et al. CAMTA1, a 1p36 tumor suppressor candidate, inhibits growth and activates differentiation programs in neuroblastoma cells. Cancer Res. 2011;71(8):3142–3151. doi: 10.1158/0008-5472.CAN-10-3014. [DOI] [PubMed] [Google Scholar]
- 11.Schraivogel D, et al. CAMTA1 is a novel tumour suppressor regulated by miR-9/9* in glioblastoma stem cells. EMBO J. 2011;30(20):4309–4322. doi: 10.1038/emboj.2011.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakai K, Miyazaki Ji. A transgenic mouse line that retains Cre recombinase activity in mature oocytes irrespective of the cre transgene transmission. Biochem Biophys Res Commun. 1997;237(2):318–324. doi: 10.1006/bbrc.1997.7111. [DOI] [PubMed] [Google Scholar]
- 13.Tronche F, et al. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23(1):99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- 14.Engleka KA, et al. Insertion of Cre into the Pax3 locus creates a new allele of Splotch and identifies unexpected Pax3 derivatives. Dev Biol. 2005;280(2):396–406. doi: 10.1016/j.ydbio.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 15.Lundin M, Nehlin JO, Ronne H. Importance of a flanking AT-rich region in target site recognition by the GC box-binding zinc finger protein MIG1. Mol Cell Biol. 1994;14(3):1979–1985. doi: 10.1128/mcb.14.3.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou Y, Zhang X, Klibanski A. MEG3 noncoding RNA: A tumor suppressor. J Mol Endocrinol. 2012;48(3):R45–R53. doi: 10.1530/JME-12-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fleming CE, Saraiva MJ, Sousa MM. Transthyretin enhances nerve regeneration. J Neurochem. 2007;103(2):831–839. doi: 10.1111/j.1471-4159.2007.04828.x. [DOI] [PubMed] [Google Scholar]
- 18.Stein TD, Johnson JA. Lack of neurodegeneration in transgenic mice overexpressing mutant amyloid precursor protein is associated with increased levels of transthyretin and the activation of cell survival pathways. J Neurosci. 2002;22(17):7380–7388. doi: 10.1523/JNEUROSCI.22-17-07380.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beal MF, Uhl G, Mazurek MF, Kowall N, Martin JB. Somatostatin: Alterations in the central nervous system in neurological diseases. Res Publ Assoc Res Nerv Ment Dis. 1986;64:215–257. [PubMed] [Google Scholar]
- 20.Geci C, How J, Alturaihi H, Kumar U. Beta-amyloid increases somatostatin expression in cultured cortical neurons. J Neurochem. 2007;101(3):664–673. doi: 10.1111/j.1471-4159.2006.04415.x. [DOI] [PubMed] [Google Scholar]
- 21.Goldowitz D, Hamre K. The cells and molecules that make a cerebellum. Trends Neurosci. 1998;21(9):375–382. doi: 10.1016/s0166-2236(98)01313-7. [DOI] [PubMed] [Google Scholar]
- 22.Battaglia A. Del 1p36 syndrome: A newly emerging clinical entity. Brain Dev. 2005;27(5):358–361. doi: 10.1016/j.braindev.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 23.Muller-Borer B, et al. Calcium dependent CAMTA1 in adult stem cell commitment to a myocardial lineage. PLoS ONE. 2012;7(6):e38454. doi: 10.1371/journal.pone.0038454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller LA, et al. CAMTA1 T polymorphism is associated with neuropsychological test performance in older adults with cardiovascular disease. Psychogeriatrics. 2011;11(3):135–140. doi: 10.1111/j.1479-8301.2011.00357.x. [DOI] [PubMed] [Google Scholar]
- 25.Bolger TA, Zhao X, Cohen TJ, Tsai CC, Yao TP. The neurodegenerative disease protein ataxin-1 antagonizes the neuronal survival function of myocyte enhancer factor-2. J Biol Chem. 2007;282(40):29186–29192. doi: 10.1074/jbc.M704182200. [DOI] [PubMed] [Google Scholar]
- 26.Heidenreich KA, Linseman DA. Myocyte enhancer factor-2 transcription factors in neuronal differentiation and survival. Mol Neurobiol. 2004;29(2):155–166. doi: 10.1385/MN:29:2:155. [DOI] [PubMed] [Google Scholar]
- 27.Moreno JA, et al. Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature. 2012;485(7399):507–511. doi: 10.1038/nature11058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443(7113):780–786. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 29.van Roon-Mom WM, Reid SJ, Faull RL, Snell RG. TATA-binding protein in neurodegenerative disease. Neuroscience. 2005;133(4):863–872. doi: 10.1016/j.neuroscience.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 30.Nakamura K, et al. SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet. 2001;10(14):1441–1448. doi: 10.1093/hmg/10.14.1441. [DOI] [PubMed] [Google Scholar]
- 31.Montgomery RL, Hsieh J, Barbosa AC, Richardson JA, Olson EN. Histone deacetylases 1 and 2 control the progression of neural precursors to neurons during brain development. Proc Natl Acad Sci USA. 2009;106(19):7876–7881. doi: 10.1073/pnas.0902750106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qin S, Liu M, Niu W, Zhang CL. Dysregulation of Kruppel-like factor 4 during brain development leads to hydrocephalus in mice. Proc Natl Acad Sci USA. 2011;108(52):21117–21121. doi: 10.1073/pnas.1112351109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.