

Significance
Canonical transient receptor potential (TRPC) channels are a family of polymodal cation channels. TRPC7 was the last member of the TRPC family cloned, and the functional role of TRPC7 in the brain is completely unknown. Here, we show that genetic ablation of TRPC7 disrupts acute severe seizures induced by pilocarpine in mice. This disruption is associated with a reduction in pilocarpine-induced increase in gamma wave activity that precedes the acute seizures. At the cellular level, loss of TRPC7 shows reduced long-term potentiation at cornu ammonis (CA) 3 recurrent collateral synapses, which is critical for the generation of epileptiform burst firing in the CA3 region of the hippocampus. Our findings reveal a critical role for TRPC7 in the generation of seizures.
Keywords: epilepsy, gamma oscillation, hippocampal circuitry, synaptic plasticity
Abstract
Status epilepticus (SE) is a life-threatening disease that has been recognized since antiquity but still causes over 50,000 deaths annually in the United States. The prevailing view on the pathophysiology of SE is that it is sustained by a loss of normal inhibitory mechanisms of neuronal activity. However, the early process leading to the initiation of SE is not well understood. Here, we show that, as seen in electroencephalograms, SE induced by the muscarinic agonist pilocarpine in mice is preceded by a specific increase in the gamma wave, and genetic ablation of canonical transient receptor potential channel (TRPC) 7 significantly reduces this pilocarpine-induced increase of gamma wave activity, preventing the occurrence of SE. At the cellular level, TRPC7 plays a critical role in the generation of spontaneous epileptiform burst firing in cornu ammonis (CA) 3 pyramidal neurons in brain slices. At the synaptic level, TRPC7 plays a significant role in the long-term potentiation at the CA3 recurrent collateral synapses and Schaffer collateral-CA1 synapses, but not at the mossy fiber-CA3 synapses. Taken together, our data suggest that epileptiform burst firing generated in the CA3 region by activity-dependent enhancement of recurrent collateral synapses may be an early event in the initiation process of SE and that TRPC7 plays a critical role in this cellular event. Our findings reveal that TRPC7 is intimately involved in the initiation of seizures both in vitro and in vivo. To our knowledge, this contribution to initiation of seizures is the first identified functional role for the TRPC7 ion channel.
Status epilepticus (SE) is a life-threatening condition in which patients suffer from continuous or rapidly repeating seizures. Although SE has been recognized since antiquity, our understanding of its pathophysiology remains incomplete (1). The generation of SE is a complex process involving a gradual loss of normal GABA inhibition (2) and a buildup of overexcitation of glutamatergic pathways (3, 4).
Canonical transient receptor potential (TRPC) channels are a family of nonselective cation channels expressed widely in neurons and glia in the brain (5). There are seven members (TRPC1 to -7) in the mammalian TRPC family, and six (TRPC1 and TRPC3 to -7) are expressed in humans (6). TRPCs can be divided into TRPC1, TRPC4/5, and TRPC3/6/7 subgroups based on sequence homology and functional properties. TRPC channels can be either homomeric channels or heteromeric channels, often but not always formed by members in the same subgroup (7, 8). The expression pattern and functional roles of TRPC7 are largely unknown. In humans, the expression of TRPC7 is restricted to the brain whereas, in rodents, TRPC7 mRNA has been detected outside the brain. Here, we present evidence to suggest that TRPC7 plays a critical role in the generation of acute seizures in a Pilocarpine (Pilo)-induced in vivo model of SE. The lack of SE in TRPC7 knockout (KO) mice results from a reduction of Pilo-induced increase in electroencephalogram (EEG) gamma wave activity, and a reduction of long-term potentiation at cornu ammonis (CA) 3 recurrent collateral synapses.
Results
To induce SE, mice were given a single dose of Pilo (280 mg/kg, i.p.), a muscarinic agonist. In most WT mice (seven of eight mice), Pilo induced SE (seizures did not reach SE state in the remaining mouse). Our EEG recordings (Fig. S1) showed that baseline brain activity was first suppressed after Pilo injection (Fig. 1A, latent phase). This suppression is consistent with the presynaptic inhibition of glutamatergic synaptic transmission by muscarinic receptors. A brief burst of electrographic seizures then appeared, followed by a stronger suppression of EEG activity and a run-up to a longer burst of seizures (Fig. 1A). The SE state appeared ∼15–20 min after the first burst of seizures was detected. This pattern held true for every WT mouse in which Pilo induced SE. It should be noted that the true start of the SE phase occurs 40–50 min after Pilo injection. This starting point is much later than the first observed occurrence of stage V convulsive seizures, which occurred during the transition between the latent phase and the SE. Furthermore, the true start of SE was not discernible by behavioral observation. In five of seven TRPC7 KO mice, the same dose of Pilo induced only suppression of EEG activity, and there were little Pilo-induced electrographic seizures (Fig. 1B and Fig. S2). Sustained seizures that partially meet the definition of SE (lasting longer than 30 min) were induced by Pilo in the remaining two TRPC7 KO mice. However, the root mean square (RMS) peak power of SE in these two mice (688,102 and 689,734 μV2) falls below the normal range of variation (713,533–1,181,504 μV2) in WT mice, and the seizure activity disappeared before the end of the recording period. Assuming that these two mice exhibited SE, there was still a highly significant difference between the WT and TRPC7 KO mice (Fig. 1C) (P = 0.02, χ2 test, two-tail). Interestingly, the suppression of EEG activity in TRPC7 KO mice disappeared about 3 h after Pilo injection, which is in agreement with the reported nearly 90% elimination of Pilo from the brain after 3 h in mice (9). Consistent with the reduction of occurrence of SE in TRPC7 KO mice, the mortality after administration of Pilo was also reduced in TRPC7 KO mice (Fig. 1D). Taken together, our data indicate that TRPC7 plays a critical role in the generation of Pilo-induced SE in mice.
Fig. 1.

TRPC7 plays a critical role in Pilocarpine-induced SE. (A and B) Root-mean-square (rms) power analysis of representative EEG signals from WT and TRPC7 KO mice using the full bandwidth (0–1 kHz) and a 10-s window and averaged for each minute were plotted over 5 h after the administration of a single dose of Pilo (280 mg/kg; i.p.). Note that, in a WT mouse (A), the SE (continuous seizures lasting more than 30 min) induced by Pilo was preceded by an initial decrease of brain activity and two main peaks of increases corresponding to the bursts of seizures (*) in the EEG (shown in the Inset; the total time span was 60 min, and the y scale is from 2,500 μV to −2500 μV). However, in a TRPC7 KO mouse (B), there was only a decrease in brain activities after Pilo injection, and the EEG activity returned to baseline at the end of the recording period. (C) Contingency table analysis of the role of TRPC7 in Pilo-induced SE. Note the significant reduction of SE in TRPC7 KO mice (P = 0.02, χ2 test, two-tail). (D) Mortality in the first 24 h following Pilo injections was reduced in TRPC7 KO mice (n = 7, 5, 5) compared with WT mice (n = 24, 10, 23).
To better understand how seizures begin and transition to the SE state in the Pilo model, we conducted the RMS power analysis of EEG signals (Fig. 2). In WT mice, after Pilo injection, all frequency bands were initially inhibited. As time progressed, the gamma wave activity recovered first and increased significantly above the baseline by the end of the latent phase whereas the remaining wave bands remained significantly inhibited (Fig. 2 A and B). This increase in gamma wave activity was associated with convulsive behaviors corresponding to stage I–III seizures on the Racine scale. In contrast, alpha, beta, and gamma wave activities were all significantly increased above the baseline during the SE phase (Fig. 2C). Thus, Pilo-induced electrographic seizures were preceded by a selective increase in gamma oscillation, suggesting that the cortical seizures were driven by a Pilo-induced increase in gamma oscillation. In TRPC7 KO mice, this Pilo-induced increase in gamma oscillation was significantly reduced (Fig. 2 D and E). However, activities in the other wave bands were comparable between WT and TRPC7 KO mice (Fig. 2E). To determine whether the reduced gamma activity during the latent phase in TRPC7 KO mice reflected a defect in the generation of gamma oscillation, we analyzed the baseline gamma activity in WT and TRPC7 KO mice. The mean RMS power of the gamma oscillation in WT mice was 7,203.5 ± 614.9 μV2 (mean ± SEM; n = 6) whereas the mean RMS power of the gamma oscillation in TRPC7 KO mice was 11,802 ± 2,240 μV2 (n = 7; P > 0.10; unpaired t test). Thus, the ability to generate gamma waves was normal in TRPC7 KO mice. These observations suggest that TRPC7 plays a critical role in the Pilo-induced increase in gamma oscillation.
Fig. 2.

Selective reduction of Pilo-induced increase in gamma wave activity in TRPC7 KO mice. (A) The rms power analysis of EEG signals from representative WT mice using a 10-s window and averaged for each minute were plotted over 70 min after the administration of a single dose of Pilo (280 mg/kg; i.p.). The normalized full bandwidth, the delta wave (0.5–4.5 Hz), the theta wave (5–7.5Hz), the alpha wave (8–12 Hz), the beta wave (12–30 Hz), and the gamma wave (30–50 Hz) were plotted. Note that, during the latent phase, there was a noticeable increase in gamma wave activity whereas EEG activities in other wavebands remained inhibited. However, once the SE state was reached, there was an increase in multiple wavebands. (B) Pooled data plotting the rms power measured over a 5-min period just before the first appearance of cortical seizures in WT mice (n = 5). Note that, with the exception of the gamma wave, all other waves remained significantly inhibited by Pilo at this time point (**P < 0.01; ##P < 0.01, one-sample t test). (C) Pooled data plotting the rms power during the SE state measured at 60 min after Pilo injection in WT mice (n = 5). Note that the full bandwidth and the alpha, the beta, and the gamma waves were all significantly increased above the pre-Pilo baseline (*P < 0.05; **P < 0.01, one-sample t test). (D) Pooled data plotting the rms power of the gamma wave after Pilo injection in WT and TRPC7 KO mice (n = 5 and 5, respectively). Note the significant reduction in TRPC7 KO mice (***P < 0.001 for genotype effects, two-way ANOVA). (E) Pooled data plotting the rms power measured over a 5-min period just before the first appearance of cortical seizures in WT mice (n = 5) and during the same time period in TRPC7 KO mice (n = 5). Note the highly significant reduction in gamma wave activity in TRPC7 KO mice (***P < 0.001, t test).
A previous study suggested that the behavioral manifestation of seizures after kindling was associated with an increase in gamma oscillation in the hippocampus (10). There are at least two gamma wave generators in the hippocampus: one located in the dentate gyrus and the other located in the CA3–CA1 region (11). The intrahippocampal gamma wave is generated by the extensive recurrent collaterals (RCs) in the CA3 region, and the gamma activity then entrains the CA1 region via its interneurons (11). The RCs also play a critical role in the initiation of epileptiform burst firing in the entorhinal cortex (EC)-hippocampus circuit (12, 13), and the epileptiform bursts may be the source of gamma waves (14). These reports motivated us to investigate the role of TRPC7 in spontaneous epileptiform bursts in the CA3 region of the hippocampus. We recorded CA3 pyramidal neurons in horizontal slices in which the EC-hippocampal connections are preserved and epileptiform burst firing can be induced by either high K+ or bicuculline (12, 13). In WT mice, the spontaneous firing pattern of CA3 pyramidal neurons was converted from single spikes to burst firing with a plateau potential during a 30-min bath application of 20 μM bicuculline, and the epileptiform bursts persisted for at least 25 min after washout of bicuculline (Fig. 3A). Each spontaneous burst contained more than 20 spikes at very high frequency and was followed by a slow and large afterhyperpolarization potential. Membrane resistance, resting membrane potential, and firing threshold of CA1and CA3 pyramidal neurons were normal (Figs. S3 and S4), and the field excitatory postsynaptic potentials (EPSPs) as a function of stimulus intensity were also comparable with that of the WT mice (Fig. S5). Thus, loss of TRPC7 does not cause abnormal firing or faulty wiring in the hippocampus. However, the same bicuculline treatment resulted in significantly reduced spontaneous epileptiform bursts in slices from TRPC7 KO mice, with a mean frequency of 1.16 ± 0.20 burst per min in WT (n = 4) and 0.27 ± 0.11 burst per min in TRPC7 KO (n = 7) (Fig. 3 A and B). In addition to inducing spontaneous epileptiform bursts, bicuculline also resulted in long-lasting hyperexcitability. A subthreshold stimulus of the mossy-fiber (MF) pathway before bicuculline treatment elicited epileptiform bursts afterward (Fig. 3C). The duration of evoked epileptiform bursts measured by the number of spikes per burst was 24.3 ± 4.4 in WT (n = 4) and 12.1 ± 0.9 in TRPC7 KO (n = 7), and the amplitude of the plateau potential was 36.1 ± 1.5 mV in WT and 26.6 ± 1.9 mV in TRPC7 KO (Fig. 3 C–E). Both parameters were significantly reduced in TRPC7 KO mice (P < 0.01). This reduction of spontaneous epileptiform bursts in TRPC7 KO is consistent with the reduced gamma wave activity and reduced SE after administration of Pilo.
Fig. 3.

TRPC7 contributes to epileptiform burst firing in CA3 pyramidal neurons. (A) Spontaneous epileptiform bursts were induced by bath application of 20 μM bicuculline for 30 min. Note that there were more spontaneous epileptiform bursts (*; with the characteristic long-lasting afterhyperpolarization) in WT mice than TRPC7 KO mice. (B) The frequency of spontaneous epileptiform bursts after bicuculline in WT and TRPC7 KO mice (n = 4 and 6, respectively). Note the significant reduction in TRPC7 KO mice (**P < 0.01, unpaired t test). (C) Representative traces showing evoked epileptiform bursts by MF stimulations in CA3 pyramidal neurons after bath application of bicuculline for 30 min (n = 6, 7 for WT and TRPC7 KO). Note that the bursts in TRPC7 KO mice were shorter and the amplitude of the plateau was also reduced. (D and E) Quantitative analysis of evoked epileptiform burst firing by MF stimulations in CA3 pyramidal neurons after bath application of bicuculline for 30 min (n = 4, 7 for WT and TRPC7 KO). Note the significant reduction of action potentials per burst, which indicates a shorter burst duration in TRPC7 KO mice (**P < 0.01, unpaired t test).
Spontaneous epileptiform bursts can also be induced in the CA1 area by bath application of muscarinic or mGluR agonists, and we have shown previously that heteromeric TRPC1/4 channels were critical (15). Interestingly, spontaneous burst firing induced by mGluR agonists in CA1 pyramidal neurons in brain slices was unaltered by genetic ablation of TRPC7 (Fig. 4). This observation is consistent with our previous finding that epileptiform bursting in CA1 correlates poorly with seizure activities in vivo (15).
Fig. 4.

TRPC7 KO mice exhibit normal mGluR agonist-induced spontaneous burst firing in the CA1 area. (A) Representative current-clamp recordings showing the spontaneous burst firing induced by 30 μM (1S,3R)-1-Aminocyclopentane-1,3-dicarboxylic acid (1S,3R-ACPD) in CA1 pyramidal neurons in adult WT and TRPC7 KO mice. (B) The amplitude of the plateau underlying the burst is comparable in the WT and TRPC7 KO mice. Pooled data (mean ± SEM) were plotted (n = 5, 6 for WT and 7 KO mice). (C) The duration of each burst was quantified by the number of action potentials within each burst, and three random bursts from each CA1 pyramidal neuron were analyzed to obtain the average number of spikes per burst. Pooled data (mean ± SEM) were plotted (n = 5, 6 for WT, and TRPC7 KO mice).
The spontaneous epileptiform bursts in the CA3 region in brain slices are generated by an increase in strength of RC synapses that depends on activation of both NMDA receptors and mGluRs (12, 13). Thus, reduced epileptiform bursts in TRPC7 KO mice may be caused by an impaired synaptic plasticity at the RC synapses. To test this hypothesis, we investigated synaptic plasticity in the CA3 and CA1 regions of the hippocampus. We recorded the field EPSP at MF-CA3 synapses. Data were included in analysis only after we confirmed that the evoked field EPSP was blocked by 1 μM (2S,2'R,3′R)-2-(2',3′-Dicarboxycyclopropyl)glycine (DCG-IV), a group II mGluR agonist (16). In WT mice, high-frequency stimulation (HFS) induced long-term potentiation (LTP), which was 115.9 ± 41.5% above baseline 30 min after HFS (n = 6) and frequently a large, slow component that resembles the paroxysmal depolarization shift (Fig. 4A). This finding is consistent with previous reports that high-frequency stimulation induces epileptiform burst firing in the CA3 region of the hippocampus. In TRPC7 KO mice, the HFS-induced MF LTP appeared to be normal (96.6 ± 38.1% above baseline 30 min after HFS; n = 5) (Fig. 5 A and B). However, we failed to observe the slow component after HFS. At the CA3 RC synapses, which were insensitive to DCG-IV, HFS-induced LTP was significantly reduced in TRPC7 KO mice (121.2 ± 4.7% above baseline 30 min after HFS; n = 6) relative to WT mice (164.3 ± 7.8% above baseline 30 min after HFS; n = 6) (Fig. 5 C and D). Similarly, the HFS-induced LTP at Schaffer collateral (SC)-CA1 synapses was also significantly reduced (Fig. 5 E and F). The field EPSP was 42.0 ± 6.5% above baseline 30 min after HFS in WT mice (n = 14), and 112.1 ± 6.4% above baseline 30 min after HFS in TRPC7 KO mice (n = 12). These results indicate that TRPC7 plays a critical role in synaptic plasticity at selected synapses in the principal trisynaptic circuit in the hippocampus. By reducing HFS-induced LTP at RC synapses, genetic ablation of TRPC7 hinders the generation of epileptiform burst firing in CA3 pyramidal neurons. The reduction of LTP at SC-CA1 synapses may also contribute to the reduction of Pilo-induced increase in gamma wave activity.
Fig. 5.
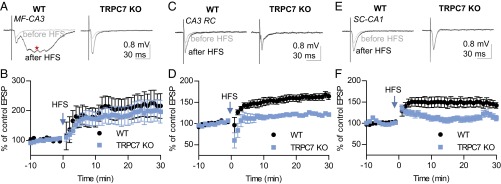
TRPC7 contributes to HFS-induced LTP at CA3 RC synapses and SC-CA1 synapses. (A) Representative traces of MF field EPSP recorded before and 30 min after high-frequency stimulation (HFS; 100 Hz, 1 s; repeated three times with a 20-s interval) in WT and TRPC7 KO mice. Traces shown were the average of 12 consecutive recordings collected at 0.1 Hz. Note the slow component resembling a paroxysmal depolarization shift after HFS (*). (B) Field EPSP slopes for each minute were determined by averaging six consecutive field EPSP recordings in each mouse, and the normalized means and SEs were plotted. There were no significant genotype effects (P > 0.05 for genotype effects, two-way ANOVA; n = 6, 5 for WT and TRPC7 KO mice). (C) Representative traces of CA3 RC field EPSP recorded before and 30 min after high-frequency stimulation (HFS; 100 Hz, 1 s; repeated three times with a 20-s interval) in WT and TRPC7 KO mice. Traces shown were the average of 12 consecutive recordings collected at 0.1 Hz. (D) Field EPSP slopes for each minute were determined by averaging six consecutive field EPSP recordings in each mouse, and the normalized means and SEs were plotted (P < 0.01 for genotype effects, two-way ANOVA; n = 6, 6 for WT and TRPC7 KO mice). (E) Representative traces of SC-CA1 field EPSP recorded before and 30 min after HFS (100 Hz, 1 s; repeated three times with a 20-s interval) in WT and TRPC7 KO mice. Traces shown were the average of 12 consecutive recordings collected at 0.2 Hz. (F) Field EPSP slopes for each minute were determined by averaging 12 consecutive field EPSP recordings in each mouse, and the normalized means and SEs were plotted (P < 0.01 for genotype effects, two-way ANOVA; n = 14, 12 for WT and TRPC7 KO mice).
Discussion
The main finding of this study is that TRPC7, the least understood member of the TRPC family, plays a critical role in the generation of seizures both in vitro and in vivo. Its contribution to synaptic plasticity, in particular at CA3 RC synapses, is central to the generation of epileptiform bursts in the CA3 region of the hippocampus in brain slices (Fig. 6). These bursts in turn sustain the epileptiform bursts in the EC-hippocampus circuit (13). Although other TRPC channels contribute both to the generation of seizures and seizure-induced neuronal cell death, TRPC7 is the only member required for the Pilo-induced increase in gamma wave activity that precedes cortical seizures in vivo.
Fig. 6.

TRPC7’s role in the initiation of seizures. We propose that TRPC7 is critical for mGluR1/mGluR5-mediated enhancement of NMDA receptor-dependent long-term potentiation at SC synapses in CA1 and RC synapses in CA3. The enhanced LTP at RC synapses in CA3 contributes to the initiation of epileptiform bursts in CA3 whereas the enhanced LTP at SC synapses contributes to hyperexcitability and the generation of ictal activities in CA1. EC, entorhinal cortex; DG, dentate gyrus; MF, mossy fiber; RC, recurrent collateral; SC, Schaffer collateral; Sub, subiculum; PF, perforant path.
Our data reveal the importance of increased gamma wave activity in the initiation of Pilo-induced SE. This increase precedes the first burst of cortical electrographic seizures by several minutes and is associated with stage I–III convulsive seizures on the Racine scale. It may provide the excitatory drive to an apparent “kindling” process that leads to the appearance of brief electrographic seizures. However, a causal relationship between the gamma wave activity and the subsequent SE has yet to be firmly established. There is a long transition period (more than 10 min) between the first burst of seizure to the SE state. This transition period is characterized by bursts of seizures followed by a silent period with greater inhibition of EEG activity of all wave bands, and this process repeats two to three times until the appearance of SE. It appears that both excitation and inhibition are enhanced during this transition period and it is the loss of inhibition after several repeats of this cycle that leads to the SE state.
Gamma waves play a critical role in learning and memory and are also implicated in a host of pathological conditions. The normal gamma wave is generated by the interplay between the inhibitory GABA interneurons and excitatory principal neurons. Because the baseline gamma wave appears to be normal in TRPC7 KO mice, it is unlikely that genetic ablation of TRPC7 significantly altered the normal neural network that permits the generation of gamma wave activity. The fact that the Pilo-induced increase in gamma wave is significantly reduced in TRPC7 KO mice suggests that a distinct mechanism underlies the gamma wave activity that precedes the generation of seizures. This distinction makes TRPC7 a highly attractive pharmacological target for antiepileptic drugs.
Genetic ablation of TRPC7 also disrupts LTP at CA3 RC synapses. It has been shown previously that epileptiform bursts in CA3 pyramidal neurons are generated by a NMDA receptor-dependent, mGluR5-dependent enhancement of synaptic strength at CA3 RC synapses (11, 12). Our findings suggest that TRPC7 plays a critical role in this process. The plateau potential underlying epileptiform burst firing was thought to be mediated by NMDA receptors (11). Therefore, one may expect that the amplitude and duration of the plateau potential will not be significantly reduced in TRPC7 KO mice. Contrary to expectations, our data indicates that the duration and the amplitude of the plateau are significantly reduced in TRPC7 KO mice. This finding suggests that TRPC7 may also amplify NMDA receptor-mediated responses.
The only previously known critical player in the generation of gamma waves and seizures is GluR6, a kainate receptor specifically expressed in the CA3 region of the hippocampus (17). Our data indicate that TRPC7, a little known member of TRPC family, plays a comparable role in the Pilo-induced SE model. Although the involvement of kainate receptors in the generation of seizures is well-known and long-suspected, the involvement of TRPC7 comes as a surprise because previous studies have suggested that other TRPC family members are more probable candidates. It is tempting to hypothesize an interaction between GluR6 and TRPC7, given the similar phenotypes of the knockout mice of GluR6 and TRPC7. Any evidence for such an interaction awaits future studies.
Materials and Methods
Animal experimental protocols were approved by the institutional animal care and use committee at University of Arkansas for Medical Sciences.
EEG Recording and Analysis.
Screws serving as EEG electrodes were placed on top of the dura through small holes drilled in the skull, and wires connected to each screw were soldered to a head mount secured to the skull using dental cement. A preamplifier connected the headmount to the AD/DA box (8200 series; Pinnacle Technology) mounted on a swivel plate to allow mice to move freely. EEG signals, sampled at 400 Hz, were recorded with integrated video and analyzed using Sirenia Seizure Pro software (Pinnacle Technology).
Electrophysiological Recordings.
Transverse or horizontal slices of adult mouse brain containing the hippocampus were obtained from 2- to 5-mo-old WT and TRPC7 KO mice (18) in a 129Sv/C57Bl6 mixed genetic background. The mice were anesthetized with ketamine (80 mg/kg) followed by decapitation. Serial 400-μm-thick sections were cut with a Vibraslice (WPI), as described previously (19), and allowed to recover in oxygenated artificial cerebrospinal fluid (ACSF) for at least 1 h at room temperature before recording. For intracellular recordings, glass microelectrodes filled with 3M sodium acetate were used as described previously (15). For field potential recordings, glass pipettes were pulled from filamented borosilicate glass filled with ACSF (15). Field EPSPs were recorded in the current-clamp mode with an Axoclamp 2B amplifier (Molecular Devices) and digitized using a model 1322A Digidata interface and pClamp 10 (Molecular Devices). For LTP experiments, the stimulus intensity was adjusted to produce a field EPSP approximately the half of maximal amplitude (Fig. S5).
Supplementary Material
Acknowledgments
This work was supported in part by the Intramural Research Program of the National Institutes of Health (Project Z01-ES-101864, to L.B.), by National Institute of Neurological Disorders and Stroke Grant NS 050381 (to F.Z.), and by the University of Arkansas for Medical Sciences Bridging Fund (F.Z.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1411442111/-/DCSupplemental.
References
- 1.Chin RFM, Neville BGR, Scott RC. A systematic review of the epidemiology of status epilepticus. Eur J Neurol. 2005;11(12):800–810. doi: 10.1111/j.1468-1331.2004.00943.x. [DOI] [PubMed] [Google Scholar]
- 2.Kapur J, Macdonald RL. Rapid seizure-induced reduction of benzodiazepine and Zn2+ sensitivity of hippocampal dentate granule cell GABAA receptors. J Neurosci. 1997;17(19):7532–7540. doi: 10.1523/JNEUROSCI.17-19-07532.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mazarati AM, Wasterlain CG. N-methyl-D-asparate receptor antagonists abolish the maintenance phase of self-sustaining status epilepticus in rat. Neurosci Lett. 1999;265(3):187–190. doi: 10.1016/s0304-3940(99)00238-4. [DOI] [PubMed] [Google Scholar]
- 4.Rice AC, DeLorenzo RJ. N-methyl-D-aspartate receptor activation regulates refractoriness of status epilepticus to diazepam. Neuroscience. 1999;93(1):117–123. doi: 10.1016/s0306-4522(99)00132-3. [DOI] [PubMed] [Google Scholar]
- 5.Birnbaumer L. The TRPC class of ion channels: A critical review of their roles in slow, sustained increases in intracellular Ca(2+) concentrations. Annu Rev Pharmacol Toxicol. 2009;49:395–426. doi: 10.1146/annurev.pharmtox.48.113006.094928. [DOI] [PubMed] [Google Scholar]
- 6.Birnbaumer L, Yildirim E, Abramowitz J. A comparison of the genes coding for canonical TRP channels and their M, V and P relatives. Cell Calcium. 2003;33(5-6):419–432. doi: 10.1016/s0143-4160(03)00068-x. [DOI] [PubMed] [Google Scholar]
- 7.Hofmann T, Schaefer M, Schultz G, Gudermann T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci USA. 2002;99(11):7461–7466. doi: 10.1073/pnas.102596199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goel M, Sinkins WG, Schilling WP. Selective association of TRPC channel subunits in rat brain synaptosomes. J Biol Chem. 2002;277(50):48303–48310. doi: 10.1074/jbc.M207882200. [DOI] [PubMed] [Google Scholar]
- 9.Mazzuferi M, Kumar G, Rospo C, Kaminski RM. Rapid epileptogenesis in the mouse pilocarpine model: Video-EEG, pharmacokinetic and histopathological characterization. Exp Neurol. 2012;238(2):156–167. doi: 10.1016/j.expneurol.2012.08.022. [DOI] [PubMed] [Google Scholar]
- 10.Ma J, Leung LS. Metabotropic glutamate receptors in the hippocampus and nucleus accumbens are involved in generating seizure-induced hippocampal gamma waves and behavioral hyperactivity. Behav Brain Res. 2002;133(1):45–56. doi: 10.1016/s0166-4328(01)00445-4. [DOI] [PubMed] [Google Scholar]
- 11.Csicsvari J, Jamieson B, Wise KD, Buzsáki G. Mechanisms of gamma oscillations in the hippocampus of the behaving rat. Neuron. 2003;37(2):311–322. doi: 10.1016/s0896-6273(02)01169-8. [DOI] [PubMed] [Google Scholar]
- 12.Bains JS, Longacher JM, Staley KJ. Reciprocal interactions between CA3 network activity and strength of recurrent collateral synapses. Nat Neurosci. 1999;2(8):720–726. doi: 10.1038/11184. [DOI] [PubMed] [Google Scholar]
- 13.Stoop R, Conquet F, Zuber B, Voronin LL, Pralong E. Activation of metabotropic glutamate 5 and NMDA receptors underlies the induction of persistent bursting and associated long-lasting changes in CA3 recurrent connections. J Neurosci. 2003;23(13):5634–5644. doi: 10.1523/JNEUROSCI.23-13-05634.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Funahashi M, Stewart M. Properties of gamma-frequency oscillations initiated by propagating population bursts in retrohippocampal regions of rat brain slices. J Physiol. 1998;510(Pt 1):191–208. doi: 10.1111/j.1469-7793.1998.191bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phelan KD, et al. Canonical transient receptor channel 5 (TRPC5) and TRPC1/4 contribute to seizure and excitotoxicity by distinct cellular mechanisms. Mol Pharmacol. 2013;83(2):429–438. doi: 10.1124/mol.112.082271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwon H-B, Castillo PE. Long-term potentiation selectively expressed by NMDA receptors at hippocampal mossy fiber synapses. Neuron. 2008;57(1):108–120. doi: 10.1016/j.neuron.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mulle C, et al. Altered synaptic physiology and reduced susceptibility to kainate-induced seizures in GluR6-deficient mice. Nature. 1998;392(6676):601–605. doi: 10.1038/33408. [DOI] [PubMed] [Google Scholar]
- 18.Perez-Leighton CE, Schmidt TM, Abramowitz J, Birnbaumer L, Kofuji P. Intrinsic phototransduction persists in melanopsin-expressing ganglion cells lacking diacylglycerol-sensitive TRPC subunits. Eur J Neurosci. 2011;33(5):856–867. doi: 10.1111/j.1460-9568.2010.07583.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Phelan KD, et al. Heteromeric canonical transient receptor potential 1 and 4 channels play a critical role in epileptiform burst firing and seizure-induced neurodegeneration. Mol Pharmacol. 2012;81(3):384–392. doi: 10.1124/mol.111.075341. [DOI] [PMC free article] [PubMed] [Google Scholar]