

Significance
Nonreceptor tyrosine kinases are major players in cell signaling. Among them, focal adhesion kinase (FAK) is the key integrator of signals from growth factors and cell adhesion. In cancer, FAK is frequently overexpressed, and by promoting adhesion to the tumor stroma and ECM, FAK provides important signals for tumor invasion and metastasis. Although autoinhibitory mechanisms have previously been described and the players involved in FAK regulation are largely known, on a mechanistic level, FAK activation is currently not understood. Here, we present a multidisciplinary approach demonstrating a multistep mechanism resulting in FAK activation. This mechanistic insight enables the design of alternative strategies for the discovery of potential anticancer drugs that inhibit both catalytic and scaffolding functions of FAK with high specificity.
Keywords: cell signaling, phosphoinositides
Abstract
Focal adhesion kinase (FAK) is a nonreceptor tyrosine kinase (NRTK) with key roles in integrating growth and cell matrix adhesion signals, and FAK is a major driver of invasion and metastasis in cancer. Cell adhesion via integrin receptors is well known to trigger FAK signaling, and many of the players involved are known; however, mechanistically, FAK activation is not understood. Here, using a multidisciplinary approach, including biochemical, biophysical, structural, computational, and cell biology approaches, we provide a detailed view of a multistep activation mechanism of FAK initiated by phosphatidylinositol-4,5-bisphosphate [PI(4,5)P2]. Interestingly, the mechanism differs from canonical NRTK activation and is tailored to the dual catalytic and scaffolding function of FAK. We find PI(4,5)P2 induces clustering of FAK on the lipid bilayer by binding a basic region in the regulatory 4.1, ezrin, radixin, moesin homology (FERM) domain. In these clusters, PI(4,5)P2 induces a partially open FAK conformation where the autophosphorylation site is exposed, facilitating efficient autophosphorylation and subsequent Src recruitment. However, PI(4,5)P2 does not release autoinhibitory interactions; rather, Src phosphorylation of the activation loop in FAK results in release of the FERM/kinase tether and full catalytic activation. We propose that PI(4,5)P2 and its generation in focal adhesions by the enzyme phosphatidylinositol 4-phosphate 5-kinase type Iγ are important in linking integrin signaling to FAK activation.
Cell attachment to the ECM is mediated via integrin transmembrane receptors on the cell surface. Integrin engagement to ECM components results in activation and clustering of integrins. In response to integrin activation, a large number of proteins are recruited to their cytoplasmic tails, resulting in the formation of focal adhesions (FAs) (1). On the one side, FAs are anchoring points for actomyosin stress fibers, which allow tension forces to build up when contracting fibers exert their pulling force via FAs against the ECM. On the other hand, integrin activation and the generation of tension trigger intricate signaling cascades. A central signaling component in FAs is the nonreceptor tyrosine kinase (NRTK) focal adhesion kinase (FAK). FAK is activated downstream of integrins, and its signaling is important for cell migration, proliferation, and survival (2, 3). FAK contains numerous binding sites for other signaling and adaptor proteins, and it has been identified as a hub in the focal adhesion (FA) interactome (4). In addition to its role as a signaling kinase, FAK is therefore thought to function as a signaling scaffold. FAK is required for diverse processes in development, wound healing, and disease (5–7). FAK KO mice are not viable due to mesodermal defects (8), and early studies with FAK KO cells indicated that FAK is important in FA turnover by inhibiting Rho activity (9, 10). Subsequent studies portray a more complex picture (11) and indicate that FAK is also involved in increasing adhesion strength, particularly in response to tension forces (12, 13). FAK is frequently overexpressed in various human cancers (14). Its overexpression highly correlates with tumor invasiveness; hence, FAK is widely pursued as a drug target for cancer therapy (15, 16).
FAK is a 120-kDa multidomain protein containing an N-terminal 4.1, ezrin, radixin, moesin homology (FERM) domain, followed by a 50-residue linker, a central kinase domain, an ∼220 residue low-complexity proline-rich region, and a C-terminal focal adhesion targeting (FAT) domain (Fig. 1A). Whereas the FAT domain is important for targeting FAK to FAs through interactions with paxillin (17–19), the FERM domain is responsible for regulating catalytic activity (20). In the autoinhibited state, the FERM domain docks onto the kinase domain, which results in catalytic inhibition and sequesters regulatory phosphorylation sites (21). FAK activation initially results in autophosphorylation of Y397 in the linker between the FERM and kinase domains (22), a process reported to occur in trans (23). Phosphorylated Y397 provides a docking site for the Src homology 2 (SH2) domain of the Src kinase, and recruited Src phosphorylates several tyrosines in FAK. Two of them (Y576 and Y577) are located in the activation loop of FAK, and their phosphorylation confers full catalytic activity (24). The activated FAK/Src complex phosphorylates several FA proteins, including paxillin (25) and p130Cas (26).
Fig. 1.

FAK interacts with PI(4,5)P2 via the basic patch in the FERM domain. (A) Domain structure of FAK with the main phosphorylation sites indicated and a ribbon diagram of the FK-FAK crystal structure as reported by Lietha et al. (21) (PDB ID code 2J0J). In the zoom window, the interaction between the FERM F2 lobe and the kinase C-lobe is shown, with residues Y180 and M183 at the interface colored green and the basic KAKTLRK residues (K216, K218, R221, and K222) colored magenta. (B) Lipid binding specificity of FK-FAK was studied using vesicle pull-down assays with PC vesicles containing 6% (mol/mol) of the indicated phospholipids. Phosphorylation of the D4 and D5 positions of the inositol head group confers full binding affinity. PS, phosphatidyl serine. (C) Vesicle pull-downs with 6% (mol/mol) PI(4,5)P2 vesicles and GST-fused F-FAKwt (FERM = FAK31-405) or F-FAK180/183A (Y180 and M183 mutated to alanine). The mutations do not affect PI(4,5)P2 binding. GST fusions were used to obtain higher readouts. (D) PI(4,5)P2 vesicle pull-downs with FK-FAKwt, FK-FAK180/183A, FK-FAK-KAKTLRK (all KAKTLRK basic residues are mutated to alanine), or FL-FAKwt. Y180/M183A mutations result in ∼2.5-fold higher affinity to PI(4,5)P2, whereas the KAKTLRK mutations abolish binding. FL-FAK binds with similar affinity to FK-FAK. ND, not determined. (E) Vesicle pull-downs with 1.5% (mol/mol), 6% (mol/mol), or 12% (mol/mol) PI(4,5)P2 and FK-FAKwt. Increasing the PI(4,5)P2 density on vesicles results in higher affinity for FK-FAKwt, indicating an avidity effect. (C–E) Error bars represent SD from three independent experiments and are shown if larger than the symbol. Kd values are determined by fitting a one-site binding model (cooperative fitting is shown in Fig. S1 B and C).
Many stimuli have been reported to initiate FAK activation (reviewed in ref. 27), such as integrin signaling and engagement of growth factor receptors (28–30), but molecular details remain elusive. Further, we showed that acidic phosphoinositides, such as phosphatidylinositol-4,5-bisphosphate [PI(4,5)P2], interact with FAK and play a role in FAK activation (31). PI(4,5)P2 is well established as a modulator of FA maturation and adhesion strength, but its role in adhesion signaling is less clear. PI(4,5)P2 promotes formation of mature FAs by binding talin and vinculin and inducing their open state, where binding sites to other FA proteins and actin are exposed (32, 33). PI(4,5)P2 is locally generated in FAs by the enzyme phosphatidylinositol 4-phosphate 5-kinase type Iγ (PIP5KIγ) (34, 35), which adds the 5-phosphate to PI(4)P. PIP5KIγ exists as two splice variants (PIP5KIγ661 and PIP5KIγ635 in mice), where the longer form (PIP5KIγ661, PIP5KIγ668 in humans) contains extra C-terminal residues that target the enzyme to FAs by interacting with the talin head domain (36, 37).
Although the main players involved in FAK activation have been identified at a mechanistic level, it is not known how their concerted action is orchestrated to achieve FAK activation. Here, we probe the molecular mechanism of FAK activation using a multidisciplinary approach, including biochemical, structural, in vitro FRET, molecular dynamics (MD) simulation, and cell biology studies, and we present evidence that PIP5KIγ and its product, PI(4,5)P2, are important mediators of the integrin/FAK signaling link. Intriguingly, we find that PI(4,5)P2 binding to a basic region on the FAK FERM domain results in clustering of FAK on the lipid membrane, a process likely to enhance integrin clustering as well as the scaffolding function of FAK. PI(4,5)P2 binding further induces conformational changes between FERM and kinase domains without causing domain dissociation. In vitro FRET experiments, together with MD simulations, support a scenario where distal changes at the PI(4,5)P2 binding site result in domain opening and exposure of linker regions, which, together with clustering, promote efficient autophosphorylation of Y397 within the linker. FAK autophosphorylation recruits Src, and, in turn, Src is responsible for full activation of FAK and FERM release by phosphorylating the FAK activation loop. The mechanistic insight we present here can aid the design of novel classes of therapeutics targeting both catalytic and scaffolding functions of FAK.
Results
FAK Binds PI(4,5)P2 via Basic Residues in the FERM F2 Lobe.
To characterize the FAK–PI(4,5)P2 interaction in detail, we performed in vitro binding studies. Using vesicle pull-down experiments, we initially analyzed the phosphoinositide specificity and find that bis-phosphorylation on the D4 and D5 positions of the inositol ring is required for full binding affinity, whereas additional phosphorylation on the D3 position has no effect (Fig. 1B). This finding suggests the enzyme PIP5KIγ as a key enzyme regulating FAK signaling because it is the enzyme generating PI(4,5)P2 in FAs. Other phospholipids, such as PI, phosphatidyl serine, or phosphatidyl choline (PC), only display background levels of binding. The FERM domain of FAK (F-FAK) is sufficient for full PI(4,5)P2 binding affinity (Fig. 1C), and binding is mediated via a conserved basic region in the FERM F2 lobe, as shown by mutation of the basic KAKTLRK sequence (all K/R to A = FAK-KAKTLRK; Fig. 1 A and D). Interestingly, the FERM + kinase fragment of FAK (FK-FAK) interacts with approximately twofold lower affinity than F-FAK (Fig. 1 C and D). In addition, a mutant form of FK-FAK, where residues Y180 and M183 in the FERM F2 lobe are mutated to alanine (FK-FAK180/183A), displays, like F-FAK, higher PI(4,5)P2 affinity than WT (FK-FAKwt) (Fig. 1 A and D). This observation is supported by the differential association of FK-FAKwt and FK-FAK180/183A to PI(4,5)P2 vesicles in surface plasmon resonance experiments (Fig. S1A). The residues Y180 and M183 are located at the region of the FERM F2 lobe that is responsible for kinase binding and autoinhibition (21) (Fig. 1A). Using small-angle X-ray scattering, we show that the 180/183A mutant of FK-FAK adopts a monomeric and open conformation with FERM and kinase domains dissociated (Fig. S2 and Table S1), whereas FK-FAKwt adopts a closed conformation (Fig. S2 and ref. 21). We therefore conclude that the reduced affinity of closed FK-FAKwt (Fig. 1D and Fig. S1A) is due to an energetically costly conformational change required to bind PI(4,5)P2. Further, we find that C-terminal regions of FAK do not affect PI(4,5)P2 binding, because full-length FAK (FL-FAK) exhibits a similar PI(4,5)P2 affinity as FK-FAK (Fig. 1D). Importantly, the PI(4,5)P2 affinity of FAK is altered by the PI(4,5)P2 density on lipid vesicles, indicating an avidity effect (Fig. 1E). For simplicity, we use a one-site model to fit our pull-down data presented in Fig. 1 C–E; however, the data fit well with a cooperative model, indicating positive cooperatively with a Hill coefficient of ∼2 (Fig. S1 B and C).
PI(4,5)P2 Enhances FAK Autophosphorylation Without Increasing Catalytic Turnover.
Next, we tested the effect of PI(4,5)P2 on FAK activity. Using an autophosphorylation assay, we find that PI(4,5)P2 vesicles strongly increase the autophosphorylation efficiency, as shown by immunoblotting using an antibody against the autophosphorylation site Y397 (Fig. 2 A and B), whereas PI(4)P, PI(5)P, and PI(3,4)P2 have no effect (Fig. S3A). As is the case for PI(4,5)P2 binding, enhanced autophosphorylation requires the basic KAKTLRK region. Using soluble PI(4,5)P2 [with eight carbons in the acyl chain, C8-PI(4,5)P2], we measured autophosphorylation by ELISA and find that C8-PI(4,5)P2 increases the autophosphorylation efficiency of FK-FAKwt to similar levels as observed for FK-FAK180/183A (Fig. 2C). Although the PI(4,5)P2 head group is necessary for this effect [compare C8-PI(4,5)P2 vs. the C8-PC plot in Fig. 2C], the head group alone [Ins(1,4,5)P3] is not sufficient. Remarkably, neither soluble nor vesicle-embedded PI(4,5)P2 affects the catalytic activity, as measured by ATP turnover in a kinetic assay with an exogenous substrate (Fig. 2D and Fig. S3 C and D). The fact that dissociation of FERM and kinase domains by mutation does increase ATP turnover, as shown with the FK-FAK180/183A mutant (Fig. 2D and Fig. S3 C and D), suggests that PI(4,5)P2 binding to FAK does not dissociate the FERM from the kinase domain.
Fig. 2.

PI(4,5)P2 mediates FAK autophosphorylation but not catalytic turnover. (A) Autophosphorylation time course of FL-FAK, FK-FAKwt, and FK-FAK-KAKTLRK in the absence (buffer) or presence of PI(4,5)P2 or PC vesicles was monitored by immunoblotting using an anti-pY397 antibody. (Lower) Loading controls stained by Coomassie blue. Note that FL-FAK stains stronger because of its higher molecular weight. (B) Quantifications of blots from A relative to loading controls [using ImageJ (National Institutes of Health)]. For FL-FAK and FK-FAKwt, autophosphorylation is significantly faster in the presence of PI(4,5)P2 vesicles, whereas mutations in the basic patch of FK-FAK-KAKTLRK abrogate this effect. (C) Autophosphorylation efficiency of FK-FAK (wt or 180/183A mutant) was assessed using an ELISA method. The presence of C8-PI(4,5)P2, but not C8-PC or the head group Ins(1,4,5)P3, enhances autophosphorylation of FK-FAKwt to levels similar to FK-FAK180/183A, which was not affected by PI(4,5)P2. (D) Catalytic steady-state activity was assayed for the indicated FAK proteins using a kinase assay, which couples ADP production to NADH consumption (Methods). Whereas dissociation of FERM/kinase domains by mutation (180/183A) activates FAK, none of the tested lipids increase catalytic turnover. (C and D) Error bars represent SD from three experiments.
PI(4,5)P2 Induces FAK Clustering.
Because FAK has been shown to autophosphorylate efficiently in trans (23), we considered the possibility that PI(4,5)P2-induced autophosphorylation might be mediated via FAK oligomers. Using negative-stain transmission EM, we show that FAK forms clusters when bound to PI(4,5)P2 vesicles, as well as bound to soluble C8-PI(4,5)P2 (Fig. 3A). We performed reference-free 2D class averaging of 574 selected FL-FAK/C8-PI(4,5)P2 particles, which likely represent a main cluster population (Fig. 3B). Based on 3D volumes generated from 2D averages or particle dimensions, we determined an approximate particle size of 900 kDa, from which we estimate the presence of approximately six to eight FL-FAK molecules per cluster. Like PI(4,5)P2 binding, formation of clusters requires the basic KAKTLRK region on the F2 lobe (Fig. 3C, Right). We further analyzed clustering of F-FAK and FK-FAK by dynamic light scattering (DLS), which confirms a clear increase in molecular weight in the presence of PI(4,5)P2, both for F-FAK and FK-FAK (Fig. S4 and Table S2). DLS also shows that PI(4,5)P2 induces significant polydispersity, indicating that the determined size of clusters by EM likely represents a main population induced by C8-PI(4,5)P2 but that the relevant FAK-PI(4,5)P2 complex possibly does not adopt a defined oligomerization state. We note that C8-PI(4,5)P2–induced clusters do not form as a consequence of micelle formation, because we determined a critical micelle concentration (CMC) for C8-PI(4,5)P2 of 2 mM, an order of magnitude higher than used in our study.
Fig. 3.

EM reveals PI(4,5)P2-induced FAK clustering. (A) Transmission electron micrographs of FL-FAK with lipid vesicles (Upper) or soluble lipids (Lower) imaged by negative staining. PI(4,5)P2 vesicles and soluble C8-PI(4,5)P2 mediate the formation of FAK clusters. Clusters on vesicles are indicated by arrowheads. (B) Reference-free 2D class averages of 574 FL-FAK/C8-PI(4,5)P2 clusters, representing a main cluster population, suggest a circular arrangement of FAK molecules in clusters. Volume calculations suggest that clusters consist of six to eight FL-FAK molecules per cluster. (C) FK-FAKwt and the 180/183A and KAKTLRK mutants were imaged in the presence of C8-PI(4,5)P2 or C8-PC. FK-FAKwt and FK-FAK180/183A, but not the KAKTLRK mutant, display clustering in the presence of PI(4,5)P2. (Scale bars: A and C, 50 nm.)
PI(4,5)P2 Prevents Formation of a Fully Closed Conformation.
As described above, our data indicate that PI(4,5)P2 does not induce dissociation of FERM and kinase domains. However, the fact that the open FK-FAK180/183A mutant exhibits higher affinity for PI(4,5)P2 than closed FK-FAKwt (Fig. 1D and Fig. S1A) suggests that closed FK-FAK requires a rearrangement of FERM and kinase domains to allow PI(4,5)P2 binding. To monitor conformational changes in a controlled environment, we used in vitro FRET experiments utilizing a conformational sensor of FAK. A similar sensor was used previously in cellular studies to demonstrate that FAK undergoes conformational changes in FAs (31). The sensor is based on intramolecular FRET by fusing CFP and citrine N-terminal to the FERM and kinase domains, respectively, and it is designed to report relative domain positions, with autoinhibited (closed) FAK exhibiting high FRET and open forms displaying lower FRET signals. We used three purified versions of the sensor that contain the FERM and kinase regions of FAK (Fig. 4A): (i) a sensor with CFP and citrine inserted into the WT FAK sequence (CYFAKwt); (ii) a sensor that is mutated to adopt an open conformation (CYFAK180/183); and (iii) a high-FRET control sensor (CYFAK-HFC) that has the FRET labels in tandem at the N terminus, and hence displays high-FRET signals independent of the intramolecular FAK conformation. Changes in FRET levels of the CYFAK-HFC sensor indicate nonconformational effects, such as trans-FRET or fluorescence quenching (which, for example, could be caused by clustering). As expected, we find lower FRET levels for the open CYFAK180/183A sensor than for CYFAKwt and high-FRET levels for CYFAK-HFC [Fig. 4B (leftmost bars in each graph) and Fig. S5A]. The addition of C8-PC does not affect FRET levels; however, C8-PI(4,5)P2 induces a reduction in FRET signals for all three sensors (Fig. 4 B and C). Because PI(4,5)P2 also affects CYF-HFC, which reports nonconformational effects, we conclude that under these conditions (without ATP, see below), PI(4,5)P2 does not significantly alter the conformation of FAK.
Fig. 4.
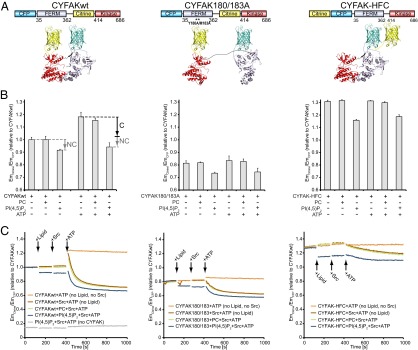
PI(4,5)P2 induces partial and Src induces full domain opening of the FAK FRET sensor in the presence of ATP. (A) Schematic illustration of the domain structure (Upper) and expected structural arrangement (Lower) of the conformational FRET sensors used. (B) Emission ratios of citrine (EmCitrine) and CFP (EmCFP) are plotted relative to CYFAKwt as a measure of FRET. The presence of C8-PI(4,5)P2 causes a reduction in FRET levels for all three sensors, suggesting that this effect is not conformational (labeled NC). The presence of ATP significantly increases FRET levels for CYFAKwt; however, this increase is reversed in the presence of PI(4,5)P2. This effect of ATP and PI(4,5)P2 is not seen for CYF-HFC, and is therefore likely conformational (labeled C). Error bars represent SD from a minimum of three experiments. (C) Relative FRET levels were monitored in real time, initially of the sensors alone and then following addition of (i) lipid [C8-PC (green plots) and C8-PI(4,5)P2 (blue plots)] or no lipid (brown and orange plots), (ii) Src (brown, green, and blue plots) or no Src (orange plots), and (iii) ATP/Mg2+ (all plots). As in B, PI(4,5)P2 reduces FRET levels of all sensors in all states (before/after phosphorylation), indicating a nonconformational effect. ATP results in a FRET spike only with CYFAKwt. If active Src is present, the spike is followed by a switch to the open conformation (lower FRET levels). In contrast, inactive SrcK298M induces only a modest FRET decrease (Fig. S5C). (Left) Gray control plot is without the CYFAK sensor to verify that PI(4,5)P2, Src, and ATP do not exhibit intrinsic fluorescence.
Interestingly, when we added ATP and Mg2+ to CYFAKwt, the FRET signal increased almost to the level of CYF-HFC (Fig. 4 B and C). This effect is not seen with CYFAK-HFC, and is therefore likely conformational, with higher FRET signals suggesting a more closed conformation in the presence of ATP. This increase in FRET is not due to a phosphorylation event, because the same effect is seen with the nonhydrolyzable ATP analog 5′-adenylylimidodiphosphate (AMP-PNP) (Fig. S5B). Importantly, the high-FRET state with ATP is not observed in the presence of PI(4,5)P2 (Fig. 4 B and C, Left) and, in fact, is reverted in a concentration-dependent manner by PI(4,5)P2 if added after ATP (Fig. S5D). This indicates a conformational effect of PI(4,5)P2 in the presence of ATP. Because ATP is present in the cell at similar concentrations that we used in our experiments (1 mM), the high-FRET state observed in the presence of ATP likely represents the basal conformation of FAK, which is affected by PI(4,5)P2. When comparing FRET levels of CYFAKwt in the presence of ATP with and without PI(4,5)P2, we can partition the FRET change into a nonconformational effect of PI(4,5)P2, which is also seen without ATP (NC in Fig. 4B), and a larger conformational effect (C in Fig. 4B). The nonconformational effect is linear (CYFAK-HFC plot in Fig. S5D, Left), which can be corrected to observe only the conformational contribution of PI(4,5)P2 (Fig. S5D, Right). At high PI(4.5)P2 concentrations, CYFAKwt in the presence of ATP approaches FRET levels without ATP. The FRET change even at high PI(4,5)P2 concentrations does, however, not correspond to full domain dissociation (see below). Together, these data suggest that in the presence of ATP, PI(4,5)P2 induces partial domain opening.
In experiments where Src was added to CYFAKwt and ATP, the initial increase in FRET is followed by a switch to an open conformation (lower FRET signals; Fig. 4C, Left), whereas only a minor effect is observed for the CYFAK-HFC control. The kinase-dead mutant SrcK298M has a small effect on FRET levels in presence of ATP (Fig. S5C), indicating that Src phosphorylation is mainly responsible for switching CYFAKwt to the open conformation and that Src binding has a minor effect. This is consistent with observations that Src phosphorylation of the FAK activation loop is incompatible with FERM inhibition (21). Comparing FRET signals of different states corroborates that PI(4,5)P2 does not induce full FERM/kinase dissociation, which is observed only upon Src phosphorylation or 180/183A mutation.
Allosteric Effects of PI(4,5)P2 on the FERM/Kinase Interface.
Because the two opposite effects of PI(4,5)P2 and ATP appear to be related, we first proceeded to understand the effect of ATP better. Using MD simulations, we monitored backbone fluctuations of the solvated FAK domain in the presence or absence of ATP over 1,500 ns. We find that the presence of ATP in the active site has the largest stabilizing effect in the αC- and αG-helices of the FAK domain (Fig. 5A). Strikingly, these two sites exactly map the autoinhibitory interaction sites with the FERM domain, as seen in the crystal structure of FK-FAK (21) (Fig. 5B). In accordance with FRET and the simulation data, we therefore propose that binding of ATP to FAK induces a tightly closed FERM/kinase conformation by rigidifying the interaction interfaces. Moreover, by performing an elastic network-based allosteric connectivity analysis of the FAK domain, we find, by a completely independent computational approach (38), a strong allosteric coupling between αC-and αG-helices despite their distal locations (Fig. S6).
Fig. 5.

Effects of ATP and PI(4,5)P2 on FAK conformation. (A) Root mean square fluctuations (RMSFs) from unbiased MD simulations of the FAK kinase domain are shown for a window of 700 ns (discarding the first 200 ns of equilibration) for FAK alone (blue plot) and for FAK with ATP/Mg2+ (red plot). ATP binding stabilizes the αC- and αG-helices. (B) RMSF values from A are color-mapped onto the FAK structure. RMSFs range from blue (low values) to red (high values). Stabilizing ATP effects on αC- and αG-helices map autoinhibitory interaction sites as seen in the FK-FAK crystal structure (21). Neutralization of the basic patch by PI(4,5)P2 binding (C) or by mutation (D) interferes with a set of FERM F2 lobe-stabilizing salt bridges. MD simulations suggest that salt bridges between K218/R221 in the basic patch and E195/E198 are formed in the absence of PI(4,5)P2 (C, Right) but not in the presence of PI(4,5)P2 (C, Left), leading to a partial destabilization of the FERM F2 lobe and an altered force distribution, as shown in Fig. S7A. (D) Minimum distances for the residues K218/R221 and residue pair E195/E198 are shown during MD simulations with FK-FAK alone (MD1 and MD2), FK-FAK bound to PI(4,5)P2, or the basic patch mutant FK-FAK-KAKTLRK. In the two independent MD simulations of FK-FAKwt, these two pairs of oppositely charged residues strongly interact with each other. PI(4,5)P2 binding or KAKTLRK mutations to alanines significantly increase the minimum distances and fluctuations. (E) Crystal structure of the basic patch mutant FERM domain (F-FAK-KAKTLRK; PDB ID code 3ZDT; full structure is shown in Fig. S7C). In contrast to F-FAKwt (Lower, PDB ID code 4CYE), the structure of F-FAK-KAKTLRK (Upper) exhibits no electron density for the loop (cyan) containing the autoinhibitory residues Y180/M183 (green), indicating that this loop is disordered. The 2Fo-Fc electron density maps are shown as gray mesh countered at 1σ.
We then proceeded to probe conformational effects of PI(4,5)P2 by performing MD simulations of (i) FK-FAK alone, (ii) FK-FAK bound to soluble PI(4,5)P2 [C2-PI(4,5)P2], or (iii) FK-FAK with the basic patch mutant FK-FAK-KAKTLRK. Interestingly, we find that PI(4,5)P2 binding results in altered interresidue forces within the FERM F2 lobe that originate at the basic patch and propagate to the interface with the kinase C-lobe (Fig. S7A, Upper). In particular, PI(4,5)P2 binding interferes with a set of stabilizing salt bridges between the basic patch (K218/R221) and the adjacent helix at the domain interface (E195/E198, Fig. 5 C and D). These PI(4,5)P2-induced changes also affect the loop containing the autoinhibitory residues Y180 and M183. This effect was reproduced in MD simulations of FK-FAK-KAKTLRK [Fig. 5D and Fig. S7 A (Lower) and B], suggesting that neutralizing the charges of the basic KAKTLRK residues by mutation has a similar effect on the FERM F2 lobe interatomic forces as PI(4,5)P2 binding. We validated our model for the PI(4,5)P2–FERM F2 lobe interaction by performing MD simulations with PI(4,5)P2 embedded in a lipid bilayer, and we find that the mode of the FERM–PI(4,5)P2 interaction is highly similar. PI(4,5)P2 remains embedded in the bilayer (at least within the time scale of the simulations) such that the PI(4,5)P2–FAK interaction is restricted to the head group; therefore, like with short-chain lipids, MD simulations primarily provide details on a charge neutralization effect.
In the absence of a FAK-PI(4,5)P2 cocrystal structure, which is complicated by clustering, as seen in Fig. 3 and Fig. S4, we pursued a crystal structure of the basic patch mutant FERM domain (F-FAK-KAKTLRK, crystallographic Table S3). Although the FERM domain retains a very similar conformation to F-FAKwt overall, in both independent molecules in the asymmetrical unit cell, no electron density is observed for the loop Y180-K190 (containing the autoinhibitory residues Y180/M183), indicating that the loop is disordered (Fig. 5E and Fig. S7C). This is in striking agreement with observations from the MD simulations described above, which find a destabilized F2 lobe upon neutralization of the basic region in the FERM F2 lobe (Fig. 5 C and D and Fig. S7 A and B). We note that the disorder could potentially be due to less stable crystal contacts, and, in fact, one F-FAKwt structure [Protein Data Bank (PDB) ID code 2AL6] does exhibit high B-factors in this region; however, the most similar packing environment is observed in F-FAKwt with PDB ID codes 4CYE and 2AEH (chain A), where the loop is ordered (Fig. S7D). In conclusion, we propose that neutralization of the basic patch by specific binding of PI(4,5)P2 destabilizes the FERM F2 lobe, resulting in an altered FERM F2/kinase C-lobe interface, which allows partial domain opening.
PI(4,5)P2 Enhances Src-Mediated FAK Activation.
FAK activation proceeds through a multistep mechanism. PI(4,5)P2-induced FAK autophosphorylation is followed by Src recruitment to the autophosphorylation site and phosphorylation of the activation loop of FAK (residues Y576 and Y577) by Src. Because Src recruitment requires FAK autophosphorylation, it can be expected that PI(4,5)P2 should also enhance Src-mediated phosphorylation of the activation loop of FAK. To test this, we monitored Src phosphorylation of Y576 in the presence or absence of PI(4,5)P2 by immunoblotting, using an antibody against phospho-Y576, following an autophosphorylation step (Fig. 6). To uncouple autophosphorylation from Src phosphorylation, autophosphorylation was stopped after 2 min with the specific FAK inhibitor TAE226 before starting Src reactions (details are provided in Methods). As expected, we find that Y576 phosphorylation is more efficient in the presence of PI(4,5)P2, whereas other tested phosphoinositides have no significant effect (Fig. S3B). In part, this effect is due to enhanced FAK autophosphorylation, because in the absence of autophosphorylation (for FK-FAK Y397F), the Y576 phosphorylation efficiency of Src is reduced. However, PI(4,5)P2 also enhances Src phosphorylation of the FK-FAK Y397F mutant, suggesting an autophosphorylation-independent effect. These data are consistent with PI(4,5)P2 inducing a partially open conformation that allows easier access to Y576.
Fig. 6.

PI(4,5)P2 enhances Src phosphorylation of Y576 in FAK. (A) Time course of Src phosphorylation of FK-FAK in the absence or presence of PI(4,5)P2 or PC vesicles as monitored by Western blotting using an anti-pY576 antibody following a 2-min autophosphorylation reaction (Methods). Mutation of the autophosphorylation site (Y397F) significantly reduces the Y576 phosphorylation rate, whereas PI(4,5)P2 enhances Y576 phosphorylation for FK-FAKwt and FK-FAK-Y397F, indicating a combined effect of more efficient Src recruitment to the FAK autophosphorylation site and PI(4,5)P2-induced conformational changes. (Lower) Coomassie blue-stained loading controls are shown. (B) Quantifications of blots in A.
PI(4,5)P2 in FAs Promotes FAK Signaling and Cell Adhesion.
We have previously proposed acidic phospholipids to be involved in FAK activation in cells (31); however, the lipid specificity (and hence the relevant upstream components) was not defined. Because we demonstrate the requirement of D4 and D5 phosphorylation of phosphoinositides (Fig. 1B), and because PIP5KIγ is known to be responsible for PI(4,5)P2 generation in FAs, we performed PIP5KIγ loss-of-function experiments in HeLa cells to establish the role of PI(4,5)P2 in FAK signaling in FAs. The expression of PIP5KIγ was knocked down using a specific shRNA against isoform 2 of PIP5KIγ (the longer PIP5KIγ668 isoform), and two different stable knockdown (KD) clones were selected (KD1 and KD2). The KD efficiency was very high, as observed by Western blotting and immunofluorescence (Fig. 7A and Fig. S8A). The total levels of FAK are not changed in the absence of PIP5KIγ (Fig. 7B). However, Y397 autophosphorylation and Y576/Y577 Src phosphorylation are significantly reduced at early stages of focal adhesion formation, compared with the scramble controls (Fig. 7B). Overall, these data suggest that PIP5KIγ may function in the proper activation of FAK. To validate the causal role of PIP5KIγ in the activation of FAK, we performed rescue experiments using vectors encoding either a WT (PIP5KIγ-WT) or kinase-dead (PIP5Kγ-D316A) form of PIP5KIγ. As shown in Fig. 7C, on a PIP5Kγ-deficient background, expression of PIP5KIγ-WT not only restores but increases the phosphorylation levels of pFAKY397 compared with the scramble controls. By contrast, the expression of the PIP5Kγ-D316A kinase-dead mutant in PIP5Kγ-deficient cells is not able to rescue pFAKY397 levels fully (Fig. 7C). Overall, these data provide evidence that PIP5Kγ activity is required for the activation of FAK, at least in certain cellular contexts.
Fig. 7.

PIP5KIγ KD decreases cellular FAK activity and cell attachment. (A) Expression levels of PIP5KIγ668 (isoform 2) in HeLa cells after KD are shown for two different clones (KD1 and KD2) and scramble control (Ctr). (Upper) Representative immunoblot is shown. (Lower) Quantification of PIP5KIγ levels observed by immunoblotting is depicted in the histogram. Quantifications are from four blots (two independent experiments, each in duplicate). (B) Effect of PIP5KIγ reductions on the total level of FAK and on the total cellular levels of pFAK Y397, pFAK Y576, and pFAK Y577. A representative immunoblot and quantifications from four blots (two independent experiments, each in duplicate) are shown. (C) pFAKY397 levels in HeLa cells of Ctr or KD1 and KD2 cells transfected with WT (PIP5KIγ-WT) or a kinase-dead (PIP5Kγ-D316A) form of PIP5KIγ. Immunoblots and quantifications of two independent experiments are shown, with each performed twice. Note that PIP5KIγ-WT more than rescues but the kinase-dead mutant does not fully rescue FAK phosphorylation levels in KD cells compared with controls. The dashed line merges different lanes of the same immunoblot experiment. (D) Immunofluorescence staining of pFAK397 (red) and total FAK (green) in PIP5KIγ KD1 and KD2 clones and Ctr, and quantification of the immunofluorescence intensity of pFAK397 relative to total FAK signals specifically in FAs at 15 min and 2 h after stimulation with serum and fibronectin. (E) Functional cell adhesion assay to determine the ability of PIP5KIγ-deficient cells to attach to fibronectin (FN) compared with Ctr. Quantification of the number of attached cells for three independent experiments in triplicate is shown. Data represent the mean value ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 (unpaired Student t test).
Immunofluorescence analysis revealed that the effect of PIP5Kγ KD on the activation of FAK is not due to impaired recruitment of FAK to FAs (Fig. S8B). FAK is recruited to FAs in KD cells from early time points of stimulation, although the pFAKY397 levels are reduced compared with controls (Fig. 7D). Interestingly, KD cells display smaller FAs (Fig. S8C). To analyze the functional relevance of PIP5Kγ in FAs, we further evaluated the ability of PIP5Kγ-deficient cells to attach to fibronectin-coated plates at early stages of cell adhesion and spreading. In comparison to controls, fewer PIP5Kγ-deficient cells attach to the plates (Fig. 7E), indicating that PIP5Kγ is required to promote the adequate adhesive properties of FAs. Globally, these data establish PIP5KIγ and its product PI(4,5)P2 as upstream activators of FAK in FAs and suggest they play important roles in FA maturation, cell attachment, and spreading.
Discussion
All NRTKs use remarkably similar concepts to switch kinase activity off. In all known cases, regulatory domains N-terminal to the tyrosine kinase domain bind the kinase to induce autoinhibition, and this sequesters regulatory phosphorylation sites in many cases. Also, initial activation steps often follow similar principles. For Src, Abl, and Syk family tyrosine kinases, activation is initiated by activator binding to their regulatory domains, which induces release of the regulatory domain from the kinase, resulting in activation. In the case of FAK, autoinhibition is achieved, as for other NRTKs, by the regulatory FERM domain docking onto the kinase domain (21). It was widely assumed that activation would also follow the canonical path of ligand-induced release of the FERM domain. This was partially based on studies showing that FAK undergoes large conformational changes in FAs (31). Here, we show that with respect to the activating PI(4,5)P2 ligand, the situation is different for FAK. FAK is in many ways an outsider among NRTKs, in that it is localized to a highly dense and clustered environment (FAs), where it has been proposed to act not only as an active kinase but also as a scaffolding protein. Our data indicate that PI(4,5)P2-induced activation of FAK follows concepts distinct from other NRTKs that are adapted to a highly crowded environment and allows concerted scaffolding and catalytic function.
Our findings support a rather complex model of sequential FAK activation, presented in Fig. 8. First, we establish PI(4,5)P2, generated in FAs by PIP5KIγ, as an important signaling messenger upstream of FAK activation. We had previously shown that acidic phospholipids, including PI(4,5)P2, bind FAK (31); however, the phospholipid specificity was not established. Here, we show that both the D4 and D5 phosphates in PI(4,5)P2 are required for full binding affinity (Fig. 1B). Although PI(3,4,5)P3 exhibits a similar affinity, PI(4,5)P2 is likely the relevant signaling lipid in FAs due to its abundance. It has been a long-standing question in the field as to how integrin signaling links to FAK activation. Because integrin signaling specifically results in PIP5KIγ recruitment and local PI(4,5)P2 production (34, 35), and we show that PIP5KIγ is an upstream activator of FAK in HeLa cells (Fig. 7), we propose that the path of the integrin-FAK signaling link leads via PIP5KIγ and generation of PI(4,5)P2 (Fig. 8, Left). However, we note that due to the complexity of FAK signaling, the exact upstream signaling events likely vary in different tissue contexts. In platelets, for example, FAK signaling does not depend on PIPKIγ (39). We note that PI(4,5)P2 levels are generally not as tightly regulated as, for example, PI(3,4,5)P3 levels; however, the property of the FAK–PI(4,5)P2 interaction appears well suited to the characteristics of PI(4,5)P2 levels in the cell and the localized generation of PI(4,5)P2. The avidity effect we observe with increased PI(4,5)P2 density (Fig. 1E and Fig. S1C) will prevent responding to basal PI(4,5)P2 levels (average estimates in the cell are 1% of total lipid) but allows a sharp and specific response to highly localized PI(4,5)P2 production in FAs by PIP5KIγ. On the other hand, Legate et al. (34) propose that bulk PI(4,5)P2 can partially substitute for local PI(4,5)P2 production, which may explain the differential effects of PIP5KIγ depletion in different cell types (39). We note that lipid involvement in FAK activation fits well with superresolution optical microscopy studies showing that FAK in FAs resides in close proximity to the cell membrane (40).
Fig. 8.
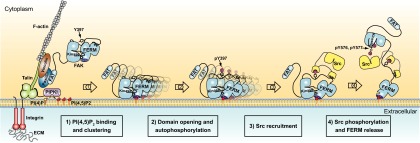
Schematic model for integrin-mediated FAK activation by PI(4,5)P2. (Left) Cell adhesion via integrin receptors to the ECM results in integrin clustering and the recruitment of FA proteins (as illustrated here for talin, vinculin, paxillin, FAK, and PIP5KIγ) to form adhesion structures that link integrins to the actin cytoskeleton. Recruitment of PIP5KIγ results in a local increase of PI(4,5)P2 levels in FAs. PI(4,5)P2 in FAs binds FAK via the basic patch (dark blue) in the FERM domain of FAK, resulting in FAK clustering at the cell membrane (step 1). PI(4,5)P2-induced FAK clustering results in a relaxed FERM/kinase conformation, with the kinase N-lobe dissociated from the linker and FERM F1 lobe. PI(4,5)P2-induced clustering and conformational relaxation allow efficient autophosphorylation of Y397 (step 2) and Src recruitment via SH2 and SH3 domains (step 3). Recruited Src phosphorylates the activation loop residues Y576/Y577 of FAK, which results in full activation and release of the kinase from the membrane-clustered FERM domain (step 4).
We demonstrate that binding to PI(4,5)P2 induces FAK clustering [Fig. 3 and Figs. S4 and S8 (step 1)]. Together with findings showing FAK autophosphorylation to occur in trans (23), this suggests FAK colocalization as an important mechanism promoting FAK transautophosphorylation, similar to what is proposed for receptor tyrosine kinases (41). Further, because FAK oligomers are multivalent PI(4,5)P2 binders, clustering likely explains the avidity effect we observe (Fig. 1E and Fig. S1C), although we cannot rule out multiple PI(4,5)P2 binding sites on a single FAK molecule. Clustering together with observed conformational changes raises the possibility that PI(4,5)P2 binding occurs in a cooperative mode. Indeed, our vesicle pull-down data fit well with a cooperative model, indicating positive cooperativity with a Hill coefficient close to 2 (Fig. S1 B and C). The observed cooperativity could be due to induced conformational changes that increase PI(4,5)P2 affinity or could occur because the induced clustering increases affinity due to the avidity effect. FAK clusters with soluble PI(4,5)P2 appear globular in shape (Fig. 3B), and we note that 2D averaged images suggest that FAK molecules are circularly arranged with less density at the center and an opening at one side of the circle. The size of FAK clusters on lipid vesicles appears more extended. Possibly, in the case of FAK clustering on a 2D membrane, the circular FAK arrangement seen with soluble PI(4,5)P2 is opened, allowing further propagation of FAK clusters along the membrane. It is currently unclear how FAK clustering is mediated, but because we observe clustering with soluble PI(4,5)P2 below its CMC concentration, direct protein–protein interactions among FAK molecules are likely involved. A potential site is the conserved W266 and a neighboring pocket involving L327, which are involved in lattice contacts in almost all FAK crystals containing the FERM domain. Interestingly, a recent report suggests W266 to be involved in activating FAK dimer formation (42). Potentially, this dimer could be stabilized in PI(4,5)P2-induced clusters; however, it is likely that additional contacts are required to form the more extended clusters we observe.
As suggested from the multiple binding partners of FAK, the clustering also underlines the importance of the scaffolding function of FAK and suggests that FAK may play an important role in FA architecture, and perhaps aids integrin clustering. Consistent with this notion, we observe that PIP5KIγ KD results in smaller FAs (Fig. S8C) and weaker cell adhesion (Fig. 7E), although this effect is not necessarily related to FAK, because PI(4,5)P2 affects several other focal adhesion proteins, such as talin. A role of FAK in FA maturation and adhesion strength would seem to contradict early studies that implicated FAK in FA turnover (8, 10). However, it is becoming clear that adhesion dynamics are highly complex, and more recent studies find an important role for FAK in increasing adhesion strength (43), particularly in response to rigid substrates and increased tension forces (13).
The PI(4,5)P2-induced reduction in FRET of CYFAKwt from a closed state in the presence of ATP to an intermediate FRET level (Fig. 4 and Fig. S5D) indicates that PI(4,5)P2, in addition to clustering, induces partial FERM/kinase domain opening. These FRET data can be explained by two possible scenarios: (i) PI(4,5)P2 induces an alternative conformation, which is partially open but with FERM and kinase domains still associated, or (ii) PI(4,5)P2 partially shifts the equilibrium between the fully open and fully closed states. Our activity data suggests that model i is correct, because a shift in equilibrium toward the open state should result in increased catalytic turnover, which we do not observe (Fig. 2D and Fig. S3 C and D). Because domain association is controlled by the FERM F2/kinase C-lobe interface (Fig. S2), we propose that this interface is intact upon PI(4,5)P2 binding. Taking all our data together, we propose that domain opening occurs at the FERM F1/linker/kinase N-lobe linkage. Opening at this site is expected to increase the distance of the FRET labels, without inducing dissociation of the FERM and kinase domains. Further, it will expose the linker for efficient autophosphorylation (Fig. 8, step 2). A recent study by Ritt et al. (44) finds that modifying the linker length greatly affects Y397 autophosphorylation without having a large effect on catalytic activity. This illustrates that similar to what we find for PI(4,5)P2, autophosphorylation and catalytic activity can be decoupled and that the linker conformation has an important effect on autophosphorylation efficiency. An intriguing question is how binding of PI(4,5)P2 to the distal basic patch on the FERM F2 lobe could affect the FERM F1/linker/kinase N-lobe interface. Our studies support a scenario where neutralization of the KAKTLRK region by PI(4,5)P2 binding results in partial destabilization of the FERM F2 lobe (Fig. 5 C–E and Fig. S7), allowing higher mobility at the FERM F2/kinase C-lobe interface, which, affects the kinase N-lobe linkage to the linker and FERM F1 lobe through increased domain movement or an allosteric coupling (Fig. S6). We note that because neither KAKTLRK mutation nor head group binding alone induces enhanced autophosphorylation (Fig. 2 A–C), it appears that, although necessary, charge neutralization is not sufficient, and it is likely that other parts of the lipid, such as triglyceride parts or acyl chains, are involved in clustering and/or promoting conformational changes. Therefore, our MD simulations with mainly charge-based PI(4,5)P2 interactions appear not to capture the full effect of PI(4,5)P2 but rather an initial charge neutralization of PI(4,5)P2 phosphates binding to the basic KAKTLRK residues. We do currently not understand how other parts of the PI(4,5)P2 lipid are involved in FAK conformational changes and clustering.
If the FERM F2/kinase C-lobe interface stays intact upon PI(4,5)P2 binding, this raises the question of how autophosphorylation can occur efficiently in a state where steady-state kinetics are inhibited. As described by Grant and Adams (45), in kinase–substrate complexes and in scenarios where the enzyme/substrate ratio is close to 1 (both are the case here), phosphorylation rates are not determined by steady-state kinetics but rather by presteady-state kinetics [reviewed by Taylor et al. (46)]. In such conditions, each enzyme only has to phosphorylate one substrate and can then stay attached to the substrate (off rate is irrelevant). Accordingly, we propose that in PI(4,5)P2-induced clusters, FAK molecules arrange themselves in an ideal position to phosphorylate their neighboring FAK molecule on the linker Y397 site.
Once FAK is autophosphorylated, Src is recruited via its SH2 and SH3 domains (Fig. 8, step 3). Our data presented in Fig. 6 show that PI(4,5)P2, in addition to mediating FAK autophosphorylation, enhances Src phosphorylation of Y576 in the activation loop of FAK. Our data indicate that this is achieved by a combined effect of increased recruitment through autophosphorylation and easier access to Y576 due to conformational changes. Using our FRET sensor, we show that Src-mediated phosphorylation of the FAK activation loop results in full dissociation of the FERM domain [Figs. 4C and 8 (step 4)], which is fully consistent with the observed structural incompatibility of FERM inhibition and Y576 phosphorylation (21). Therefore, PI(4,5)P2, via promoting autophosphorylation and Src phosphorylation, can trigger full FAK activation. However, it is clear that additional FAK activators are likely to be important, perhaps depending on the cellular setting. Cytoplasmic portions of several growth factor receptors have been reported to activate FAK (28–30), and direct mechanisms have been proposed for some of them that involve the KAKTLRK region of FAK (28, 29). Phosphorylated tails of the c-Met receptor are reported to interact with the KAKTLRK region, resulting in phosphorylation of Y194 in FAK (47). Intriguingly, Y194 is completely buried within the FERM F2 lobe, indicating that perhaps phospho-Met tail interactions could induce similar destabilization of the FERM F2 lobe as we describe for PI(4,5)P2 (Fig. 5 C–E and Fig. S7), hence exposing Y194 for phosphorylation. However, details of growth factor-mediated FAK activation remain to be determined, particularly whether they also involve PI(4,5)P2. Interestingly, EGF-stimulated cell migration was reported to depend on PIP5KIγ, indicating an important role for PI(4,5)P2 generation in the case of EGF receptor signaling (48). On the other hand, using FRET sensors similar to the ones used in our study, Ritt et al. (44) demonstrate that FAK can be conformationally activated by increased pH, which could be a route by which cancer cells could activate FAK ligand independently. Yet another signal involved in FAK activation could be tension forces generated by actomyosin contraction, because FAK was described as a force sensor (12, 13). It is probable that different routes resulting in FAK activation are active, depending on the cellular context. The mechanism we propose is likely restricted to situations where basal PI(4,5)P2 concentrations are limiting FAK signaling, and activation can therefore be triggered by local PI(4,5)P2 production.
In conclusion, the combination of our experimental approach with computational simulations has allowed us to propose a complex and detailed multistep activation mechanism for FAK. We propose that PI(4,5)P2 activates FAK via a combination of clustering and conformational changes that promote efficient autophosphorylation; Src recruitment; and, in turn, full activation by Src phosphorylation. Our findings fit well with observations from numerous cell biology studies that have described FAK signaling at the cell membrane in a highly crowded environment with dual catalytic and scaffolding functions. Our insights into the allosteric regulation and intramolecular communication of FAK could have major implications for the development of new classes of allosteric cancer therapeutics targeting FAK regulatory mechanisms that affect both catalytic and scaffolding functions of FAK.
Methods
Additional methods, including structure solution, MD simulations, docking of P(4,5)P2, and cell biology, are provided in SI Methods.
Vesicle Pull-Downs.
Lipid vesicles were prepared by mixing dissolved phospholipids (Avanti Polar Lipids) to obtain 6% (mol/mol) phosphoinositide in PC (or as indicated). Solvent was removed on a rotary evaporator; lipid films were hydrated with 20 mM Hepes (pH 7.5), 150 mM NaCl, and 1 mM Tris (2-carboxyethyl) phosphine (TCEP), and were sonicated until resuspended. Vesicle pull-downs were performed with 2 μM FAK proteins and serial dilution of vesicles. After 2 h of incubation, vesicles were centrifuged for 30 min at 16,100 × g at 4 °C. Supernatants were collected, and pelleted vesicles were washed and resuspended. Proteins in pellets and supernatants were quantified by the Bradford method (Biorad), reading absorption at 590 nm. Because F-FAK proteins at 2 μM yield low readings, we used GST fusion proteins of F-FAK. For all samples (including GST fusions), buffer blanks were subtracted.
Kinase Activity Assays.
Autophosphorylation was monitored by Western blotting using an anti-pY397 antibody and colorimetric staining with 4CN (Biorad). Alternatively, autophosphorylation levels were measured by ELISA using a PY20 antibody conjugated with HRP and development with TMB (Calbiochem). An enzyme-coupled assay using poly(E4Y) as a substrate was used to determine ATP turnover, as described by Lietha et al. (21). Plotted in Fig. 2D are negative slopes of NADH depletion. Lipid concentrations are at 100 μM C8-lipids or 1.5 mM lipid vesicles [only PC or 6% (mol/mol) PI(4,5)P2, equaling 45 μM PI(4,5)P2 per leaflet]. Src phosphorylation of Y576 was monitored by Western blotting. To prevent confounding results due to low levels of Y576 autophosphorylation, autophosphorylation and Src phosphorylation reactions were performed in two separate steps. Initially, FAK was allowed to autophosphorylate for 2 min. Autophosphorylation was stopped with the specific FAK inhibitor TAE226, and Src phosphorylation was started by adding Src. Blots were developed using an anti-pY576 antibody and colorimetric staining with 4CN.
EM.
Proteins were prepared in the presence of 200 μM C8-PC, C8-PI(4,5)P2, or polymerized PC liposomes containing 0% or 5% (mol/mol) PI(4,5)P2 PolyPIPosomes (Echelon Biosciences). Samples were negatively stained on carbon-coated grids with uranyl acetate and imaged using a Tecnai G2 Spirit electron microscope (FEI) operated at 120 kV and a Slow Scan CCD (Gatan) or TemCam-F416 4,096 × 4,096-pixel camera (TVIPS GmbH).
FRET Measurements.
FRET measurements were performed using a CFP excitation wavelength of 410 nm and recording emissions at 475 nm (CFP) and 525 nm (citrine), and relative FRET levels were obtained by the 525-nm/475-nm emission ratio.
Supplementary Material
Acknowledgments
We thank staff at European Synchrotron Radiation Facilities beamline ID14-4 and Swiss Light Source beamline X06SA and Liang Guo at Advanced Photon Source beamline 18ID for assistance. We also thank Diego Megias, head of the Confocal Microscopy Unit at the Centro Nacional de Investigaciones Oncológicas, for his help with FA quantifications. The research was supported by Spanish Ministerio de Ciencia e Innovacion (now Ministerio de Economía y Competitividad) Grants BFU2010-15923 (to D.L.) and BFU2009-11885 (to M.P.-M.), Comunidad Autónoma de Madrid Grant S2010/BMD-2457 (to D.L.), Bundesministerium für Bildung und Forschung Grant, New Methods in Systems Biology (to F.G.), and a grant from the Klaus Tschira Foundation (to F.G.). D.L. is also the recipient of awards from the Ramón y Cajal program (Grant RYC-2010-06948) and the Volkswagen Foundation.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 3ZDT and 4CYE).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1317022111/-/DCSupplemental.
References
- 1.Schiller HB, Fässler R. Mechanosensitivity and compositional dynamics of cell-matrix adhesions. EMBO Rep. 2013;14(6):509–519. doi: 10.1038/embor.2013.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schaller MD. Cellular functions of FAK kinases: Insight into molecular mechanisms and novel functions. J Cell Sci. 2010;123(Pt 7):1007–1013. doi: 10.1242/jcs.045112. [DOI] [PubMed] [Google Scholar]
- 3.Zhao X, Guan JL. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv Drug Deliv Rev. 2011;63(8):610–615. doi: 10.1016/j.addr.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zamir E, Geiger B. Molecular complexity and dynamics of cell-matrix adhesions. J Cell Sci. 2001;114(Pt 20):3583–3590. doi: 10.1242/jcs.114.20.3583. [DOI] [PubMed] [Google Scholar]
- 5.Ashton GH, et al. Focal adhesion kinase is required for intestinal regeneration and tumorigenesis downstream of Wnt/c-Myc signaling. Dev Cell. 2010;19(2):259–269. doi: 10.1016/j.devcel.2010.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lim ST, et al. Knock-in mutation reveals an essential role for focal adhesion kinase activity in blood vessel morphogenesis and cell motility-polarity but not cell proliferation. J Biol Chem. 2010;285(28):21526–21536. doi: 10.1074/jbc.M110.129999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peng X, et al. Cardiac developmental defects and eccentric right ventricular hypertrophy in cardiomyocyte focal adhesion kinase (FAK) conditional knockout mice. Proc Natl Acad Sci USA. 2008;105(18):6638–6643. doi: 10.1073/pnas.0802319105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ilić D, et al. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377(6549):539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- 9.Chen BH, Tzen JT, Bresnick AR, Chen HC. Roles of Rho-associated kinase and myosin light chain kinase in morphological and migratory defects of focal adhesion kinase-null cells. J Biol Chem. 2002;277(37):33857–33863. doi: 10.1074/jbc.M204429200. [DOI] [PubMed] [Google Scholar]
- 10.Ren XD, et al. Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J Cell Sci. 2000;113(Pt 20):3673–3678. doi: 10.1242/jcs.113.20.3673. [DOI] [PubMed] [Google Scholar]
- 11.Tomar A, Schlaepfer DD. Focal adhesion kinase: Switching between GAPs and GEFs in the regulation of cell motility. Curr Opin Cell Biol. 2009;21(5):676–683. doi: 10.1016/j.ceb.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tilghman RW, Parsons JT. Focal adhesion kinase as a regulator of cell tension in the progression of cancer. Semin Cancer Biol. 2008;18(1):45–52. doi: 10.1016/j.semcancer.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang HB, Dembo M, Hanks SK, Wang Y. Focal adhesion kinase is involved in mechanosensing during fibroblast migration. Proc Natl Acad Sci USA. 2001;98(20):11295–11300. doi: 10.1073/pnas.201201198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009;28(1-2):35–49. doi: 10.1007/s10555-008-9165-4. [DOI] [PubMed] [Google Scholar]
- 15.Parsons JT, Slack-Davis J, Tilghman R, Roberts WG. Focal adhesion kinase: Targeting adhesion signaling pathways for therapeutic intervention. Clin Cancer Res. 2008;14(3):627–632. doi: 10.1158/1078-0432.CCR-07-2220. [DOI] [PubMed] [Google Scholar]
- 16.Schultze A, Fiedler W. Therapeutic potential and limitations of new FAK inhibitors in the treatment of cancer. Expert Opin Investig Drugs. 2010;19(6):777–788. doi: 10.1517/13543784.2010.489548. [DOI] [PubMed] [Google Scholar]
- 17.Arold ST, Hoellerer MK, Noble ME. The structural basis of localization and signaling by the focal adhesion targeting domain. Structure. 2002;10(3):319–327. doi: 10.1016/s0969-2126(02)00717-7. [DOI] [PubMed] [Google Scholar]
- 18.Gao G, et al. NMR solution structure of the focal adhesion targeting domain of focal adhesion kinase in complex with a paxillin LD peptide: Evidence for a two-site binding model. J Biol Chem. 2004;279(9):8441–8451. doi: 10.1074/jbc.M309808200. [DOI] [PubMed] [Google Scholar]
- 19.Hayashi I, Vuori K, Liddington RC. The focal adhesion targeting (FAT) region of focal adhesion kinase is a four-helix bundle that binds paxillin. Nat Struct Biol. 2002;9(2):101–106. doi: 10.1038/nsb755. [DOI] [PubMed] [Google Scholar]
- 20.Cooper LA, Shen TL, Guan JL. Regulation of focal adhesion kinase by its amino-terminal domain through an autoinhibitory interaction. Mol Cell Biol. 2003;23(22):8030–8041. doi: 10.1128/MCB.23.22.8030-8041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lietha D, et al. Structural basis for the autoinhibition of focal adhesion kinase. Cell. 2007;129(6):1177–1187. doi: 10.1016/j.cell.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schaller MD, et al. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol Cell Biol. 1994;14(3):1680–1688. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toutant M, et al. Alternative splicing controls the mechanisms of FAK autophosphorylation. Mol Cell Biol. 2002;22(22):7731–7743. doi: 10.1128/MCB.22.22.7731-7743.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: A role for Src family kinases. Mol Cell Biol. 1995;15(2):954–963. doi: 10.1128/mcb.15.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schaller MD, Parsons JT. pp125FAK-dependent tyrosine phosphorylation of paxillin creates a high-affinity binding site for Crk. Mol Cell Biol. 1995;15(5):2635–2645. doi: 10.1128/mcb.15.5.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tachibana K, et al. Tyrosine phosphorylation of Crk-associated substrates by focal adhesion kinase. A putative mechanism for the integrin-mediated tyrosine phosphorylation of Crk-associated substrates. J Biol Chem. 1997;272(46):29083–29090. doi: 10.1074/jbc.272.46.29083. [DOI] [PubMed] [Google Scholar]
- 27.Frame MC, Patel H, Serrels B, Lietha D, Eck MJ. The FERM domain: Organizing the structure and function of FAK. Nat Rev Mol Cell Biol. 2010;11(11):802–814. doi: 10.1038/nrm2996. [DOI] [PubMed] [Google Scholar]
- 28.Chen SY, Chen HC. Direct interaction of focal adhesion kinase (FAK) with Met is required for FAK to promote hepatocyte growth factor-induced cell invasion. Mol Cell Biol. 2006;26(13):5155–5167. doi: 10.1128/MCB.02186-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Plaza-Menacho I, et al. Focal adhesion kinase (FAK) binds RET kinase via its FERM domain, priming a direct and reciprocal RET-FAK transactivation mechanism. J Biol Chem. 2011;286(19):17292–17302. doi: 10.1074/jbc.M110.168500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sieg DJ, et al. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2(5):249–256. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- 31.Cai X, et al. Spatial and temporal regulation of focal adhesion kinase activity in living cells. Mol Cell Biol. 2008;28(1):201–214. doi: 10.1128/MCB.01324-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goksoy E, et al. Structural basis for the autoinhibition of talin in regulating integrin activation. Mol Cell. 2008;31(1):124–133. doi: 10.1016/j.molcel.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palmer SM, Playford MP, Craig SW, Schaller MD, Campbell SL. Lipid binding to the tail domain of vinculin: Specificity and the role of the N and C termini. J Biol Chem. 2009;284(11):7223–7231. doi: 10.1074/jbc.M807842200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Legate KR, et al. Integrin adhesion and force coupling are independently regulated by localized PtdIns(4,5)2 synthesis. EMBO J. 2011;30(22):4539–4553. doi: 10.1038/emboj.2011.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McNamee HP, Liley HG, Ingber DE. Integrin-dependent control of inositol lipid synthesis in vascular endothelial cells and smooth muscle cells. Exp Cell Res. 1996;224(1):116–122. doi: 10.1006/excr.1996.0118. [DOI] [PubMed] [Google Scholar]
- 36.Di Paolo G, et al. Recruitment and regulation of phosphatidylinositol phosphate kinase type 1 gamma by the FERM domain of talin. Nature. 2002;420(6911):85–89. doi: 10.1038/nature01147. [DOI] [PubMed] [Google Scholar]
- 37.Ling K, Doughman RL, Firestone AJ, Bunce MW, Anderson RA. Type I gamma phosphatidylinositol phosphate kinase targets and regulates focal adhesions. Nature. 2002;420(6911):89–93. doi: 10.1038/nature01082. [DOI] [PubMed] [Google Scholar]
- 38.Balabin IA, Yang W, Beratan DN. Coarse-grained modeling of allosteric regulation in protein receptors. Proc Natl Acad Sci USA. 2009;106(34):14253–14258. doi: 10.1073/pnas.0901811106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, et al. Platelets lacking PIP5KIγ have normal integrin activation but impaired cytoskeletal-membrane integrity and adhesion. Blood. 2013;121(14):2743–2752. doi: 10.1182/blood-2012-07-445205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kanchanawong P, et al. Nanoscale architecture of integrin-based cell adhesions. Nature. 2010;468(7323):580–584. doi: 10.1038/nature09621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103(2):211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 42.Brami-Cherrier K, et al. FAK dimerization controls its kinase-dependent functions at focal adhesions. EMBO J. 2014;33(4):356–370. doi: 10.1002/embj.201386399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fabry B, Klemm AH, Kienle S, Schäffer TE, Goldmann WH. Focal adhesion kinase stabilizes the cytoskeleton. Biophys J. 2011;101(9):2131–2138. doi: 10.1016/j.bpj.2011.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ritt M, Guan JL, Sivaramakrishnan S. Visualizing and manipulating focal adhesion kinase regulation in live cells. J Biol Chem. 2013;288(13):8875–8886. doi: 10.1074/jbc.M112.421164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grant BD, Adams JA. Pre-steady-state kinetic analysis of cAMP-dependent protein kinase using rapid quench flow techniques. Biochemistry. 1996;35(6):2022–2029. doi: 10.1021/bi952144+. [DOI] [PubMed] [Google Scholar]
- 46.Taylor SS, Keshwani MM, Steichen JM, Kornev AP. Evolution of the eukaryotic protein kinases as dynamic molecular switches. Philos Trans R Soc Lond B Biol Sci. 2012;367(1602):2517–2528. doi: 10.1098/rstb.2012.0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen TH, Chan PC, Chen CL, Chen HC. Phosphorylation of focal adhesion kinase on tyrosine 194 by Met leads to its activation through relief of autoinhibition. Oncogene. 2011;30(2):153–166. doi: 10.1038/onc.2010.398. [DOI] [PubMed] [Google Scholar]
- 48.Sun Y, Ling K, Wagoner MP, Anderson RA. Type I gamma phosphatidylinositol phosphate kinase is required for EGF-stimulated directional cell migration. J Cell Biol. 2007;178(2):297–308. doi: 10.1083/jcb.200701078. [DOI] [PMC free article] [PubMed] [Google Scholar]