

Significance
The retinoblastoma protein (pRb) is a key regulator of cell cycle progression and the DNA damage response. Its importance in these processes is highlighted by the fact that it is mutated or functionally inactivated in almost all human tumors. Its activity is finely regulated by a number of post-translational modifications, including phosphorylation and methylation, which act to recruit “reader” proteins that mediate signaling events. Here, to our knowledge for the first time, we describe the methyl-dependent interaction between pRb and the tudor domain containing tumor protein p53 binding protein 1 (53BP1) and describe how this interaction integrates pRb cell cycle control with the DNA damage response. Our results therefore widen the repertoire of cellular targets for 53BP1 and suggest a new role in regulating pRb tumor suppressor activity.
Abstract
The retinoblastoma tumor suppressor protein pRb is a key regulator of cell cycle progression and mediator of the DNA damage response. Lysine methylation at K810, which occurs within a critical Cdk phosphorylation motif, holds pRb in the hypophosphorylated growth-suppressing state. We show here that methyl K810 is read by the tandem tudor domain containing tumor protein p53 binding protein 1 (53BP1). Structural elucidation of 53BP1 in complex with a methylated K810 pRb peptide emphasized the role of the 53BP1 tandem tudor domain in recognition of the methylated lysine and surrounding residues. Significantly, binding of 53BP1 to methyl K810 occurs on E2 promoter binding factor target genes and allows pRb activity to be effectively integrated with the DNA damage response. Our results widen the repertoire of cellular targets for 53BP1 and suggest a previously unidentified role for 53BP1 in regulating pRb tumor suppressor activity.
The retinoblastoma tumor suppressor protein pRb is either directly mutated or functionally inactivated in the vast majority of human tumors (1). One of its principle roles in cells is to regulate transcription, and the E2 promoter binding factor (E2F) family of transcription factors represents one of its most important targets (2). E2F acts to regulate the expression of a variety of genes connected with cell cycle progression and cell fate (including apoptosis, senescence, and differentiation), and the physical interaction between pRb and E2F hinders transcriptional activation by E2F, which coincides with cell cycle arrest (2, 3).
The tumor suppressor activity of pRb and its interaction with E2F is regulated by posttranslational modifications (4). For example, multiple cyclin-dependent kinase (Cdk) phosphorylation events occur within pRb, and pRb phosphorylation is temporally regulated as cells progress through the cell cycle, which disrupts the interaction between pRb and E2F. Other types of modification, such as acetylation in the C-terminal region, are also known to influence pRb activity (5).
More recently, a role for lysine (K) methylation in pRb control has been described (6). Thus, residue K810 undergoes methylation mediated by SET [su(var), enhancer-of-zeste, trithorax] domain containing lysine methyltransferase 7 (Set7/9) (also known as SETD7/KMT7) (6). Monomethylation at K810 holds pRb in the hypophosphorylated growth suppressing state, which occurs at a mechanistic level by inhibiting the physical association of Cdk complexes with pRb and thereby blocks Cdk-dependent phosphorylation (6). This is because, in the unmethylated state, K810 acts as the essential basic residue in the Cdk consensus phosphorylation site S807 (namely SPLK, Fig. 1A), which is an early pRb phosphorylation event during cell cycle control (7). Significantly, methylation of K810 hinders recognition and subsequent phosphorylation of S807 by cyclin/Cdk complexes (6).
Fig. 1.

Identification of a reader protein for pRb methylated at K810. (A) Schematic representation of the pRb and 53BP1 protein. The N-terminal, C-terminal, and pocket domain (A + B) of pRb are indicated. The amino acid sequence around residue K810 (highlighted in red) has been expanded to indicate the Cdk consensus motif SPLK (boxed in gray). A methyl-dependent interaction with the tandem Tudor domain of 53BP1 is indicated by dashed lines. BRCT, BRCA1 carboxyl-terminal domain; GAR, glycine and arginine rich motif; OD, oligomerization domain; UDR, ubiquitylation-dependent recruitment motif. (B) CADOR array probed with anti-GST (Top), biotinylated pRb peptide (Middle), or biotinylated pRb peptide with monomethylated K810 (Bottom). The gray-boxed regions demarked and enlarged show binding of the methylated pRb peptide to two independent samples of recombinant 53BP1 Tudor domain. The additional green spots on the array represent CADOR–peptide interactions that were not validated in cells. (C) Peptide binding assay in which avidin-immobilized unmethylated RbK810 peptide (0) or monomethylated RbK810 (1) was incubated with recombinant GST-53BP1 Tudor protein. The left side displays flow-through from the assay, whereas the right side displays the remaining eluted protein. n = 3. (D) As above, although unmodified (0), monomethylated (1), dimethylated (2), and trimethylated (3) RbK810 peptides were used in the binding assay. n = 2.
Here, we have elucidated a previously unidentified level of regulation imposed on pRb mediated by the methylation event at K810. In addition to inhibiting Cdk-dependent phosphorylation of pRb, we found that methylated K810 is “read” by the tandem tudor domain containing tumor protein p53 binding protein 1 (53BP1), which enables 53BP1 to form a stable interaction with pRb. This interaction was further exemplified by the cocrystal structure of the 53BP1 tandem tudor domain in complex with the methylated K810 peptide, and further by the presence of 53BP1 bound to pRb on E2F target genes. An established function of 53BP1 is in DNA double strand break (DSB) repair (8). Significantly, the methylation-dependent binding of 53BP1 to K810 allows pRb activity to be effectively integrated with the DNA damage response. The ability of 53BP1 to read pRb methylation at K810 thus links the DNA damage response with pRb, and suggests a previously unidentified level of functional interplay between 53BP1, pRb, and cell cycle control.
Results
pRb Lysine Methylation Is Read by 53BP1.
Residue K810 in pRb is modified by the Set7/9 methyltransferase (Fig. 1A), and methylation of K810 is up-regulated in DNA-damage-treated cells (6). We reasoned that in addition to its DNA-damage-dependent impact on pRb and E2F-dependent transcription, the methylated form of pRb might also play a direct mechanistic role in the DNA-damage response through its recognition by an accessory protein. To explore this idea, we first investigated the possibility that methylated K810 is recognized by an appropriate “reader” protein (9). We therefore prepared biotinylated pRb peptides that were either unmodified or methylated at K810, and used the fluorescently labeled steptavidin conjugate to screen the chromatin-associated domain array (CADOR). CADOR is a protein array platform developed to identify protein domains that bind to modified peptides, and includes a large number of reader domains involved with chromatin and transcriptional control (10). We identified a “hit” protein in the screen, the tandem tudor protein 53BP1, which bound to the methylated, but not unmethylated, pRb peptide (Fig. 1B).
53BP1 is a DNA-damage repair protein in which the tandem tudor domain is known to bind to methylated K382 in tumor protein p53 and K20 in histone H4 (11, 12). 53BP1 binds to either mono- or dimethylated H4K20 with a KD around 50 μM and 20 μM, respectively, as measured by isothermal titration calorimetry (ITC) (11). It is involved in repairing DNA DSBs and contributes to nonhomologous end joining. Its recruitment to γH2AX enriched chromatin is caused in part by a direct interaction between 53BP1 and methylated H4K20 (11). In cells, methylation of K20 is mediated by Set8 (PR-Set7/KMT5A), which monomethylates H4K20 (13), and the enzymes MMSET/WHSC1 (14) and Suv4-20 (h1 and h2; KMT5B and KMT5C) (15), which convert the methylation event to the di- and trimethylated form respectively.
We established that the interaction between 53BP1 and pRb was methylation- dependent using in vitro binding assays and cell-based approaches. In a peptide binding assay, only the methylated (RbK810me) and not the unmethylated RbK810 peptide bound to 53BP1 (Fig. 1C); both mono- and dimethyl RbK810 exhibited similar binding efficiencies, in contrast to the trimethylated RbK810 peptide, which failed to bind to 53BP1 (Fig. 1D). In transfected cells expressing ectopic pRb, 53BP1 coimmunoprecipitated with wild-type pRb but not with the lysine-to-arginine (K810R) mutant derivative (Fig. 2A), and this interaction was enhanced in the presence of DNA damage, because cells treated with etoposide displayed a stronger pRb-53BP1 association than unperturbed cells (Fig. S1A). An interaction between endogenous 53BP1 and pRb together with E2F-1 was also apparent in a number of cancer cell lines, and again, the interaction was enhanced under DNA-damage conditions (doxorubicin treatment and UV exposure) (Fig. 2B and Fig. S1B). By biolayer interferometry, 53BP1 bound to either mono- or dimethylated (but not the trimethylated peptide) K810, with a dissociation constant in the order of 42 µM for the dimethylated peptide (Fig. 2 C and D); this compares favorably with 53BP1 binding to H4K20 with a dissociation constant of 20 µM (Fig. S1C) (a value comparable to the reported KD as measured by ITC; ref. 11). Moreover, tudor domain recognition of methylated K810 was selective for 53BP1, as other members of the tudor domain family such as UHRF1 failed to bind to the methylated pRb K810 peptide, despite being able to bind to its methylated histone H3K9 target (Fig. S1 D and E).
Fig. 2.

53BP1 binds to methylated pRb. (A) U2OS cells were transfected with 2 μg of HA-pRb, HA-pRb-K810R, or empty vector (−). Cells were also treated with 20 μM etoposide for the last 16 h of the experiment. An immunoprecipitation was performed using anti-HA antibody and coprecipitating 53BP1 was detected by immunoblot. n = 2. (B) 293T cells were exposed to 50 J/m2 UV light where indicated, and a pRb immunoprecipitation was performed. Coprecipitating 53BP1 and E2F-1 were detected by immunoblot. (C) Biolayer interferometry real-time kinetic analysis of immobilized unmodified (RbK810me0), monomethylated (RbK810me1), dimethylated (RbK810me2), and trimethylated RbK810 (RbK810me3) peptides bound to His-53BP1 Tudor domain (1459–1599). (D) As above, but showing the concentration dependent binding of 53BP1 Tudor with the RbK810me2 peptide. A KD value of 42 μM was calculated from these data.
Structural Basis for Recognition of meK810.
To understand the recognition of methylated pRb by 53BP1, we determined the 3D structure of the 53BP1 tandem tudor domain in complex with the RbK810me2 peptide using X-ray crystallography at 2.35-Å resolution. We used the dimethylated peptide rather than the monomethylated peptide because of the higher observed KD (Fig. 2 C and D); the crystallographic statistics are given in Table S1. The crystals contained two copies of the 53BP1 tandem tudor domain in the asymmetric unit with each of them participating in peptide binding. Their overall structure was largely similar with a root-mean-square deviation value of 0.49 Å between C-alpha positions, although molecule A and its peptide were better defined by electron density. This structural similarity was also reflected by the different average B-factors for the two molecules: 67.9 Å2 for molecule A and 81.1 Å2 for molecule B. Each tudor domain was comprised of six antiparallel β strands, with tudor domain 1 composed of β1 to β6 and tudor 2 β7 to β12 (Fig. 3A). The C-terminal α-helix interacted with the tudor 1 domain by two hydrogen bonds formed between the backbone carbonyl of Y1600 and the backbone amide of L1534, and between L1602 and I1532.
Fig. 3.
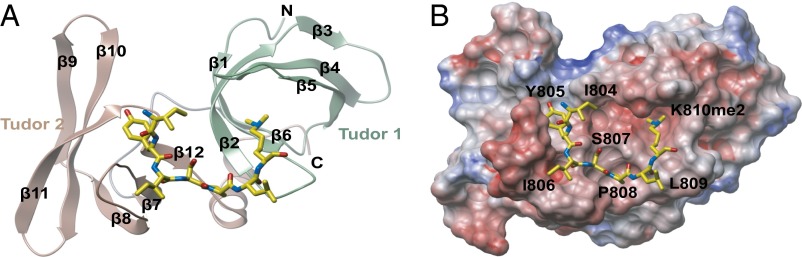
3D structure of 53BP1 tandem tudor domain in complex with a dimethylated pRb peptide. (A) Ribbon representation of the 53BP1 tandem tudor domain in complex with an RbK810me2 peptide encompassing residues G802-G818 (yellow sticks). (B) Electrostatic surface potential of 53BP1 tandem tudor domain viewed from the same direction as in A, and the RbK810me2 peptide is shown in yellow sticks.
The RbK810me2 peptide bound to a pocket encompassing both tudor domains (Fig. 3 A and B), and residues I804 to K810me2 were clearly visible in the density map of the complex (Fig. 4A). The modified K810me2 residue occupied the aromatic cage in tudor domain 1, composed of Y1502, F1519, D1521, Y1523, and W1495. Hydrogen bonds were formed between the peptide carbonyl of L809 and the side chain amine nitrogen of N1498, and also between the peptide backbone nitrogen of I806 and the backbone carbonyl of S1548 (Fig. 4A). A RbK810me1 peptide would be expected to interact with 53BP1 in a similar fashion, and specificity for mono- and dimethylated pRb over the trimethylated form would be dictated by the direct interaction of the K810me amino proton to the carboxylate group of D1521. This mechanism of K810me2 recognition is very similar to the previously reported 53BP1 complexes with p53 (16) and histone 4 (11) peptides. However, the present structure of the pRb-53BP1 complex had unambiguous electron density of the bound peptide outside of the dimethyl lysine (Fig. 4A). This observation is in sharp contrast to previously reported crystal structures where only one or two amino acid residues were defined by electron density (11, 16). The S-P-L sequence preceding the methylated lysine in the pRb peptide is also different from the R-H-K/R motif that accounts for specificity in p53K382me2 and H4K20me2, showing closer similarity to the S-H-L motif present in p53K370me2 (16). Superimposition with the NMR complex of 53BP1-H4K20me2 (PDB ID code 2LVM) revealed that tudor domain binding to pRbK810me2 occurs in a different orientation to that observed for H4K20me2 (Fig. 4C). The structured I804-K810 region of RbK810me2 formed extensive hydrophobic interactions with both tandem tudor domains, and the conformation of the peptide was stabilized by intramolecular hydrogen bonds between the side chain of S807 and the backbone carbonyl of I806 (Fig. 4 A and B). This interaction, as well as the inherent rigidity of P808, which lies in a shallow pocket between the two tudor domains defined by Y1500, T1545, F1553, I1587, and S1589, probably explains the structured nature of the pRb peptide apparent in the crystals (Figs. 3B and 4 A and B). Interestingly, I804-S807 occupies a groove in the second tudor domain composed of E1549, D1550, E1551, K1582 and M1584 (Fig. 4 A and B), the three acidic residues being completely conserved in 53BP1 across all vertebrate species (17). This conservation likely highlights the importance of these residues in the tudor 2 domain for determining substrate specificity.
Fig. 4.

Detailed 3D structure of RbK810me2 binding site in the 53BP1 tandem tudor domain. (A) Close up view of the RbK810me2 peptide binding site in the 53BP1-RbK810me2 complex. The mFc- DFc omit map of the peptide is contoured to 2.5σ level and shown in blue. Dashed lines indicate hydrogen bonds. (B) Close up view of the peptide binding pocket. White represents neutral regions, green represents hydrophobic regions, blue represents a hydrogen bond donor, and red represents hydrogen bond acceptors. (C) 53BP1 tandem tudor domains in complex with a pRbK810me2 peptide (yellow sticks) superimposed with the 53BP1-H4K20me2 NMR complex (PDB ID code 2LVM). The tandem tudor domains in 53BP1 and the pRb peptide are colored as in Fig. 3A, whereas the H4K20me2 backbone is shown in orange.
Role of pRb and 53BP1 in DNA Repair and Senescence.
53BP1 accumulates at DSBs (18), and is involved in DNA repair through its essential role in nonhomologous end joining (8, 19). We reasoned that the interaction between 53BP1 and methylated K810 may contribute to the role of pRb in DNA repair. Initially, therefore, we studied the DNA-damage response in Rb+/+ compared with Rb−/− mouse embryo fibroblasts (MEFs) upon exposure to etoposide, which causes the appearance of DSBs. We analyzed the occurrence of phosphorylated γH2AX, which is a marker for the appearance of DSBs that coincides with 53BP1 binding to H4K20me (20, 21). We found that the magnitude and duration of the γH2AX response was elevated in Rb−/− relative to Rb+/+ cells (Fig. 5A and Fig. S2C). Furthermore, although the levels of p53 and p21 increased in both cell lines in response to etoposide exposure, Rb−/− MEFs displayed a modest reduction compared with Rb+/+ MEFs, suggesting that checkpoint signaling in response to DNA damage was compromised in the absence of pRb (Fig. S2C). Moreover, the expression of ectopic pRb in Rb−/− cells confirmed the importance of K810 in the DNA-damage-response effect, as the expression of wild-type pRb suppressed the appearance of γH2AX in comparison with the K810R mutant, where γH2AX remained elevated, similar to vector-transfected cells (Fig. 5B and Fig. S2 A and B).
Fig. 5.

pRb and 53BP1 in the DNA-damage response. (A) Rb−/− and Rb+/+ MEFs were treated with 20 μM etoposide for 1 h. Etoposide was washed out with PBS, and fresh media was reapplied. Cells were harvested at the indicated time points, with time = 0 (indicated by −) being the point of etoposide addition. Extracts were used in immunoblots with the indicated antibodies. Numbers below the blot represent relative phospho-γH2AX levels after quantitation. n = 3. (B) Rb−/− MEFs were transfected with 2 μg of HA-pRb, HA-pRb-K810R, or empty vector. Cells were then treated as detailed above, and harvested for immunoblotting at the indicated time points. Numbers below the blot represent relative γH2AX levels after quantitation. n = 3. (C) U2OS cells were transfected with 50 nM nontargeting siRNA (N), Set7/9 siRNA (S), 53BP1 siRNA (53), or a combination of the two (S/53) for 72 h. Cells were also treated with 20 μM etoposide for the last 16 h of the experiment where indicated (Et). Extracts were used for immunoblotting with the indicated antibodies. n = 3.
We then addressed whether the influence of pRb in DNA-damaged cells might relate to the activity of 53BP1. To test this idea, we studied the phosphorylation of pRb, which previous studies have established to be inversely related to methylation at K810 (6). In 53BP1 siRNA-treated cells, an increase in pRb phosphorylation was evident (Fig. 5C), which is similar to what had previously been observed under conditions of Set7/9 depletion (Fig. 5C; ref. 6). Furthermore, when 53BP1 and Set7/9 were codepleted, no further effect on pRb phosphorylation was observed (Fig. 5C), suggesting that Set7/9 and 53BP1 binding act through a common pathway to regulate pRb phosphorylation. In support of this idea, we identified 53BP1 by chromatin immunoprecipitation (ChIP) on a number of E2F responsive target genes, including thymidine kinase (TK), thymidine synthase (TS), apoptotic peptidase activating factor 1 (Apaf-1), Cdc6, and E2F-1, where its presence coincided with pRb (Fig. 6A and Fig. S3 A-C). Moreover, we detected 53BP1 and pRb in a chromatin-bound complex on E2F target genes, including TK, TS, and Apaf-1 (Fig. 6A and Fig. S3 B and C). Significantly, the level of 53BP1 in complex with chromatin associated pRb was enhanced, relative to untreated cells, in cells that had been exposed to etoposide (Fig. 6A and Fig. S3 B and C); this is the anticipated outcome based on the DNA-damage-dependent methylation of K810 (6) and the interaction detected by immunoprecipitation in DNA-damaged cells (Fig. 2B and Fig. S1 A and B). These results strongly suggest that 53BP1 and pRb biochemically and functionally interact in cells, which contributes to the ability of pRb to regulate cell cycle progression and influence the DNA-damage response.
Fig. 6.
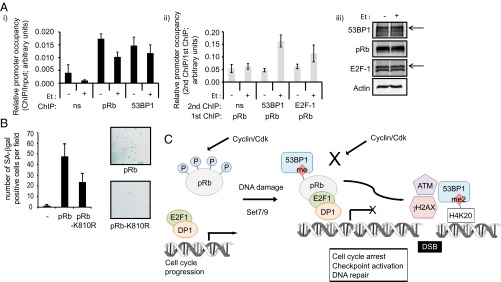
pRb and 53BP1 interact on the chromatin. (A) Extracts from U2OS cells treated with 20 μM etoposide for 1 h (+), or untreated cells (−) were immunoprecipitated with control IgG (ns), pRb, or 53BP1 antibodies (graph shown in black). Chromatin was isolated and analyzed by quantitative PCR using primers targeting the E2F responsive thymidine kinase (TK) promoter (i). Ten percent of the total chromatin fraction used for the immunoprecipitation was also included as an input control and was used to normalize primary ChIPs. The pRb-immunoprecipitated chromatin was then reimmunoprecipitated a second time by IgG (ns), 53BP1 or E2F-1 antibodies, as indicated (graph shown in gray). Secondary ChIPs were normalized to the inputs provided from the primary pRb ChIP (ii). Means with SD are shown from an experiment with triplicate samples. Expression levels of 53BP1, pRb, and E2F-1 in the extracts used for ChIP are also displayed. Arrows indicate the relevant specific bands (iii). (B) SAOS2 cells were stably transfected with HA-pRb, HA-pRb-K810R, or empty vector (−) as indicated. After 2 wk of selection in G418 to remove untransfected cells, 104 transfected cells were reseeded onto coverslips and flat cells were detected by staining for SA-β-gal as described. The graph displays the mean number of SA-β-gal–positive cells per field of view, with SD shown (calculated from three independent experiments). Phase contrast micrographs of SA-β-gal–stained SAOS2 flat cells are included for HA-pRb– and HA-pRb-K810R–transfected cells (magnification: 5×). (C) Model for 53BP1 assembly with methylated pRb on chromatin. Growth control by pRb is influenced by its posttranslational modifications, with Cdk-dependent phosphorylation events releasing E2F activity and driving cell cycle progression. In response to DNA damage, Set7/9-directed methylation of pRb leads to the recruitment of 53BP1 to chromatin-bound pRb–E2F complexes, where it acts to maintain pRb in its hypophosphorylated, growth-regulating state. The 53BP1–pRb interaction also appears to be important for integrating pRb activity with the DNA-damage response, because pRb methylation at K810 influences γH2AX levels after the occurrence of DSBs. Because 53BP1 impacts on p53 activity, checkpoint activation and DNA repair, it likely modulates multiple pathways to facilitate growth regulation in response to DNA damage.
Another property of pRb that is believed to relate to its tumor suppressor activity is its ability to induce cellular senescence (22). When ectopically expressed in SAOS2 cells, pRb can, under appropriate culture conditions, induce a flat cell phenotype that exhibits similarities with the properties of senescing cells (22). Indeed, characteristic flat cell morphology and senescence-associated β-galactosidase (SA-β-gal) expression were evident in cells expressing wild-type pRb and the K810R mutant derivative (Fig. 6B). However, K810R was less efficient at inducing senescent cells than wild-type pRb (Fig. 6B). This finding suggests a biological role for K810 methylation, and consequently 53BP1 binding, in the growth-regulating effects of pRb.
Discussion
One of the principal roles for the pRb tumor suppressor protein is to regulate progression through the early stages of the cell cycle, and this activity is mediated in part by influencing the activity of the E2F family of transcription factors (2). The pRb–E2F interaction is highly significant because the ability of pRb to bind E2F coincides with growth inhibition and cell cycle delay and the pathway is under aberrant control in most human tumor cells (2, 3).
Growth control by pRb is influenced by its posttranslational modifications, with Cdk-dependent phosphorylation being the most widely described (3). The classic view has always been that, under conditions of cellular stress, such as in response to DNA damage, Cdk activity is inhibited, enforcing the hypophosphorylated pRb state and permitting pRb-directed cell cycle arrest, although a recent report suggests that cells undergoing a DNA-damage response retain monophosphorylated pRb (23). Inhibition of Cdk activity reflects the combined function of several signaling pathways, including the induction of Cdk inhibitors like p21 via increased p53 activity (24). However, a more direct substrate-based mechanism also exists, in which residue K810 in pRb is methylated in a DNA-damage-dependent fashion by the methyltransferase Set7/9 (6).
Here, we have uncovered an additional and important level of control by showing that methylated K810 acts to recruit the tandem tudor domain protein 53BP1 (8). It has been implicated in a number of cellular processes, including checkpoint signaling, DSB repair pathway choice, and the long-range DNA end-joining that occurs during V(D)J recombination and class switch Ig gene recombination (8). In the context of its interaction with pRb, 53BP1 appears to be important for integrating pRb activity with the DNA-damage response, because cells lacking pRb and specifically K810 show elevated levels of γH2AX after exposure to etoposide, and 53BP1 impacts on the level of pRb phosphorylation. These observations suggest that K810 methylation, and its subsequent recognition by 53BP1, is important for maintaining pRb in its hypophosphorylated growth regulating state, which in turn contributes to the DNA-damage response (Fig. 6C). Indeed, the crystal structure of the tandem tudor domain of 53BP1 in complex with a RbK810me2 peptide indicated that once the interaction has formed, phosphorylation of the neighboring S807 residue would be unlikely to occur, as the serine residue is engaged in the intramolecular interaction and is part of the stable complex interface (Figs. 3 and 4).
53BP1 is a multifunctional protein that acts not only to promote DSB repair, but also to mediate checkpoint signaling in response to DNA damage (8). For example, 53BP1 is required for the efficient phosphorylation of ATM targets, such as Chk2, p53, and E2F-1 (25, 26), and loss of 53BP1 leads to checkpoint defects and genomic instability. 53BP1 is also known to associate with p53 which, like pRb, is methylated on lysine residues in response to DNA damage (12, 27). This interaction enables a proper accumulation of p53 protein and p53-dependent transcription in conditions of cellular stress (12, 27). Our results suggest that, because 53BP1 can interact with pRb, in addition to p53, it is likely to play an important role in modulating growth control, and perhaps its interaction with both pRb and p53 pathways facilitates growth regulation in response to DNA damage.
Materials and Methods
CADOR Array Screening.
The generation of protein microarrays has been described (9), and a list of the protein domains on the array has been published (10). Peptides were synthesized as biotin-PEG-GNIYISPLKSPYKISEG and biotin-PEG-GNIYISPLK[me]SPYKISEG. Biotinylated peptides were labeled as described (9).
Biolayer Interferometry.
Biolayer interferometry experiments were performed on a 16-channel ForteBio Octet RED384 instrument at 25 °C in 25 mM Hepes, pH 7.5, and 100 mM NaCl buffer. Biotinylated pRb peptide GNIYISPLK*SPYKISEG (where K* indicates unmodified, mono-, di-, or trimethylated lysine) was attached to streptavidin coated biosensors by incubation for 3 min at 3 μM concentration. Reference sensors were blocked with biocytin. 53BP1 Tudor sample was prepared in seven 2.5-fold dilutions starting from 200 μM. Measurements were performed using 300-s association step followed by a 300-s dissociation step on a black 384-well plate (Greiner). Baseline was stabilized for 120 s before association. Signal from reference sensors was subtracted before KD calculations using Analysis software (ForteBio).
Crystallization of 53BP1 Tudor Domain with Methylated-pRb Peptide.
Crystals of 53BP1 tudor domains encompassing residues 1480–1568 were obtained using the sitting drop vapor diffusion method at 20 °C in a final volume of 150 nL. The protein was concentrated to 25 mg/mL and preincubated with the RbK810me2 peptide (residue G802–G818) at a molar ratio of 1:2. The protein–peptide mixture was transferred to crystallization plates and crystals used for X-ray diffraction were obtained from a solution containing 5% (wt/vol) PEG10K, 0.15 MgCl2, and 0.1 M Tris, pH 7.5. Crystals were cryoprotected with reservoir solution supplemented with 25% (vol/vol) ethylene glycol before flash freezing in liquid nitrogen for storage and data collection.
Structure Solution and Refinement.
Crystals of 53BP1 in complex with RbK810me2 peptide belong to space group P6122 with unit cell parameters a = b = 105.8 Å, c = 156.2 Å. Diffraction data were collected on beamline I04 at the Diamond Light Source. X-ray data were integrated with MOSFLM (28) and scaled with AIMLESS (29). The structure was solved by molecular replacement with PHASER (30) using the human 53BP1 tandem tudor domain (PDB ID code 3LH0) as a search model. Refinement was done with PHENIX (31) and after several cycles of manual rebuilding with COOT (32), the model converged to a Rcryst/Rfree of 19.4% and 23.0%, respectively. The quality of the model was validated with MOLPROBITY (33) with 97.5% of the residues being in the favored region of the Ramachandran plot and no outliers. Coordinates and structure factors have been deposited in the Protein Data Bank with the accession code 4CRI.
Additional materials and methods are described in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank the Diamond Light Source for access to beamline I04 (mx8421). This work was supported by Cancer Research U.K. Programme Award 300/A13058 and a Medical Research Council (MRC) grant (to N.B.L.T.). M.T.B. is supported by National Institutes of Health Grant DK062248 and Cancer Prevention Research Institute of Texas funding (RP110471). The Protein Domain Microarray Core is supported by the Centre for Environmental and Molecular Carcinogenesis at MD Anderson. O.F., C.J., and T.K. are grateful for support by the Structural Genomics Consortium, a registered charity (no. 1097737) that receives funds from the Canadian Institute for Health Research, the Canada Foundation for Innovation, Genome Canada, GlaxoSmithKline, Pfizer, Eli Lilly, Takeda, AbbVie, the Novartis Research Foundation, the Ontario Ministry of Research and Innovation, and the Wellcome Trust (092809/Z/10/Z).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 4CRI).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1403737111/-/DCSupplemental.
References
- 1.Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8(9):671–682. doi: 10.1038/nrc2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stevens C, La Thangue NB. E2F and cell cycle control: A double-edged sword. Arch Biochem Biophys. 2003;412(2):157–169. doi: 10.1016/s0003-9861(03)00054-7. [DOI] [PubMed] [Google Scholar]
- 3.Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12(15):2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- 4.Munro S, Carr SM, La Thangue NB. Diversity within the pRb pathway: Is there a code of conduct? Oncogene. 2012;31(40):4343–4352. doi: 10.1038/onc.2011.603. [DOI] [PubMed] [Google Scholar]
- 5.Chan HM, Krstic-Demonacos M, Smith L, Demonacos C, La Thangue NB. Acetylation control of the retinoblastoma tumour-suppressor protein. Nat Cell Biol. 2001;3(7):667–674. doi: 10.1038/35083062. [DOI] [PubMed] [Google Scholar]
- 6.Carr SM, Munro S, Kessler B, Oppermann U, La Thangue NB. Interplay between lysine methylation and Cdk phosphorylation in growth control by the retinoblastoma protein. EMBO J. 2011;30(2):317–327. doi: 10.1038/emboj.2010.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ren S, Rollins BJ. Cyclin C/cdk3 promotes Rb-dependent G0 exit. Cell. 2004;117(2):239–251. doi: 10.1016/s0092-8674(04)00300-9. [DOI] [PubMed] [Google Scholar]
- 8.Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol. 2014;15(1):7–18. doi: 10.1038/nrm3719. [DOI] [PubMed] [Google Scholar]
- 9.Kim J, et al. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 2006;7(4):397–403. doi: 10.1038/sj.embor.7400625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Y, et al. TDRD3 is an effector molecule for arginine-methylated histone marks. Mol Cell. 2010;40(6):1016–1023. doi: 10.1016/j.molcel.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Botuyan MV, et al. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127(7):1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kachirskaia I, et al. Role for 53BP1 Tudor domain recognition of p53 dimethylated at lysine 382 in DNA damage signaling. J Biol Chem. 2008;283(50):34660–34666. doi: 10.1074/jbc.M806020200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oda H, et al. Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Mol Cell. 2010;40(3):364–376. doi: 10.1016/j.molcel.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pei H, et al. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature. 2011;470(7332):124–128. doi: 10.1038/nature09658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schotta G, et al. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004;18(11):1251–1262. doi: 10.1101/gad.300704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roy S, et al. Structural insight into p53 recognition by the 53BP1 tandem Tudor domain. J Mol Biol. 2010;398(4):489–496. doi: 10.1016/j.jmb.2010.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang J, et al. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat Struct Mol Biol. 2013;20(3):317–325. doi: 10.1038/nsmb.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anderson L, Henderson C, Adachi Y. Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol Cell Biol. 2001;21(5):1719–1729. doi: 10.1128/MCB.21.5.1719-1729.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakamura K, et al. Genetic dissection of vertebrate 53BP1: A major role in non-homologous end joining of DNA double strand breaks. DNA Repair (Amst) 2006;5(6):741–749. doi: 10.1016/j.dnarep.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 20.Celeste A, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296(5569):922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146(5):905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Templeton DJ, Park SH, Lanier L, Weinberg RA. Nonfunctional mutants of the retinoblastoma protein are characterized by defects in phosphorylation, viral oncoprotein association, and nuclear tethering. Proc Natl Acad Sci USA. 1991;88(8):3033–3037. doi: 10.1073/pnas.88.8.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Narasimha AM, et al. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. eLife. 2014:e02872. doi: 10.7554/eLife.02872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vidal A, Koff A. Cell-cycle inhibitors: Three families united by a common cause. Gene. 2000;247(1-2):1–15. doi: 10.1016/s0378-1119(00)00092-5. [DOI] [PubMed] [Google Scholar]
- 25.Lin WC, Lin FT, Nevins JR. Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev. 2001;15(14):1833–1844. [PMC free article] [PubMed] [Google Scholar]
- 26.Shiloh Y, Ziv Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14:197–210. [PubMed] [Google Scholar]
- 27.Huang J, et al. p53 is regulated by the lysine demethylase LSD1. Nature. 2007;449(7158):105–108. doi: 10.1038/nature06092. [DOI] [PubMed] [Google Scholar]
- 28.Leslie AGW, Powell HR. Processing diffraction data with MOSFLM. Nato Sci. Ser. Ii. Math. 2007;245:41–51. [Google Scholar]
- 29.Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr. 2013;69(Pt 7):1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen VB, et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]