

Abstract
Substrates of Insulin-Degrading Enzyme are numerous and share little homology, like amyloid-beta and insulin. Small molecules binding both at the permanent exosite and at the discontinuous, conformational catalytic site, were discovered and co-crystallized with Insulin-Degrading Enzyme. Selective inhibition of amyloid-beta degradation over insulin hydrolysis was possible. Neuroblastoma cells treated with the optimized compound display a dose-dependent increase in amyloid-beta levels.
Keywords: enzymes, medicinal chemistry, Structure-activity relationships, amyloid-beta peptides, inhibitors, X-ray diffraction
1. Introduction
Insulin-degrading enzyme (IDE) is an atypical, highly conserved and ubiquitous metalloprotease of the M16 family [1,2]. It is mainly intracellular [3] but a small amount is trafficked extracellularly [4]. IDE shows a unique structure [5,6]: it is organized in two 56 kDa halves which are joined by a 28 amino acid residue loop [7], and enclose a large, almost spherical, catalytic chamber, named “crypt”.
Using X-ray crystallography Tang et al. have shown that substrates bind to two distant sites in the chamber. To let substrates in, the enzyme opens. The binding of substrates to the exosite, 30 Å away from the catalytic Zinc, promotes a conformational change of the substrate that is critical for cleavage at the catalytic site [8,9] Also, recently, via X-ray crystallography, an unexpected displacement (swinging-door) of a subdomain of IDE that creates an 18Å opening to the chamber, that permits the entry of short peptides [10].
Along with insulin, substrates of IDE include amyloid-β (Aβ) [11], Insulin-Growth Factor-II IGF-II [12,13], glucagon [11], somatostatin [14] most of which are amyloidogenic [15] (Fig. 1A). Ubiquitin [16] and CGRP [17] are also shown as IDE substrates. IDE prefers hydrophobic or basic residues at P1 and P’1 and substrates that lack a positive charge at the C-terminus [1]. Interestingly, beside its peptidolytic role, IDE interacts with and regulates the proteasome complex [18]. Also, the activity of IDE can be modulated by the binding of ATP to the catalytic chamber [19].
Fig. 1.

A) IDE’s substrates, B) structure of 1 (BDM41367) inhibitor of labelled Aβ16–23 hydrolysis C) by IDEwt (● IC50 Aβ16–23 = 2.9 µM).
Silencing IDE expression with siRNA reduces insulin-mediated protein degradation [20]. In several animal models, deletion of Ide gene or mutations in the gene result in elevated insulin levels and glucose intolerance, associated with elevated Aβ in the brain [21]. Also, transgenic overexpression of IDE in neurons results in significantly reduced levels of Aβ in brain and retards plaque formation in amyloid precursor protein (APP) transgenic mice [22]. In addition, Ide gene was linked to type-2 diabetes (T2D) and Alzheimer’s disease (AD) in humans [23–24].
Small organic molecules are complementary to genomic or transcriptomic interventions, because they are systems modulators and not “erasers” of protein activity. They help to understand the target’s function and can be transcribed into therapeutically agents by modulation of the proteolytic profile (i.e. inhibition or activation), the distribution pattern and the chronically or temporarily inhibition of IDE. Leissring et al. described the first substrate-based zinc-binding hydroxamate inhibitors of IDE [25]. However, their hydroxamate group [26] combined with an arginine residue limit their use as pharmacological probes. The poorly bioavailable suramin [27] and two other compounds identified in a cell-free assay, were reported to be activators [28]. Also, by molecular modelling, Çakir et al. have discovered compounds that activate the hydrolysis of several substrates of IDE and Kukday et al. described activators specific of the rat ortholog [29].
Herein, we describe the discovery of drug-like ligands of IDE with an atypical binding mode and report their impact on the hydrolytic profile of IDE as well as their effect on neuroblastoma cells expressing amyloid-β.
2. Results and Discussion
2.1. Hit identification
2.1.1. Screening of a 2000-member library
A library of 2000 drug-like compounds (Supporting Information Fig. S1 and [30]) was screened in a fluorescence assay for the inhibition of the hydrolysis of dual labelled substrate (Aβ16–23: Km = 100 µM) by IDE. Compounds showing dose-dependent inhibition were defined as hits. 1 (BDM41367, Fig. 1B), was the most active compound (IC50 Aβ16–23: 2.9 µM). Interestingly, the dose-response curve plateaus at 50% inhibition (Fig. 1C). A study of the enzymatic mechanism evidenced that 1 is a reversible, partial and competitive inhibitor of the hydrolysis of Aβ16–23 by IDE (Supporting Information Fig. S2).
2.1.2. Binding of the hit
Using X-ray crystallography Tang et al. have shown that substrates bind to two distant sites in the chamber. To understand how 1 interacts with IDE, we co-crystallized it catalytically active IDE enzyme and solved the structure by molecular replacement (pdb code=4DTT, Supplemental information and Table S1). Surprisingly, in the co-crystal, 1 is observed at these two binding sites (Fig. 2A) [31]. At the exosite (Fig. 2B), the imidazole ring forms a hydrogen bond with Glu341, a residue that binds the N-terminus of IDE substrates. The amide and amine functions interact with Gly361 and Gln363 main chains. At the catalytic site, 1 interacts with residues from both the N- and C-terminal domains (Fig. 2C). The carboxylate group of 1 completes the zinc coordination sphere formed by His108, His112 and Glu189. 1 also forms hydrogen bonds with side chains of Tyr831 and Asn139 and the main chain of Val833 via its imidazole ring. The benzyl group of 1 makes a hydrophobic contact with the side chain of Phe115. The negatively charged Glu111 is located in the vicinity of the ionizable imino function of 1. In line with this observation, when 1 is co-crystallized with the mutated, catalytically inactive E111Q enzyme, it is found only at the exosite (PDB code=2YPU, Supporting Information Table S1, Fig. S3). The binding of 1 to the exosite was further substantiated by an enzymatic assay using the E341A exosite mutant. In this mutant, the glutamate shown to interact with 1 in the exosite is replaced with a neutral alanine [16]. With this enzyme, 1 behaves as a full inhibitor (IC50 Aβ16–23: 5.8 µM, Fig. 3).
Fig. 2.

X-ray crystal structure of hIDE-CF co-crystallized with 1 (PDB code: 4DTT). A) general view, B) detailed interactions at the exosite C) detailed interactions at the catalytic site. Zn (red sphere); O (red); N (blue); C (cyan for IDE N-terminal domain, gray for IDE C-terminal domain, orange for 1); hydrogen contacts in dotted lines. Arrows point residues shown to be critical by mutagenesis.
Fig. 3.

1 is an inhibitor of the dual labelled Aβ16–23 substrate hydrolysis by IDEwt (● IC50 = 2.9 µM) and IDE E341A (■ IC50 = 5.3 µM).
Comparing the X-ray structures obtained with proteins that differ at the catalytic site on one hand, and enzyme inhibitions measured with protein that differ at the exosite on the other hand, we propose the following hypothesis to explain the peculiar behaviour of 1. IDE has an enclosed catalytic chamber. During catalysis, IDE needs to open to give access to substrates or modulators to the catalytic chamber [5,6]. In the open state, the non contiguous catalytic site (formed by both IDE-N and IDE-C residues) is not fully assembled. However, in this open conformation, the exosite remains as a binding site for 1. Upon capture of the substrate and closure of the catalytic chamber, 1 could dissociate and compete with the substrate either at the exosite or at the re-assembled catalytic site. The equilibrium between 1 bound to the exosite and to the catalytic site in a 1:2 ligand:site ratio could explain why 1 behaves as a partial inhibitor in the wild type IDE and as a full inhibitor in the exosite-mutated enzyme [32].
2.2. Design and synthesis of analogues
2.2.1. Synthesis
We made a limited number of variations at key interaction points of 1 with IDE to confirm its binding mode. Non commercial iminodiacetic precursors were prepared by alkylation of iminodiacetic acid with alkyl bromides (Scheme 1) or by acylation using a 3-step procedure: i. formation of the diester using MeOH and SOCl2, ii. acylation of the amine by benzoylchloride, iii. saponification of esters (Scheme 1). Synthesis of desired compounds proceeded first by in situ conversion of the corresponding N-alkylated of N-acylated iminodiacetic precursors into a cyclic anhydride with TFAA in acetic anhydride. Anhydrides are then reacted with an histidine or phenylalanine derivative to give compounds 1–3, 6 and 7 after a deprotection step if needed (Scheme 1). Finally, amide analogues 4–5 were obtained by reacting methyl esters 1 and 3 respectively with methylamine, in refluxed methanol overnight.
Scheme 1.

a Synthesis of carboxylic acids 1–7.
a Reactants and conditions : a) R-Br, MeOH, DIEA or TEA or KOH, room temp., 2–12 h b) trifluoroacetic anhydride 2% in acetic anhydride, 20–70 °C, 5h, 100%. c) histidine derivative or phenylalanine methyl ester, anhydrous DIEA, anhydrous DMF, Argon, room temp., overnight. d) TFA 5%/DCM, triisopropylsilane, 1 h, room temp. e) MeNH2/EtOH, MeOH, reflux. f) i. 80/20 MeOH/SOCl2, overnight, room temp. ii. Benzoylchloride, DIEA, CH2Cl2 overnight, room temp. iii. NaOH, MeOH, H2O.
2.2.2. Structure-activity relationships
Table 1 gathers results of the preliminary SARs. We found that homologation of the benzylalkyl chain consistently improves IC50 (3 versus 1, 5 versus 4). Also, the replacement of the methyl ester by a methylamide group led to more potent compounds (5 versus 3, 4 versus 1). Compound 5, bearing the phenylpropyl chain, is the most potent compounds (BDM43079, IC50 Aβ16–23 = 100 nM). In this series, all active compounds plateau at 50%. This is consistent with their retained ability to bind to the exosite that we observed in X-ray crystal structures (Supporting Information Fig. S3). Expectedly, the replacement of the imidazole ring by a phenyl ring (6) or the replacement of the benzyl by a benzoyl (7), completely abolish activity in line with the binding requirements described for the enzyme and the E111Q mutant.
Table 1.
Inhibition of the hydrolysis of Aβ16–23 by IDE by 1–7.
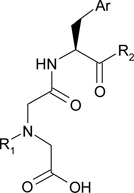 | ||||
|---|---|---|---|---|
| Cmpd | R1− | Ar | -R2 | IC50 Aβ16–23 hydrolysis [µM] [a] |
| 1 (BDM41367) | -OCH3 | 2.9 ± 1.9 | ||
| 2 | -OCH3 | 1.4 ± 1.3 | ||
| 3 | -OCH3 | 0.6 ± 0.9 | ||
| 4 | -NHCH3 | 0.6 ± 0.6 | ||
| 5 (BDM43079) | -NHCH3 | 0.1 ± 0.5 | ||
| 6 | -OCH3 | > 100 | ||
| 7 | 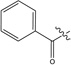 |
-OCH3 | > 100 | |
IC50 values are given as the mean standard error from 3–7 independent experiments performed in quadruplicate.
2.3. Characterization of 5
2.3.1. Substrate-dependent activity
The effect of the best inhibitor 5 on the hydrolytic profile of IDE was assessed using five unlabelled native substrates (Fig. 4). Expectedly 5 inhibits the hydrolysis of amyloid-β1–40 by IDE (IC50 Aβ1–40: 3.1 ± 2.0 µM) (Fig. 4A). However, in contrast with EDTA, the effect of 5 depends on the nature of the substrate. Indeed, it promotes the hydrolysis of insulin (EC50 insulin: 0.53 ± 0.39 µM) and IGF-II (Fig. 4B–C), and has no effect on the hydrolysis of somatostatin or glucagon (Fig. 4D–E).
Fig. 4.

Modification of hIDE peptidolytic profile by 5 (BDM43079) measured on full unlabelled native substrates.
A) amyloid-β1–40, B) insulin C) IGF-II, D) somatostatin, E) glucagon. Values are given as the mean of 3 experiments. [hIDE] = 7.5 µg/mL. [5] = 100 µM; [EDTA] = 50 mM. EDTA inhibits hIDE independently of the substrate’s nature by chelating the zinc, whereas 5 behaves either as an activator or an inhibitor depending of the substrate.
The absence of inhibition of insulin hydrolysis could be due to the much higher affinity of this substrate to the enzyme (Km = 85 nM) [33]. Also, to bind to the exosite, amyloid-β uses two acidic residues while insulin and IGF-II has rather neutral residues. The changes in the molecular surface of the exosite of 1-bound-IDE may contribute to the differences in substrate susceptibility to the ligands. The activating effect observed could also be mediated by the reduction of IDE aggregation observed in a Small-Angle X-Ray scattering experiment (Supporting Information Fig. S4).
2.3.2. Selectivity of 5
Before using 5 in cellular systems, we checked its selectivity against a panel of related metalloproteases including hNEP and hECE, two other metalloproteases implicated in the hydrolysis of amyloid-β. 5 proved to be selective with IC50s above 100µM for hNEP, hECE, hACE, hACE2, hTACE, hMMP1 and hMMP13.
2.4. Cellular activity
2.4.1. Synthesis and characterization of precursor 8
To assess the effect of 5 on cells that natively express IDE, we used SH-SY5Y neuroblastoma cells that produce amyloid-β from hAPP751 with the Swedish mutation (APPswe) or hAPP696 (APPwt) (Fig. S5) [34]. As one could expect from its zwitterionic behaviour, 5 does not cross easily cell membranes. We thus synthesized the methyl ester 8 (BDM43124, Fig. 5, Scheme 2). Compound 8 was synthesized similarly to analogues 1–7. Opening of the anhydride was performed with methanol, then the resulting monoacid compound was reacted with the histidine derivative with EDCI and HOBt. Deprotection of the trityle function in acidic conditions gave 8 (Scheme 2).
Fig. 5.

Probe profile for 5 and its methyl ester 8.
MW in g.mol−1; Solubility: in PBS from a DMSO stock solution; LogD7.4: PBS/octanol partition from a DMSO stock solution; Plasma stability: mixed gender mouse plasma lithium heparin; Cell medium stability: William’s E medium supplemented with 10 % fetal calf serum.
Scheme 2.

a Synthesis of precursor 8.
a Reactants and conditions : a) trifluoroacetic anhydride 2% in acetic anhydride, 20–70 °C, 5h, 100%. b) MeOH, DIEA, reflux, overnight; c) H-L-His(1-Trt)-NHMe, EDCI, HOBt, DIEA, DMF, room temp., overnight; d) TFA 5%/DCM, triisopropylsilane, 1 h, room temp.
The carboxylic acid is essential for binding to the catalytic site but not to the exosite. In agreement with this binding mode observed in this series, 8 is inactive regarding Aβ16–23 hydrolysis (IC50 > 100 µM) and shows some residual activation of insulin hydrolysis (50% of 5 activity at 100µM). This compound has a higher logD7.4 while retaining a good solubility and is cell permeable (0.2 10−6 cm.s−1). We checked that it hydrolyses readily to give back the active acid 5 (conversion half lives = 6 min in plasma and 11.3 h in cell culture medium) (Fig. 5) [35]
2.4.2. Cellular activity of 8
SH-SY5Y cells [36] treated with 8 display a dose-dependent increase in levels of both Aβ1–40 and 1–42 (Fig. 6) [37]. Differences between 24 and 48 hours may reflect the slow permeation kinetic of 8. No effect on APP holoproteins and on CTFs was observed, which is consistent with the inhibition of amyloid-β degradation. This confirms the implication of IDE in the catabolism of amyloid-β. [38]
Fig. 6.

Aβ Cellular responses to 8 (BDM43124) methyl ester precursor of 5.
Effect of 0.1 or 10 µM of 8 on extracellular levels of Aβ 1–40 (A, B) and Aβ1–42 (C, D) in different APP SY5Y strains at 24 h (□) or 48 h (■). A) and C) APPswe, B) and D) APPwt. 8 increases Aβ peptides levels in a dose-dependent manner. Results are normalized to Aβ levels in vehicle treated cells. *p < 0.05, **p < 0.01.
We also use β-pancreatic cells and hepatocytes which we show express IDE. 8 did neither affect the level of insulin secreted by β-pancreatic cells nor modify the repression of PEPCK by insulin on hepatocytes. This result suggests that 8 does not inhibit insulin degradation by IDE in a cellular context and confirm the substrate selectivity observed in the biochemical assays. (Supporting Information Fig. S6). The activating effect observed in a biochemical setting, which we linked to the reduction of aggregation of the enzyme, may not be relevant in a much more complex cellular context.
3. Conclusion
Altogether, we report here the discovery and characterization of small dual ligands of IDE. Unlike the previously described hydroxamate inhibitors, the lead compound 1 binds to the catalytic site and a distal exosite in the N-terminal domain of IDE 30Å away from the zinc atom [5].
The optimized compound 5 modulates the proteolytic profile of IDE in a substrate-dependent manner. In particular, it has contrasted effects on insulin and amyloid-β hydrolysis in cell-free enzymatic assays.
Altogether, this work brings new modulators of IDE that differentiate from previously described inhibitors [39] by their binding mode and their selectivity toward Amyloid beta hydrolysis, leaving intact insulin degradation in cell models. Moreover, in a cell-based assay, using 8 as a selective pharmacological reagent, we proved that IDE is involved in the degradation of amyloid-β 1–40 and 1–42. This is in line with previous implication of IDE in Alzheimer's disease by genomic and genetic experiments.
Along with the results on cell-models, the stability, solubility and selectivity properties of 5 and its precursor 8 (BDM43124), support their use as a probes for the exploration of the role of IDE in the amyloid pathway without affecting the insulin pathway.
More importantly our results strongly consolidate previous observations that pointed out IDE as an important player in the amyloid degradation and encourage the search for drugs that would either activate directly IDE, or increase its expression, to clear amyloid peptide efficiently.
4. Experimental section
4.1. Biology
4.1.1. In vitro IDE activity assay
In vitro IDE activity was measured with a quenched substrate ATTO 655- Cys-Lys-Leu-Val-Phe-Phe-Ala-Glu-Asp-Trp. Human recombinant IDE was cloned and purified as previously described. Briefly, human IDE (1.87 ng/µL) was incubated 10 min at 37 °C with compound in Hepes 50 mM, NaCl 100 mM, pH 7.4 and the enzymatic reaction is started by adding the substrate (final concentration 5 µM). After 30 min, samples (1% DMSO final) are excited at 635 nm and fluorescence emission at 750 nm is measured on a Victor3 V1420 Perkin Elmer spectrophotometer. All measurements were carried out as 8-point dose response curves and are reported as the average of at least three independent measurements. EDTA was used as a reference inhibitor (100% inhibition at 2 mM).
4.1.2. Cell culture of SH-SY5Y stable cell lines
Human neuroblastoma cell lines SH-SY5Y expressing either human APP751 with the Swedish mutation (APPSw) or human APP696 (SY5Y-APPwt) were generated as previously described (Vingtdeux, V. et al. Neurobiol Dis 20, 625–37 (2005) and Vingtdeux, V. et al. Neurobiol Dis 25, 686–96 (2007). Cells were maintained in Dulbecco’s modified Eagle medium (DMEM, GIBCO BRL, France) supplemented with 10% fetal calf serum, 2 mM L-glutamine, 1 mM non-essential amino-acids, 50 units/mL penicillin/streptomycin (Invitrogen, France) in a 5% CO2 humidified incubator at 37 °C.
4.1.3. Cellular assay in the amyloid pathway
Cells were seeded in 6-well plates (1.94 105 cells/well). 24 h later, cells were replenished with 2 ml of medium containing either vehicle or drug at 0.1 µM or 10 µM (from a stock solution at 10 mM in DMSO). Following 24 h and 48 h of drug exposure, the cell medium was collected, discarded from cell debris by centrifugation (1000 × g, 10 min) and was kept at −80°C for ELISA dosages of amyloid β 1–42 and amyloid β 1–40. For preparation of total cellular proteins, adherent cells were washed with PBS and scraped with a rubber policeman in 50 µL of RIPA buffer (Thermo Scientific) containing protease inhibitors (Complete Mini, Roche Molecular Biochemicals, Meylan, France). The cell homogenate was sonicated and stirred 10 min at 4 °C. Cell lysate was recovered in supernatant after centrifugation at 12,000 × g, 10 min at 4 °C. Protein concentration was determined using the BCA protein assay kit (Pierce) and samples were kept at −80 °C. Proteins were mixed to LDS Sample Buffer containing a reducing agent (Invitrogen NP-009) and boiled at 100°C for 10 minutes. 12 µg of total proteins were loaded onto a 4–12% Bis-Tris-SDS-polyacrylamide gel (Novex NuPAGE® Invitrogen), blotted onto nitrocellulose membrane (0.44 µm Hybond and Hybond-Phosphate from Amersham/GE Healthcare France). For APP-CTF analysis, 30 µg of total proteins were loaded on Tris-Tricine SDS-polyacrylamide gel electrophoresis (Criterion precast gel, Bio-Rad Bioresearch division, Marnes la coquette, France). Proteins were transferred to nitrocellulose membrane (0.22 µM Hybond, Amersham Biosciences). Membranes were blocked and incubated with the appropriate antibody according to Supporting Information Table 2 and then incubated with a Horseradish peroxidase-conjugated secondary antibody (Goat anti-rabbit A4914 from Sigma-Aldrich and Horse anti-mouse from Vector Laboratories). Finally, peroxidase activity was revealed with the ECL detection kit and visualized with Hyperfilm™ ECL™ (Amersham/GE Healthcare France). Quantitative determinations of amyloid β 1–42 and amyloid β 1–40 in cell medium at the end of drug treatments were performed using human amyloid β 1–42 and amyloid β 1–40 Elisa kits (Invitrogen). Following initial experiments for determining optimal dilution factors, dosages were performed with 100 µL of cell medium for amyloid β 1–42 (undiluted sample) and 10 µL of cell medium for amyloid β 1–40 (dilution factor of 1:10). The data was obtained in duplicate from three independent experiments and normalized to untreated cells.
4.2. Chemistry
4.2.1. General information
NMR spectra were recorded on a Bruker Avance 300 spectrometer. Chemical shifts are in parts per million (ppm) and were referenced to the residual proton peaks in deuterated solvents. The assignments were made using one dimensional (1D) 1H and 13C spectra (classical or Jmod) and two-dimensional (2D) HSQC, HMBC, ROESY and COSY spectra. Mass spectra were recorded with a LC−MS−MS triple−quadrupole system (Varian 1200ws) or a LCMS (Waters Alliance Micromass ZQ 2000). LCMS analysis was performed using a C18 TSK-GEL Super ODS (2 µm particle size column, dimensions 50 mm × 4.6 mm). A gradient starting from 100% H2O/0.1% formic acid and reaching 20% H2O/80% CH3CN/0.08% formic acid within 10 min (method A) or 5 min (method B) at a flow rate of 1 mL/min was used. Preparative HPLC were performed using a Varian PRoStar system using an OmniSphere 10 column C18 250 mm × 41.4 mm Dynamax from Varian, Inc. A gradient starting from 20% CH3CN/80% H2O/0.1% formic acid and reaching 100% CH3CN/0.1% formic acid at a flow rate of 80 mL/min or 20% MeOH/80% H2O/0.1% formic acid reaching 100% MeOH/0.1% formic acid was used. Purity (%) was determined by reversed phase HPLC, using UV detection (215 nm), and all compounds showed purity greater than 95%; Melting points were determined on a Büchi B-540 apparatus and are uncorrected. All commercial reagents and solvents were used without further purification. Organic layers obtained after extraction of aqueous solutions were dried over MgSO4 and filtered before evaporation under reduced pressure. Purification yields were not optimized. Thick layer chromatography was performed with Silica Gel 60 (Merck, 40–63 µm).
4.2.2. (Carboxymethyl-phenethyl-amino)-acetic acid
To a stirred solution of iminodiacetic acid (500mg, 2.7 mmol) in MeOH (30 mL) was added KOH (454 mg, 8.1 mmol) and (2-bromoethyl)benzene (549 mg, 2.97 mmol). The mixture was refluxed overnight, cooled at room temperature and filtrated. The filtrate was evaporated and the crude product was purified by preparative HPLC to give the compound as a white solid (230 mg, 36%). Purity 100%; 1H NMR (DMSO-d6) δ ppm: 7.22 (m, 5H), 3.46 (s, 4H), 2.86 (m, 2H), 2.70 (m, 2H). 13C NMR (DMSO-d6) δ ppm : 172.5, 140.0, 128.6, 128.3, 125.9, 55.8, 54.7, 33.8. LC tR=1.12 min, MS (ESI+): m/z= 238 (M+H)+.
4.2.3. [Carboxymethyl-(3-phenyl-propyl)-amino]-acetic acid
To a stirred solution of iminodiacetic acid (277 mg, 2.08 mmol) and TEA (870 µL, 6.24 mmol) in methanol (10 mL) was added 1-bromo-3-phenylpropane (348 µL, 2.28 mmol). The mixture was stirred at room temperature for 10 hours and then was refluxed for 24 hours. The solvent was evaporated and the crude product was purified by preparative HPLC to give the compound (103 mg, 27%) Purity 99%; 1H NMR (DMSO-d6) δ ppm: 1.67 (dt, J = 7.5Hz, 2H), 2.56 (t, J = 7.5Hz, 2H), 2.65 (t, J = 7.5Hz, 2H), 3.41 (s, 4H), 7.13–7.26 (m, 5H). 13C NMR (DMSO-d6) δ ppm: 172.6, 141.9, 128.3, 126.1, 125.7, 57.5, 53.7, 32.5, 28.8. LC tr = 2.76 min; MS (ESI+): m/z = 252 (M+H)+.
4.2.4. (S)-2-[2-(Benzyl-carboxymethyl-amino)-acetylamino]-3-(1H-imidazol-4-yl)-propionic acid methyl ester (1) (BDM41367)
To a stirred solution of L-histidine methyl ester dihydrochloride (324 mg, 1.34 mmol) in DMF (13.4 mL) were added DIEA (704 µL) and N-benzyliminodiacetic anhydride acid (250 mg, 1.22 mmol) in THF (2.24 mL). The mixture was stirred overnight at room temperature and the solvent was evaporated. The crude product was purified by preparative HPLC to yield compound 1 (BDM41367) as a white solid (20%). Purity 100%; 1H NMR (DMSO-d6) δ ppm: 2.99 (d, J=6.3Hz, 2H), 3.23 (s, 2H), 3.27 (s, 2H), 3.59 (s, 3H), 3.71 (d, J=13.4Hz, 1H), 3.77 (d, J=13.4Hz, 1H), 4.65 (m, 1H), 6.93 (d, J=0.9 Hz, 1H), 7.30 (m, 5H), 8.26 (d, J=0.9 Hz, 1H), 8.34 (d, J=8.0Hz, 1H); 13C NMR (DMSO-d6) δ ppm : 172.3, 171.7, 170.4, 138.2, 137.9, 129.0, 128.4, 127.4, 116.0, 57.6, 56.7, 53.6, 52.2, 51.6, 28.4. LC tR= 2.30 min, MS (ESI+): m/z= 375 (M+H)+. HRMS (m/z): [MH+] calcd. for C18H23O5N4, 375.1668; found, 375.1665.
4.2.5. (S)-2-[2-(Carboxymethyl-phenethyl-amino)-acetylamino]-3-(1H-imidazol-4-yl)-propionic acid methyl ester (2)
(Carboxymethyl-phenethyl-amino)-acetic acid (210 mg, 0.88 mmol) was stirred in a 2% TFAA in acetic anhydride solution at 40 °C during 4 h and solvent were evaporated. To a stirred solution of L-histidine methyl ester dihydrochloride (245 mg, 1.01 mmol) in anhydrous DMF (10 mL) were added dry DIEA (510 µL) and the anhydride (0.88mmol) in anhydrous DMF (2 mL). The mixture was stirred overnight at room temperature under argon and the solvent was evaporated. The crude product was purified by preparative HPLC to yield compound 2 as a white solid (200 mg, 52%). Purity 97%; 1H NMR (DMSO-d6) δ ppm:8.22 (d, J=7.8Hz, 1H), 8.14 (s, 1H, HCOOH), 7.56 (d, J=1.1Hz, 1H), 7.28-7.22 (m, 2H), 7.18-7.14 (m, 3H), 6.82 (d, J=1.0Hz, 1H), 4.52 (m, 1H), 3.58 (s, 3H), 3.38 (s, 2H), 3.25 (s, 2H), 2.92 (m, 2H), 2.76 (m, 2H), 2.66 (m, 2H); LC tR= 2.63 min, MS (ESI+): m/z= 389 (M+H)+. HRMS (m/z): [MH+] calcd. for C19H25O5N4, 389.1825; found, 389.1820.
4.2.6. (S)-2-{2-[Carboxymethyl-(3-phenyl-propyl)-amino]-acetylamino}-3-(1H-imidazol-4-yl)-propionic acid methyl ester .2TFA (3)
[Carboxymethyl-(3-phenyl-propyl)-amino]-acetic acid (0.4 mmol) was stirred in a 2% TFAA in acetic anhydride solution at 40°C during 4h and solvent were evaporated. To a stirred solution of L-(Trt)Histidine methyl ester hydrochloride (174 mg, 0.39 mmol) and DIEA (200 µL, 1.17 mmol) in anhydrous DMF (3 mL) was added the anhydride (99 mg, 0.39 mmol). The mixture was stirred overnight at room temperature under argon and the solvent was evaporated. The crude product was purified by preparative HPLC to give the compound as a white solid (142 mg, 57%). Purity 98% ;1H NMR (DMSO-d6) δ ppm: 1.62 (dt, J = 7.4Hz, 2H), 2.5 (m, 2H), 2.59 (t, J = 7.4Hz, 2H), 2.88 (m, 2H), 3.19 (s, 2H), 3.28 (s, 2H), 3.51 (s, 3H), 4.58 (ddd, J= 6Hz, 8.1Hz, 1H), 6.62 (s, 1H), 6.99–7.38 (m, 21 H), 8.49 (d, J = 8.2 Hz, 1H). 13C NMR (DMSO-d6) δ ppm: 29.3, 29.8, 32.7, 51.8, 51.8, 53.8, 55.1, 57.8, 119.0, 125.6, 127.5, 127.8, 128.0, 128.2, 129.2, 136.3, 137.6, 142.1, 170.6, 171.7, 172.7. LC tr = 5.21 min; MS (ESI+): m/z = 645 (M+H)+. The trityl group was removed in TFA 5%/DCM. Triisopropylsilane was added until the yellow color disappears. The mixture was stirred at room temperature for 1 h. The solvent was evaporated and the crude product was triturated in diethyl ether to give 3 as a white solid (69 mg, 55%). Purity 99% ;1H NMR (DMSO-d6) δ ppm: 1.79 (m, 2H), 2.57 (m, 2H), 2.90 (m, 2H), 3.06 (dd, J=8.6 and 15.4 Hz, 1H), 3.19 (dd, J=5.4 and 15.4 Hz, 1H), (m, 2H), 3.62 (s, 3H), 3.70 (s, 2H), 3.78 (s, 2H), 4.70 (m, 1H), 7.17–7.30 (m, 5H), 7.41 (s, 1H), 8.86 (d, J = 7.2Hz, H), 8.98 (s, 1H). 13C NMR (DMSO-d6) δ ppm: 26.1, 26.8, 32.2, 51.2, 52.4, 54.7, 54.8, 55.7, 117.2, 126.0, 128.3, 128.4, 128.9, 133.7, 141.1, 167.5, 169.7, 170.6. LC tr = 3.06 min; MS (ESI+): m/z = 403 (M+H)+. HRMS (m/z): [MH+] calcd. for C20H27O5N4, 403.1981; found, 403.1978.
4.2.7. (Benzyl-{[(S)-2-(1H-imidazol-4-yl)-1-methylcarbamoyl-ethylcarbamoyl]-methyl}-amino)-acetic acid. 2TFA (4)
To a stirred solution of L-His(Trt)-OMe hydrochloride (1.5 mmol) in DMF (15 mL) were added DIEA (1 eq) and N-benzyliminodiacetic anhydride acid (1.4 mmol) in THF (3 mL). The mixture was stirred overnight at room temperature and the solvent was evaporated. The crude product was purified by precipitation in H2O as a white powder (80%) Purity 97% ;1H NMR (DMSO-d6) δ ppm: 2.91–2.94 (m, 2H), 3.24 (s, 2H), 3.26 (s, 2H), 3.52 (s, 3H), 3.77 (s, 2H), 4.58–4.65 (m, 1H), 6.66 (s, 1H), 7.01–7.04 (m, 6H), 7.15–7.18 (m, 5H), 7.33 (s, 1H), 7.35–7.38 (m, 9H), 8.47(d, J = 8.1Hz, 1H). 13C NMR (DMSO-d6) δ ppm: 29.5, 51.7, 51.9, 53.1, 56.9, 57.4, 74.7, 119.2, 127.2, 127.6, 127.8, 128.1, 128.2, 128.9, 129.3, 136.1, 137.9, 138.1, 142.1, 170.1, 171.6, 172.1. LC tr = 5.08 min; MS (ESI+): m/z = 617 (M+H)+. To a stirred solution of methyl ester (0.5 mmol) in methanol (1 mL) were added 622 µL of a methylamine solution in ethanol (33%). The mixture was refluxed overnight. The solvent was evaporated and the crude product was precipitated. The product was purified by preparative HPLC to give the formiate salt of (Benzyl-{[(S)-1-methylcarbamoyl-2-(1-trityl-1H-imidazol-4-yl)-ethyl-carbamoyl]-methyl}-amino)-acetic acid (100%). Purity: 97%; LC : tr = 4.5min; MS (ESI+) : m/z = 616 (M+H)+. The trityl group was removed in TFA 5%/DCM. Triisopropylsilane was added until the yellow color disappears. The mixture was stirred at room temperature for 1 h. The solvent was evaporated and the crude product was triturated in diethyl ether to give 4 as colourless oil (70%).Purity : 98% ; 1H NMR (DMSO-d6) δ ppm : 8.95 (s, 1H), 8.39 (d, J = 8.1 Hz, 1H), 8.00 (q, J = 4.5 Hz, 1H), 7.33 (m, 5H), 7.28 (s, 1H), 4.57 (m, 1H), 3.91 (s, 2H), 3.51 (s, 2H), 3.49 (s, 2H), 3.14 (dd, J = 5.4 Hz and J = 15.3 Hz, 1H), 2.93 (dd, J = 8.1 Hz and J = 15.3 Hz, 1H), 2.60 (d, J = 4.5 Hz, 3H); 13C NMR (DMSO-d6) δ ppm : 170.8, 169.8, 168.4, 135.4, 133.9., 129.8, 129.3, 128.5, 128.1, 116.8, 57.9, 55.9, 53.7, 51.3, 27.3, 25.7; LC : tr = 2.55min; MS (ESI+) : m/z = 374 (M+H)+. HRMS (m/z): [MH+] calcd. for C18H24O4N5, 374.1828; found, 374.1823.
4.2.8. : ((((S)-2-(1H-Imidazol-4-yl)-1-methylcarbamoyl-ethylcarbamoyl)-methyl)-(3-phenylpropyl)-amino)-acetic acid. 2TFA (5) (BDM43079)
The diacid (0.4 mmol) was stirred in a 2% TFAA in acetic anhydride solution at 40°C during 4h and solvent were evaporated. To a stirred solution of L-(Trt)Histidine methyl ester hydrochloride (174 mg, 0.39 mmol) and DIEA (200 µL, 1.17 mmol) in anhydrous DMF (3 mL) was added the anhydride (0.39 mmol). The mixture was stirred overnight at room temperature under argon and the solvent was evaporated. The crude product was purified by preparative HPLC to give the compound as a white solid (63%, formiate salt). Purity 92% ;1H NMR (DMSO-d6) δ ppm: 1.62 (dt, J = 7.4Hz, 2H), 2.5 (m, 2H), 2.59 (t, J = 7.4Hz, 2H), 2.88 (m, 2H), 3.19 (s, 2H), 3.28 (s, 2H), 3.51 (s, 3H), 4.58 (ddd, J= 6Hz, 8.1Hz, 1H), 6.62 (s, 1H), 6.99–7.38 (m, 21 H), 8.49 (d, J = 8.2 Hz, 1H). 13C NMR (DMSO-d6) δ ppm: 29.3, 29.8, 32.7, 51.8, 51.8, 53.8, 55.1, 57.8, 119.0, 125.6, 127.5, 127.8, 128.0, 128.2, 129.2, 136.3, 137.6, 142.1, 170.6, 171.7, 172.7. LC tr = 5.21 min; MS (ESI+): m/z = 645 (M+H)+. To a stirred solution of methyl ester (0.25 mmol) in methanol (0.5 mL) were added 311 µL of a methylamine solution in ethanol (33%). The mixture was refluxed overnight. The solvent was evaporated and the crude product was precipitated in an aqueous solution of formic acid to give [{[(S)-1-Methylcarbamoyl-2-(1-trityl-1H-imidazol-4-yl)-ethylcarbamoyl]-methyl}-(3-phenyl-propyl)-amino]-acetic acid (49%) as the formiate salt. Purity : 97% ; LC : tr = 5.27min; MS (ESI+) : m/z = 644 (M+H)+. The trityl group was removed in TFA 5%/DCM. Triisopropylsilane was added until the yellow color disappears. The mixture was stirred at room temperature for 1 h. The solvent was evaporated and the crude product was triturated in diethyl ether to give 5 (BDM43079).2TFA as colourless oil (58%). Purity : 98% ; 1H NMR (DMSO-d6) δ ppm : 9.05 (d, J = 8.7Hz, 1H), 9.01 (s, 1H), 8.22 (q, J = 4.8Hz, 1H), 7.38 (s, 1H), 7.32-7.19 (m, 5H), 4.58 (m, 1H), 4.13 (m, 2H+1H), 4.08 (d, J = 6.6Hz, 1H), 3.17-3.13 (m, 2H+1H), 2.96 (dd, J = 8.7 Hz and J = 14.8 Hz, 1H),2.60-2.57 (m, 3H+2H), 1.92 (m, 2H); 13C NMR (DMSO-d6) δ ppm : 169.5, 167.7, 165.2, 140.5, 134.1, 129.2, 128.4, 128.3, 126.1, 116.8, 55.3, 54.9, 54.1, 52.1, 31.8, 27.0, 25.7, 25.3; LC : tr = 3.25min; MS (ESI+) : m/z = 402 (M+H)+. HRMS (m/z): [MH+] calcd. for C20H28O4N5, 402.2141; found, 402.2137.
4.2.9. (S)-2-(2-(Benzyl-carboxymethyl-amino)-acetylamino)-3-phenyl-propionic acid methyl ester. (6)
To a stirred solution of L-Phe-OMe hydrochloride (1.5 mmol) in DMF (15 mL) were added DIEA (1 eq) and N-benzyliminodiacetic anhydride acid (1.4 mmol) in THF (3 mL). The mixture was stirred overnight at room temperature and the solvent was evaporated. The crude product was purified by precipitation in H2O to give 6 as a colorless oil. Yield 40%. 1H NMR (DMSO-d6) δ ppm : 8.13 (d, J = 8.10 Hz, 1H), 7.31-7.16 (m, 10H), 4.58 (m, 1H), 3.69 (d, J = 13.2 Hz, 1H), 3.64 (d, J = 13.2 Hz, 1H), 3.61 (s, 3H), 3.22 (s, 2H), 3.16 (s, 2H), 3.08 (dd, J = 5.4 and 13.8 Hz, 1H), 2.96 (dd, J = 8.7 and 13.8 Hz, 1H); 13C NMR (MeOD) δ ppm : 173.8, 173.0, 172.5, 137.7, 130.6, 130.3, 129.6, 128.9, 128.1, 59.5, 57.7, 55.6, 54.6, 52.8, 38.3 tR,LCMS = 4.39 min, Purity 100%; MS (ESI+): m/z = 385 [M+H]+. HRMS (m/z): [MH+] calcd. for C21H25O5N2, 385.1763; found, 385.1756.
4.2.10. (S)-2-(2-(Benzoyl-carboxymethyl-amino)-acetylamino)-3-(1H-imidazol-4-yl)-propionic acid methyl ester (7)
Iminodiacetic acid was stirred overnight in a 80/20 MeOH/SOCl2 mixture. The diester obtained after evaporation under reduced pressure (300 mg, 1.52 mmol) was dissolved in CH2Cl2 (10 mL) and DIEA (792 µL, 4.56 mmol) and benzoylchloride (176 µL, 1.52 mmol) were added. The mixture was stirred overnight at room temperature and washed with HCl 0.5 N and NaHCO3 10 % solutions. The organic layer was dried over MgSO4 and evaporated to give the desired diester as a colorless oil (99%). The former compound (400 mg, 1.5 mmol) was saponified with NaOH (6 mmol) in MeOH (5 mL) and H2O (1 mL). After evaporation of MeOH under reduced pressure, HCl 1 N was added to the residue and the aqueous layer was extracted 3 times with AcOEt to give (benzoyl-carboxymethyl-amino)-acetic acid directly used in the next step. White powder. Yield 98 %. tR,LCMS = 2.53 min (5 min gradient), Purity 98%; MS (ESI+): m/z = 238 [M+H]+. The diacid (0.74 mmol) was dissolved in trifluoracetic anhydride 2% in acetic anhydride (3 mL). The reaction mixture was stirred for 4 hours at room temperature then evaporated. To a stirred solution of L-histidine methyl ester dihydrochloride (188 mg, 0.77 mmol) in DMF (8.0 mL) were added DIEA (515µL) and the anhydride (0.74 mmol) in DMF (1.0 mL). The mixture was stirred overnight at room temperature and the solvent was evaporated. The crude product was purified by preparative HPLC to give 7 as a white solid. Yield 26%. 1H NMR (DMSO-d6) δ ppm : 2.85–3.02 (m, 2H), 3.59(s, 1.2H), 3.64 (s, 1.8H), 3.88 (s, 2H), 4.02 (s, 1.2H), 4.08 (s, 0.8H), 4.55 (m, 1H), 6.86 (s, 0.6H), 6.91 (s, 0.4H), 7.31–7.44 (m, 5H), 7.69 (s, 0.6H), 7.74 (s, 0.4H), 8.78 (d, J = 7.5 Hz, 0.6 H), 8.89 (J = 7.2 Hz, 0.4 H). 13C NMR (MeOD) δ ppm 175.9, 174.9, 172.1, 172.0, 136.7, 135.7, 131.3, 129.6, 127.8, 118.7, 56.6, 55.9, 53.0, 28.3. tR,LCMS = 2.48 min; Purity 98 %; MS (ESI+): m/z = 389 [M+H]+. HRMS (m/z): [MH+] calcd. for C18H21O6N4, 389.1461; found, 389.1471.
4.2.11. ((((S)-2-(1H-Imidazol-4-yl)-1-methylcarbamoyl-ethyl-carbamoyl)-methyl)-(3-phenylpropyl)-amino)-acetic acid methyl ester. 2TFA (8) (BDM43124)
To a stirred solution of H-L-His(Trt)-OMe (2.5 g, 5.5mmol) in methanol (10 mL) were added 4mL of a solution of methylamine in ethanol (33%). The mixture was refluxed overnight. The solvent was evaporated and the crude product was precipitated in water and filtrated. The crude product was purified by preparative HPLC to give H-L-His(1-Trt)-NHMe as a white powder (1.43 g, 63%, formiate salt). Purity : 74% ; 1H NMR (DMSO-d6) δ ppm : 8.30 (s, 1H), 7.95 (q, J = 4.8Hz, 1H), 7.06–7.43 (m, 15H), 6.63 (s, 1H), 3.53 (dd, J = 5.7Hz and J = 7.2Hz, 1H), 2.77 (dd, J = 5.7Hz and J = 14.4Hz, 1H), 2.63 (dd, J = 5.7Hz and J = 14.4Hz, 1H), 2.54 (d, J = 4.8Hz, 3H); LC : tr = 4.79min; MS (ESI+) : m/z = 411 (M+H)+. To a stirred solution of H-L-His(1-Trt)-NHMe (456 mg, 1.1 mmol) in DMF (5 mL) was added the anhydride of [Carboxymethyl-(3-phenyl-propyl)-amino]-acetic acid (1 mmol), HOBt (149 mg, 1.1 mmol), EDCI (211 mg, 1.1 mmol) and DIEA (690 µL, 4 mmol). The mixture was stirred overnight. Then 213 mg of EDCI and 200 µL of DIEA were added and the resulting slurry was stirred overnight. The solvent was evaporated. The crude product was purified by preparative HPLC to give [{[(S)-1-Methylcarbamoyl-2-(1-trityl-1H-imidazol-4-yl)-ethylcarbamoyl]-methyl}-(3-phenyl-propyl)-amino]-acetic acid methyl ester (171 mg, 26%). Purity: 92%; LC: tr = 6.14 min; MS : (ESI+) : m/z = 658 (M+H)+. To a stirred solution of triisopropylsilane and DCM (0.5mL/4mL) was added the protected compound (171 mg, 0.26 mmol). Then 500 µL of TFA were added. The mixture was stirred at room temperature for 2 h. The solvents were evaporated and the crude product was washed with petroleum ether and diethyl ether to give 8.2TFA as yellow oil (165 mg, 99%). Purity : 99% ; 1H NMR (DMSO-d6) δ ppm : 8.97 (s, 1H), 8.53 (d, J = 7.9Hz, CONH),, 8.04 (q, J = 4.7Hz, CONH), 7.33-7.14 (m, 6H), 4.58 (m, 1H), 3.82 (s, 2H), 3.67 (s, 3H), 3.62 (s, 1H), 3.13 (dd, J = 5.12 Hz and J = 15.36 Hz, 1H), 2.92 (dd, J = 8.4 Hz and J = 15.3Hz, 1H), 2.87 (t, J = 7.4Hz, 2H), 2.59 (d, J = 4.2Hz, 3H), 2.53 (m, 2H), 1.77 (qt, J = 7.4Hz, 2H); 13C NMR (DMSO-d6) δ ppm : 169.8, 169.2, 167.7, 141.2, 134.4, 129.4, 128.4, 128.3 126.0, 116.9, 56.3, 54.9, 54.5, 52.1, 51.5, 32.3, 27.3, 27.1, 25.8; LC : tr = 4.07min; MS (ESI+) : m/z = 644 (M+H)+. HRMS (m/z): [M+] calcd. for C21H30O4N5, 416.2298; found, 416.2284.
Supplementary Material
Acknowledgements
The authors acknowledge assistance from C. Piveteau, S. Dassonneville, H. Drobecq and financial support from INSERM, University of Lille 2, Pasteur Institute of Lille, Region Nord Pas de Calais, EDFR, Etat (0823007, 0823008, 07-CPER 009-01, 2007-0172-02-CPER/3), ANR (11-JS07-015-01), FRM (DCM20111223046), NIH GM81539 (to WJT) and PRIM (Pôle de Recherche Interdisciplinaire pour le Médicament). We thank the LARMNS NMR laboratory, School of Pharmacy Lille. M.G. is a recipient of a fellowship from Ministère de la Recherche et de l’Enseignement Superieur. Data management was performed using Pipeline Pilot™ from Accelrys Inc.
Abbreviations used
- EtOAc
ethyl acetate
- AcOH
acetic acid
- Boc
tert-butoxycarbonyl
- CH3CN
acetonitrile
- DCE
dichloroethane
- DCM
dichloromethane
- DIEA
N, N-diisopropylethylamine
- DMF
dimethylformamide
- DMSO
dimethylsulfoxide
- EDCI
N-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- Et3N
triethylamine
- EtOH
ethanol
- HOBt
N-hydroxybenzotriazole
- MeOH
methanol
- PBS
phosphate buffered saline
- PTSA
paratoluenesulfonic acid
- rt
room temperature
- SAR
structure–activity relationship
- TFAA
Trifluoroacetic anhydride
- THF
tetrahydrofuran
- TMB
3,3’,5,5’-tetramethylbenzidine
Footnotes
Supporting Information
Supporting information for this article is available: description of the library, mechanistic studies, enzyme substrates, full protocols, protein crystallography, and NMR spectra.
References
- 1.Fernandez-Gamba A, Leal MC, Morelli L, Castano EM. Curr. Pharm. Des. 2009;15:3644–3655. doi: 10.2174/138161209789271799. [DOI] [PubMed] [Google Scholar]
- 2.Hersh LB. Cell. Mol. Life Sci. 2006;63:2432–2434. doi: 10.1007/s00018-006-6238-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leissring MA, Farris W, Wu X, Christodoulou DC, Haigis MC, Guarente L, Selkoe DJ. Biochem. J. 2004;383:439–446. doi: 10.1042/BJ20041081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glebov K, Schütze S, Walter J. J. Biol. Chem. 2011;286:22711–22715. doi: 10.1074/jbc.C110.217893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen Y, Joachimiak A, Rosner MR, Tang WJ. Nature. 2006;443:870–874. doi: 10.1038/nature05143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malito E, Hulse RE, Tang WJ. Cell. Mol. Life Sci. 2008;65:2574–2585. doi: 10.1007/s00018-008-8112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Im H, Manolopoulou M, Malito E, Shen Y, Zhao J, Neant-Fery M, Sun CY, Meredith SC, Sisodia SS, Leissring MA, Tang WJ. J. Biol. Chem. 2007;282:25453–25463. doi: 10.1074/jbc.M701590200. [DOI] [PubMed] [Google Scholar]
- 8.Noinaj N, Bhasin SK, Song ES, Scoggin KE, Juliano MA, Juliano L, Hersh LB, Rodgers DW. PLoS ONE. 2011;6:e20864. doi: 10.1371/journal.pone.0020864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manolopoulou M, Guo Q, Malito E, Schilling AB, Tang WJ. J. Biol. Chem. 2009;284:14177–14188. doi: 10.1074/jbc.M900068200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCord LA, Liang WG, Dowdell E, Kalas V, Hoey RJ, Koide A, Koide S, Tang WJ. PNAS. 2013;110:13827–13832. doi: 10.1073/pnas.1304575110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kurochkin IV, Goto S. FEBS Lett. 1994;345:33–37. doi: 10.1016/0014-5793(94)00387-4. [DOI] [PubMed] [Google Scholar]
- 12.Roth RA, Mesirow ML, Yokono K, Baba S. Endocr. Res. 1984;10:101–112. doi: 10.3109/07435808409035411. [DOI] [PubMed] [Google Scholar]
- 13.Guo Q, Manolopoulou M, Bian Y, Schilling AB, Tang W-J. J. Mol. Biol. 2010;395:430–443. doi: 10.1016/j.jmb.2009.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ciaccio C, Tundo GR, Grasso G, Spoto G, Marasco D, Ruvo M, Gioia M, Rizzarelli E, Coletta M. J. Mol. Biol. 2009;385:1556–1567. doi: 10.1016/j.jmb.2008.11.025. [DOI] [PubMed] [Google Scholar]
- 15.Kurochkin IV. FEBS Lett. 1998;427:153–156. doi: 10.1016/s0014-5793(98)00422-0. [DOI] [PubMed] [Google Scholar]
- 16.Ralat LA, Kalas V, Zheng Z, Goldman RD, Sosnick TR, Tang W-J. J. Mol. Biol. 2011;406:454–466. doi: 10.1016/j.jmb.2010.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim Y-G, Lone AM, Nolte WM, Saghatelian A. Proc. Natl. Acad. Sci. U.S.A. 2012;109:8523–8527. doi: 10.1073/pnas.1203195109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bennett RG, Fawcett J, Kruer MC, Duckworth WC, Hamel FG. J. Endocrinol. 2003;177:399–405. doi: 10.1677/joe.0.1770399. [DOI] [PubMed] [Google Scholar]
- 19.Noinaj N, Song ES, Bhasin S, Alper BJ, Schmidt WK, Hersh LB, Rodgers DW. J. Biol. Chem. 2011;287:48–57. doi: 10.1074/jbc.M111.264614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Fawcett J, Duckworth WC. Diabetologia. 2009;52:1457–1460. doi: 10.1007/s00125-009-1414-8. [DOI] [PubMed] [Google Scholar]; (b) Fawcett J, Permana PA, Levy JL, Duckworth WC. Arch. Biochem. Biophys. 2007;468:128–133. doi: 10.1016/j.abb.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 21.(a) Farris W, Mansourian S, Leissring MA, Eckman EA, Bertram L, Eckman CB, Tanzi RE, Selkoe DJ. Am. J. Pathol. 2004;164:1425–1434. doi: 10.1016/s0002-9440(10)63229-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fakhrai-Rad H, Nikoshkov A, Kamel A, Fernström M, Zierath JR, Norgren S, Luthman H, Galli J. Hum. Mol. Genet. 2000;9:2149–2158. doi: 10.1093/hmg/9.14.2149. [DOI] [PubMed] [Google Scholar]
- 22.Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X, Frosch MP, Selkoe DJ. Neuron. 2003;40:1087–1093. doi: 10.1016/s0896-6273(03)00787-6. [DOI] [PubMed] [Google Scholar]
- 23.Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, Boutin P, Vincent D, Belisle A, Hadjadj S, Balkau B, Heude B, Charpentier G, Hudson TJ, Montpetit A, Pshezhetsky AV, Prentki M, Posner BI, Balding DJ, Meyre D, Polychronakos C, Froguel P. Nature. 2007;445:881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 24.(a) Bertram L, Blacker D, Mullin K, Keeney D, Jones J, Basu S, Yhu S, McInnis MG, Go RC, Vekrellis K, Selkoe DJ, Saunders AJ, Tanzi RE. Science. 2000;290:2302–2303. doi: 10.1126/science.290.5500.2302. [DOI] [PubMed] [Google Scholar]; (b) Prince JA, Feuk L, Gu HF, Johansson B, Gatz M, Blennow K, Brookes AJ. Hum. Mutat. 2003;22:363–371. doi: 10.1002/humu.10282. [DOI] [PubMed] [Google Scholar]; (c) Qiu WQ, Folstein MF. Neurobiol. Aging. 2006;27:190–198. doi: 10.1016/j.neurobiolaging.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 25.(a) Leissring MA, Malito E, Hedouin S, Reinstatler L, Sahara T, Abdul-Hay SO, Choudhry S, Maharvi GM, Fauq AH, Huzarska M, May PS, Choi S, Logan TP, Turk BE, Cantley LC, Manolopoulou M, Tang W-J, Stein RL, Cuny GD, Selkoe DJ. PLoS ONE. 2010;5:e10504. doi: 10.1371/journal.pone.0010504. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Abdul-Hay SO, Lane AL, Caulfield TR, Claussin C, Bertrand J, Masson A, Choudhry S, Fauq AH, Maharvi GM, Leissring MA. J. Med. Chem. 2013;56:2246–2255. doi: 10.1021/jm301280p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flipo M, Charton J, Hocine A, Dassonneville S, Deprez B, Deprez-Poulain R. J. Med. Chem. 2009;52:6790–6802. doi: 10.1021/jm900648x. [DOI] [PubMed] [Google Scholar]
- 27.Hoffman F. 2006066847 La Roche AG. WO.
- 28.Cabrol C, Huzarska MA, Dinolfo C, Rodriguez MC, Reinstatler L, Ni J, Yeh LA, Cuny GD, Stein RL, Selkoe DJ, Leissring MA. PLoS One. 2009;4:e5274. doi: 10.1371/journal.pone.0005274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.(a) Cakir B, Dagliyan O, Dagyildiz E, Baris I, Kavakli IH, Kizilel S, Türkay M. PLoS ONE. 2012;7:e31787. doi: 10.1371/journal.pone.0031787. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kukday SS, Manandhar SP, Ludley MC, Burriss ME, Alper BJ, Schmidt WK. J. Biomol. Screen. 2012;17:1348–1361. doi: 10.1177/1087057112451921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.El Bakali J, Maingot L, Dumont J, Host H, Hocine A, Cousaert N, Dassonneville S, Leroux F, Deprez B, Deprez-Poulain R. Med Chem Comm. 2012;3:469–474. [Google Scholar]
- 31.The electron-density is compatible with a 1:1 ligand-enzyme ratio and a random occupancy on each site.
- 32.Deprez-Poulain, et al. EP_20100330093425. [Google Scholar]
- 33.Chesneau V, Rosner MR. Protein Expr. Purif. 2000;19:91–98. doi: 10.1006/prep.2000.1217. [DOI] [PubMed] [Google Scholar]
- 34.(a) Expression of IDE was checked by Q-RT PCR (Supporting Information Figure S5) Vingtdeux V, Hamdane M, Begard S, Loyens A, Delacourte A, Beauvillain JC, Buee L, Marambaud P, Sergeant N. Neurobiol. Dis. 2007;25:686–696. doi: 10.1016/j.nbd.2006.09.019.
- 35.Beaumont K, Webster R, Gardner I, Dack K. Curr. Drug Metab. 2003;4:461–485. doi: 10.2174/1389200033489253. [DOI] [PubMed] [Google Scholar]
- 36.{Eketjall 2013 #2765}.
- 37.Non toxic doses as revealed by MTT cell viability assay (data not shown).
- 38.Behl M, Zhang Y, Zheng W. Cerebrospinal Fluid Res. 2009;6:11. doi: 10.1186/1743-8454-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.5 differs from hydroxamic inhibitor Ii1 that binds to uniquely to Zn2+ making it a pan-substrate inhibitor. A direct comparison shows that the two compounds have similar Ligand Efficiencies (LE), (ie. 5: LE = 0.29 vs Ii1 : LE = 0.21). Abad-Zapatero, C., and Metz, J. T. Drug Discov. Today 2005 10, 464.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.