

Abstract
A paradigm shift has recently occurred in our knowledge and understanding of water in the lunar interior. This has transpired principally through continued analysis of returned lunar samples using modern analytical instrumentation. While these recent studies have undoubtedly measured indigenous water in lunar samples they have also highlighted our current limitations and some future challenges that need to be overcome in order to fully understand the origin, distribution and evolution of water in the lunar interior. Another exciting recent development in the field of lunar science has been the unambiguous detection of water or water ice on the surface of the Moon through instruments flown on a number of orbiting spacecraft missions. Considered together, sample-based studies and those from orbit strongly suggest that the Moon is not an anhydrous planetary body, as previously believed. New observations and measurements support the possibility of a wet lunar interior and the presence of distinct reservoirs of water on the lunar surface. Furthermore, an approach combining measurements of water abundance in lunar samples and its hydrogen isotopic composition has proved to be of vital importance to fingerprint and elucidate processes and source(s) involved in giving rise to the lunar water inventory. A number of sources are likely to have contributed to the water inventory of the Moon ranging from primordial water to meteorite-derived water ice through to the water formed during the reaction of solar wind hydrogen with the lunar soil. Perhaps two of the most striking findings from these recent studies are the revelation that at least some portions of the lunar interior are as water-rich as some Mid-Ocean Ridge Basalt source regions on Earth and that the water in the Earth and the Moon probably share a common origin.
Keywords: Moon, water, apatite, mare basalts, lunar highlands, hydrogen isotopes
1. Introduction
The origin of the Moon remains a subject of considerable debate, as evidenced by emergence of several new ideas [1,2] as well as refinements of previously proposed models [3,4]. However, regardless of which model is more applicable to the lunar origin, one common feature appears to be processing of the Moon-forming material in a magma disc at very high temperatures corresponding to several thousands of kelvin. It is hypothesized that the accretion of the Moon was followed by a Lunar Magma Ocean (LMO) phase, crystallization of which led to differentiation of the Moon, resulting in the planetary body as we know it today. The original depth, timing and the type of crystallization involved in the LMO have been a topic of considerable interest and debate in the lunar science community. However, it is a reasonable assumption that any inherited primordial water and other volatiles present originally in the Moon-forming material would have experienced significant loss, exchange and/or processing before being sequestered in the lunar interior [5]. This primordial volatile inventory of the Moon has almost certainly been modified through planetary impacts during the geological history of the Moon spanning at least a period of more than 4 billion years. Lunar samples returned by the US Apollo and Soviet Luna missions and lunar meteorites found at various locations on Earth provide an excellent opportunity to investigate the volatile contents of lunar materials, and, by extension, determine the volatile inventory of the Moon. Most lunar samples in our collections have been available for over four decades, but it is only recently that technological advancements in modern analytical instrumentation have permitted unambiguous detection of water and other volatiles, and investigation of their isotopic characteristics, in lunar samples. In this contribution, we provide a comprehensive review and current status of our knowledge and understanding of water in the Moon as a result of laboratory-based investigations of lunar samples. We evaluate different scenarios and models to reconcile measurements with observations. We also highlight complexities, current limitations and challenges in this area of research and discuss future prospects for improving our understanding of the history of water in the Earth–Moon system.
2. A brief summary of water on the Moon
The possibility for presence of water ice at the lunar poles was first proposed by Watson et al. [6] and was explored in some detail by Arnold [7]. The subject of lunar polar water ice received increased attention in the 1990s after Earth-based radar observations of the north polar region of Mercury suggested presence of water ice [8]. The similarity between the polar conditions at Mercury and the Moon refuelled the speculation that lunar poles might also act as a trap for water ice and other volatiles over the geological history of the Moon. During the same time period, data returned by instruments on board two NASA lunar orbiters (Clementine and Lunar Prospector) also indicated the existence of water ice in the permanently shadowed polar regions of the Moon [9–12]. However, subsequent Earth-based radar studies of the lunar poles contradicted the water ice theory, and became a topic of strong scientific controversy (see [13] and references therein), which appears to be settling in favour of the water ice theory largely as a result of more recent measurements made by instruments flown on board India's Chandrayaan-1 and NASA's LRO missions as well as direct impact experiments conducted on the lunar surface [14–20]. Reflectance spectroscopy of the lunar surface using instruments on three recent spacecraft missions (Cassini, Chandrayaan-1 and Deep Impact) have also detected surface-correlated OH/H2O species in lunar soils at low latitudes [21–23], through the presence of absorption features in the infrared region characteristic of OH/H2O molecules bonded to mineral structures or adsorbed at the surface of lunar soil particles. The production of these OH/H2O species at the lunar surface, at lower latitudes, is mainly thought to be a result of solar wind (SW) interaction with the lunar regolith. The source(s) for lunar surface water ice deposits are thought be extraneous in origin, most likely to be cometary although the migration of water molecules from lower latitudes to polar cold traps in the permanently shadowed craters is also a distinct possibility [7,14,23]. Recent analysis of spectroscopic data from the Moon Mineralogy Mapper (M3) instrument on board Chandrayaan-1 has been used to argue for the presence of hydroxyl, bound in minerals, in the central peak of the Bullialdus crater implicating magmatic sources [24]. Thus, some of the lunar water at the surface may have multiple origins including SW reduction of silicates and oxides in lunar soils and from magmatic sources.
3. Water in the Moon as revealed through studies of lunar samples
The six Apollo and three Luna missions returned about 382 kg of rock and soil samples from nine locations on the Moon between 1969 and 1974 [25]. Since then, our lunar sample inventory has been further augmented by regular discoveries of lunar meteorites from various locations on Earth. To date, approximately 177 individual named lunar meteorites have been collected on Earth, representing approximately 61 kg of lunar material. Some of the lunar meteorites have been grouped based on their compositional, mineralogical and isotopic similarities and probably originated from approximately 85 separate fireball entry events (for excellent reviews of lunar meteorites, see [26,27]; for an up-to-date list of lunar meteorites, visit Randy Korotev's website at http://meteorites.wustl.edu/lunar/moon_meteorites.htm). Laboratory-based analysis carried out on Apollo samples shortly after their arrival on Earth indicated a near absence of any water-bearing (or hydrous) mineral phases in Moon rocks [28]. In a few instances, some water-bearing phases were identified [29,30] in Apollo samples but they were subsequently attributed to terrestrial alteration [31]. In the vast majority of cases, the pristine nature of lunar samples, occurrence of Fe metal and the lack of Fe3+ in lunar minerals were used as strong pieces of evidence for the absence of water in the lunar interior.
(a). Initial bulk sample measurements
In the early 1970s, soon after Apollo astronauts brought back lunar samples to Earth, numerous workers measured water and hydrogen contents, and H isotopic compositions of lunar soils and regolith breccias [32–40]. Stepwise heating of these samples was used to analyse molecular H2 and H2O; H2 being usually extracted at higher temperature (more than 500°C) than H2O (less than 500°C). In terms of H2O content in regolith breccias and in bulk soils, no regional variations were detected, both displaying a range of H2O contents between approximately 100 and 450 ppm (figure 1a). However, the amount of H2 extracted is clearly lower in bulk soils (approx. 0–60 ppm) than in regolith breccias (approx. 35–100 ppm; figure 1a). D/H ratios, expressed using the conventional δD (‰) notation (δD=[(D/H)sample/(D/H)SMOW−1]×1000, where (D/H)SMOW is the D/H ratio of the Standard Mean Ocean Water), of H2 and H2O extracted from soils and regolith breccias range between approximately −1000 and −400‰ and −900 and −120‰, respectively (figure 1b). The δD of H2 decreases with increasing H2 content in soils and breccias, while the δD of H2O increases with increasing molar H2O/H2 ratio (figure 1b). The δD values of extracted H2O seem to converge towards −100‰. From these analyses of lunar soils and regolith breccias, it was concluded that H2 analysed consisted of D-free hydrogen implanted on the lunar surface due to interaction with SW, whereas H2O extracted from these samples was terrestrial water that has contaminated the samples (e.g. [35]).
Figure 1.

(a) Total H2 (i) and H2O (ii) contents extracted from some Apollo lunar soils and regolith breccias; (b) δD versus total extracted H2 (i) and molar H2O/H2 ratio (ii) from some Apollo lunar soils and regolith breccias. Dotted grey lines have been fitted visually through the data points. Data are from [32–40].
A few measurements of H2 and H2O abundances and H isotopic compositions have also been carried out on bulk samples of Apollo mare basalts [37,39]. Friedman et al. [37] analysed sample 12051, a low-Ti mare basalt, and Merlivat et al. [39] analysed two high-Ti mare basalts, samples 70215 and 75035. Sample 12051 contained 6 ppm H2 with a δD of 340‰ and sample 70215 contained 1.5 ppm H2 with a δD of 200‰. In sample 75035, Merlivat et al. [39] analysed two splits that yielded 0.5 and 2 ppm H2 with δD values of approximately 250 and −100‰, respectively. These authors interpreted the low δD split of 75035 as resulting from more implantation of SW H. In the three other bulk analyses conducted on mare basalts, hydrogen is characterized by a δD of about 200–300‰, which is higher than any known terrestrial material. These elevated δD values have been originally ascribed to cosmic-ray spallation processes [37].
(b). Minerals and agglutinates in soils/regolith breccias
Water content and H isotopic composition have been investigated in minerals and agglutinates from lunar soils and regolith breccias by in situ ion microprobe techniques in a couple of studies. Hashizume et al. [41] carried out analyses of H and N contents and their isotopic compositions in 500–1000-μm-sized olivine, pyroxene and ilmenite grains from regolith breccia 79035, and soil 71501, using a Cameca IMS 1270 ion probe at the Centre de Recherches Pétrographiques et Géochimiques (CRPG, CNRS) in Nancy (France). To study the effect of SW implantation on the lunar surface on the H and N isotope systematics, they selected two samples with very different SW exposure histories, regolith breccia 79035 having been exposed to SW for 1–2 Ga, whereas the soil 71501 has been irradiated for about 100 Ma [41]. They measured changes in H2O contents and D/H ratios with depth in one grain from each sample. In a silicate grain from regolith breccia 79035, H2O content decreases with depth from approximately 1000 ppm to approximately 350 over about 400 nm while the δD value increases from approximately −920 to approximately −800‰ (figure 2a). Analyses in an ilmenite grain from soil 71501 display completely different characteristics, as H2O content remains fairly constant around 20–60 ppm with depth, while its δD decreases from elevated values of approximately 200–400‰ down to approximately −200‰ (figure 2a). Analyses of several grains in regolith breccia 79035 yielded δD values down to −950‰, which indicated the presence of pure SW hydrogen. By contrast, D enrichment up to a δD value of approximately 450‰ (after correction for spallation production of D) highlights the contribution of a non-solar H component in soils recently exposed to SW.
Figure 2.
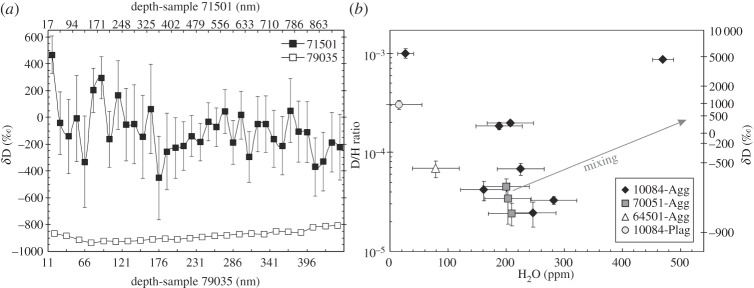
(a) Depth profiles of the variations of the D/H ratios measured in two grains extracted from soil 71501 and regolith breccia 79035 (after [41]). (b) D/H ratios versus H2O contents of agglutinitic glasses and a plagioclase extracted from three soil samples (after [42]). Arrow for mixing corresponds to the one drawn in Liu et al. [42] for mixing with cometary and meteoritic materials.
Recently, Liu et al. [42] have reported the water content and H isotopic composition of some mineral fragments, glasses and agglutinates from three different soil samples measured using a Cameca IMS 7f-GEO ion probe at Caltech, Pasadena, CA, USA. Agglutinates, which consist of regolith particles cemented by quenched melts, formed as a result of micro-meteorite impacts, contain approximately 25–500 ppm H2O (figure 2b), which represents almost all water present in these soils. Several agglutinates were analysed for their H isotopic composition. δD values range from approximately −850‰ to approximately 5000‰ (figure 2b), with the majority of the agglutinates being characterized by δD values below −500‰. According to Liu et al. [42], the H isotopic composition of water in agglutinates shows that a large proportion of this water is derived from SW-implanted protons. This ‘SW water’ can be either modified by subsequent fractionation processes or admixed with another non-SW source such as cometary and/or meteoritic materials as proposed by Liu et al. [42] to account for some analyses with elevated H2O contents and δD values measured in agglutinates in 10084 (figure 2b). However, such a mixing scenario cannot explain the low H2O and very high δD data points shown in figure 2b. It is possible that multiple processes may have modified the water contents and H isotopic composition of lunar agglutinates.
(c). Pyroclastic glasses and their melt inclusions
Unambiguous detection and quantification of water and other volatile abundances in lunar pyroclastic glasses by Saal et al. [43] heralded a new era in lunar volatiles research. They measured the volatile contents of different types of pyroclastic glass beads using two different secondary ion mass spectrometry (SIMS) instruments at the Department of Terrestrial Magmatism, Carnegie Institution of Washington (DTM-CIW), a Cameca IMS 6f and a NanoSIMS 50L. The high-Ti orange glasses contain approximately 5–13 ppm H2O, roughly correlated with abundances of other volatiles (F=9−17 ppm; Cl=0.02−−0.14 ppm; S=282−490 ppm; figure 3). The very-low-Ti green glasses are characterized by a larger range of H2O contents between approximately 0.4 and 30 ppm, which are very well correlated with abundances of other volatiles (F=2−10 ppm; Cl=0.03−−0.50 ppm; S=114−270 ppm; figure 3). Finally, only two analyses were carried out on low-Ti yellow glasses but they seem to be the richest in volatiles, having approximately 17–46 ppm H2O (F=29−40 ppm; Cl=1.3−2.0 ppm; S=518−576 ppm; figure 3). Each group of glasses display specific ranges of volatile abundances, which Saal et al. [43] linked to differences in their initial volatile contents that have not been completely erased by the degassing during melt transport and fire-fountain eruption of these pyroclastic deposits. Also, the clear correlation between H2O and other volatiles displayed by the very-low-Ti glasses (figure 3) indicates that H2O, and other volatiles, in the glasses are indigenous, and not products of laboratory contamination or SW implantation.
Figure 3.
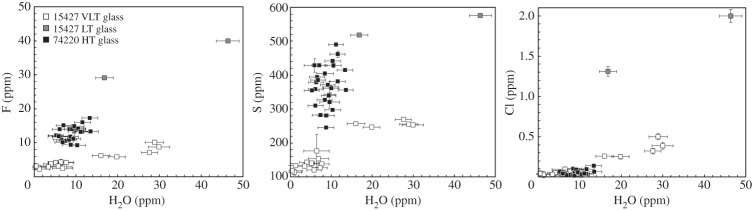
F, S and Cl abundances versus H2O contents measured in high-, low- and very-low-Ti pyroclastic glasses (after [43]).
Saal et al. [43] also measured the radial concentration profiles of H2O, F, S and Cl within a single very-low-Ti glass bead, which showed that volatile contents decrease from core to rim, from approximately 30 ppm to approximately 14 ppm for H2O. Such profiles supported the hypothesis that volatiles in these glass beads were indigenous, but were affected by degassing upon eruption. Modelling of diffusive degassing of volatiles reproduced well the measured profiles for cooling rates of 2–3 K s−1 over a period of 2–5 min between eruption and quenching, and suggested that the beads lost approximately 19% S, 45% F, 57% Cl and 98% H2O. According to Saal et al. [43], this indicates that the pre-degassing H2O content was at least 260 ppm, the best fit having been obtained for an initial H2O content of 745 ppm.
The same group followed on their initial study by reporting the volatile contents of melt inclusions trapped within olivine crystals in some of these high-Ti orange pyroclastic glass beads [44]. These new data were acquired using the DTM-CIW NanoSIMS 50L ion probe. They measured approximately 270–1200 ppm H2O in these melt inclusions, and once again H2O contents are correlated with those of F (approx. 37–72 ppm), S (approx. 450–880 ppm) and Cl (approx. 1.5–2.4 ppm), correlations that eventually point towards the volatile compositions of the highly degassed host glass beads. The melt inclusions analysed by Hauri et al. [44] are trapped in olivine crystals contained within primitive lunar volcanic glasses, and probably quenched within minutes after eruption, preventing significant post-eruptive H diffusion out of the inclusions. These measurements therefore constitute direct analyses of the volatile contents of a primary lunar magma. Volatile abundances in these lunar melt inclusions are similar to those in melt inclusions from primitive samples of some terrestrial Mid-Ocean Ridge Basalts (MORBs), suggesting that the volatile signature of the lunar mantle source of the high-Ti pyroclastic glasses is very similar to that of the upper mantle source of MORBs. Considering that these high-Ti magmas formed after 5–30% partial melting of their mantle source regions, Hauri et al. [44] estimated that these source regions contained approximately 80–400 ppm H2O, which is indeed similar to estimates for mantle sources of some terrestrial MORBs [45].
Recently, Saal et al. [46] also reported the isotopic composition of H dissolved in these lunar pyroclastic glasses and in their olivine-hosted melt inclusions in order to get insights regarding the source of the lunar magmatic water. Their measured data show that all the glass beads and melt inclusions are enriched in D compared with terrestrial ocean water (δD=0 by convention), with δD values ranging from 189‰ up to 5023‰. However, as pointed by Saal et al. [46], secondary processes that can affect D/H ratios must be accounted for before the H isotopic composition can be used as a source indicator for planetary water. Cosmic-ray spallation and magmatic degassing are two processes that may have modified water contents and D/H ratios of the pyroclastic glasses (SW implantation was negligible in these samples; [46]). The data reported by Saal et al. [46] displayed a negative correlation between δD values and H2O content (figure 4) for all the three types of glasses, pointing to a set of processes that have modified the characteristics of original magmatic water in these lunar magmas during and/or after their eruption. The total range of δD values of the pyroclastic glasses is significantly reduced (approx. −700 to approx. 2200‰, excluding one outlier with δD around 4000‰) once the data have been corrected for spallogenic production of H and D (figure 4). Excluding glasses with H2O contents below 10 ppm, as they have large uncertainties, the data still display a slightly negative trend of δD with H2O content (figure 4). For high-Ti glasses, such a relationship has been interpreted by Saal et al. [46] as reflecting kinetic degassing of H2 followed by OH-dominated diffusion at later stages in a low-pressure environment after fragmentation of the magma. In glasses with more than 10 ppm water, low- to very-low-Ti glasses display heavier D/H ratios than high-Ti glasses. This could indicate that degassing of H2 was more important in these glasses than in high-Ti glasses. As the fraction of total hydrogen present as H2 depends on several parameters (T, P, fO2, amount of H-bearing species; [47]), further interpretations are merely speculative, but considering that T, P and fO2 were similar for both high- and (very-)low-Ti glasses, a possible explanation would be that (very-)low-Ti magmas initially contained higher water contents than high-Ti magmas, as the fraction of total hydrogen present as H2 increases with the water content [47]. The elevated H2O contents of the olivine-hosted melt inclusions prevented spallation and degassing from having any significant effect on the D/H ratios, making them ideal samples to estimate the primitive H isotopic composition of the lunar water. As pointed out by Saal et al. [46], the δD value of approximately 190‰ associated with the wettest melt inclusion still represents an upper limit on the primary D/H ratios of these magmas, as there is no way to ascertain if degassing, which would have induced kinetic fractionation of hydrogen isotopes towards heavy D/H ratios, had occurred before melt inclusions were trapped. Such a δD value of 190‰ is within the range of carbonaceous chondrite δD values [48]. Altogether, the striking similarity of pre-eruptive H2O content of lunar pyroclastic glasses [44] with the H2O content of MORBs, and the similarity of D/H ratios of these pyroclastic glasses [46] with chondritic H isotopic compositions, are probably not fortuitous, and argue for a common link among chondritic, terrestrial and lunar waters.
Figure 4.

H2O contents and H isotope compositions of lunar volcanic glasses and melt inclusions. Transparent data are measured data, while those portrayed on the foreground have been corrected for spallogenic production of H and D (after [46]).
(d). Mare basalts
So far, in situ investigation of water in mare basalts has been focused on the most common volatile-bearing mineral phase present in these rocks, apatite. Apatite, with an idealized formula of Ca5(PO4)3(F,Cl,OH), can indeed contain water, structurally bound into the crystal structure as hydroxyl (OH) and is commonly associated with other volatiles such as F and Cl. Apatites form at the very late stage of magma crystallization [49] and they are the very common and main carrier of Cl and F in lunar basalts, as well as in rocks comprising the Mg and alkali suites of the lunar highlands (figure 5). McCubbin et al. [50,51] were first to estimate OH contents in lunar apatites based on stoichiometric calculations of electron microprobe data. However, since F and Cl also occupy the same crystallographic site (the X-site) in the apatite crystal structure as the OH molecule, and since the quantification of F in apatite is extremely challenging by electron microprobe, these initial reports of water in lunar apatites were not considered very robust. Besides, these calculations assumed a priori that, apart from F, Cl and OH, no other species occupied the X-site in apatite and that there was no natural vacancy at the X site. Therefore, at best, it was an indirect method of estimating OH (proxy for water) content in lunar apatites. Later SIMS analyses confirmed that apatite in mare basalts indeed contains variable and sometimes significant amounts of OH [52–57].
Figure 5.

Back-scattered electron images illustrating textural content for apatite occurrence in diverse lunar lithologies. Ap, apatite; Bd, baddeleyite; Cpx, clinopyroxene; Fa, fayalite; Fe, iron–nickel metal; Gl, glass; Ilm, ilmenite; Kfs, K-feldspar; Mer, merrillite; MI, melt inclusion; Opx, orthopyroxene; Pl, plagioclase; Sil, silica; Symp, symplectites; Tro, troilite; Trq, tranquillityite.
(i). Apollo low-Ti basalts
Direct measurement of significant amounts of OH in lunar apatites was first reported for the peculiar high-Al mare basalt sample 14053, which is known to be the most reduced rock from the Moon [58]. McCubbin et al. [56] carried out analyses of the volatile contents in apatites from mare basalt 14053 using the DTM-CIW Cameca IMS 6f ion probe. In this sample, apatite grains are typically subhedral to anhedral, ranging in size from approximately 2 μm to larger than 200 μm in their longest dimension, and are typically associated with late-stage melt pockets together with fayalite, Fe metal, troilite and silica. McCubbin et al. [56] performed four analyses on two apatite grains that yielded approximately 700–1400 ppm H2O. They also measured approximately 1700–4700 ppm Cl and approximately 2.5–2.9 wt% F in these grains. Boyce et al. [53] also analysed the volatile abundances in a single large apatite grain in sample 14053 using the Caltech (USA) Cameca IMS 7f-GEO ion probe. Their seven analyses yielded H2O contents of approximately 1600–2400 ppm. Owing to the correlation between measured 12C/18O and 16O1H/18O ratios, Boyce et al. [53] were cautious and only considered the C-free analyses and concluded that this apatite grain contained at least 1600 ppm H2O, together with approximately 310–460 ppm S and approximately 1300–3500 ppm Cl. Finally, Greenwood et al. [54] analysed the water content and H isotopic composition of several apatite grains in 14053 using a Cameca IMS 1270 ion probe at Hokkaido University (Japan), and found slightly lower H2O abundances of 570–930 ppm with δD values clustering around −200‰ (figure 6a). Greenwood et al. [54] also analysed apatites from low-Ti mare basalt 12039. These analyses yielded a sixfold variation in apatite H2O contents between approximately 1000 and 6000 ppm, with elevated δD values ranging from approximately 400 to 1000‰ (figure 6a). These pioneering apatite D/H measurements of Greenwood et al. [54] were advanced recently by four new studies [52,57,59,64]. These studies carried out analyses of the water content and D/H ratios in apatite grains using a Cameca NanoSIMS 50L ion probe at the Open University (UK). Similar to what Greenwood et al. [54] reported for apatites in sample 12039, analyses carried out by Barnes et al. [52] and Tartèse et al. [57] in low-Ti mare basalts yielded large ranges in H2O content within individual samples. In Apollo 12 samples 12064 and 12039, H2O contents of apatite ranged from approximately 700 to 2400 ppm and approximately 1500 to 3800 ppm, respectively (figure 6a). Apatites in these two samples were characterized by narrow ranges in D/H ratios and yielded similar average δD values of 896±76‰ and 873±65‰, respectively (figure 6a). In Apollo 15 samples 15058 and 15555, apatite H2O contents ranged from approximately 200 to 750 ppm and approximately 1200 to 3600 ppm, respectively, with also very similar average δD values of 581±62‰ and 597±99‰, respectively (figure 6a). On average, δD values in apatites in Apollo 12 low-Ti mare basalts are therefore approximately 300‰ heavier than those in apatites in Apollo 15 low-Ti mare basalts (figure 6a). Such large ranges in apatite H2O contents in low-Ti basalts, as directly measured by SIMS technique, are in excellent agreement with previous estimates of apatite H2O contents in Apollo 12 and 15 low-Ti basalts that were made using the ‘missing component’ method from electron probe micro-analyses covering a range from approximately 400 to 8400 ppm, with an average abundance of approximately 1600±1900 ppm [55,65].
Figure 6.
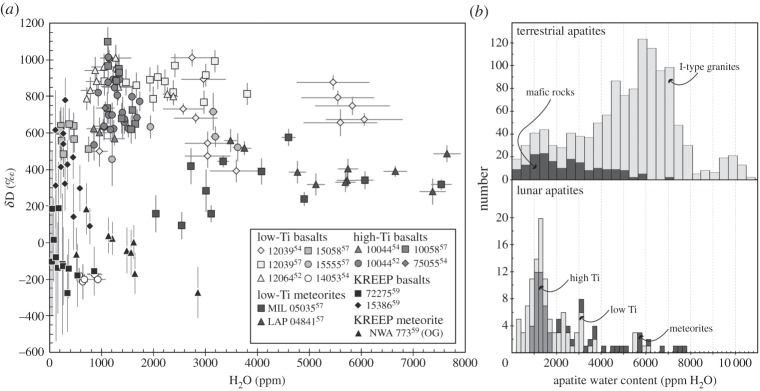
(a) D/H ratios versus H2O contents measured in apatite in low-Ti and high-Ti Apollo mare basalts and in basaltic meteorites. OG stands for olivine gabbro lithology in NWA 773. Data are from [52,54,57,59]. (b) Histograms of H2O contents in terrestrial and in lunar apatites from mare basalts. Data for terrestrial apatites are from [60–63], and for lunar apatites from [52–57].
(ii). Apollo high-Ti basalts
In situ SIMS analyses of apatite H2O content and H isotopic composition in high-Ti mare basalts have also been reported [52,54,57]. In mare basalt 10044, Greenwood et al. [54] and Barnes et al. [52] reported very consistent ranges in H2O contents of 850–1250 ppm (average=1023±169 ppm) and 856–1735 ppm (average=1271±224 ppm), respectively, associated with δD values of approximately 570–640‰ and approximately 530–1010‰, respectively (figure 6a). The large dataset (n=23) obtained by Barnes et al. [52] showed that the D/H ratios of water in apatites in basalt 10044 are highly variable, while the H2O contents display restricted variations. Tartèse et al. [57] analysed apatites in sample 10058, which are characterized by H2O contents of approximately 1100–1600 ppm (average=1350±200 ppm) and variable δD values of approximately 620–1100‰, very similar to apatites in 10044 (figure 6a). Finally, one analysis in apatite in mare basalt 75055 yielded 1070±180 ppm H2O for a δD value of 735±36‰ [54], which is in good agreement with data reported for the other high-Ti basalts 10044 and 10058 (figure 6a). McCubbin et al. [65] also calculated the apatite H2O content in some high-Ti mare basalt clasts using the ‘missing component’ method from EPMA data. In sample 10084, one analysis yielded an apatite H2O content of approximately 5000 ppm. Two Apollo 17 samples yielded contrasting results, as no ‘missing component’ was detected in apatite in 74246 while apatites in sample 79195 contained between 0 and approximately 2000 ppm H2O.
(iii). Apollo KREEP basalts
A recent study has reported the results of the first in situ analyses of water contents and H isotopic composition of apatites in Apollo KREEP basalts (samples 15386 and 72275) (figure 6a) [59]. Tartèse et al. [59] measured between approximately 50 and 850 ppm H2O in apatites from sample 72275 with an associated δD range of −277 to 187‰. In the case of apatite analysis in sample 15386, they report a range of 110–780 ppm H2O and δD values from 89 to 778‰.
(iv). Basaltic meteorites
So far, the H2O content and H isotopic composition have been reported for a very few basaltic meteorites. Tartèse et al. [57] reported OH-δD systematics for apatites from meteorites LAP 04841 and MIL 05035. In LAP 04841, 10 analyses were carried out on five separate apatite grains that yielded elevated H2O contents of approximately 3500 to 7600 ppm, with δD values ranging from approximately 280 to 560‰ (figure 6a). In MIL 05035, the H2O contents and δD values measured on four separate apatite grains ranged from approximately 2000 to 7500 ppm and approximately 100 to 570‰, respectively (figure 6a). These results are consistent with apatite H2O contents of 2000–5500 ppm and an average δD value of approximately 400±80‰, reported for another piece of MIL 05035 by Wang et al. [66]. For LAP 04841, elevated apatite H2O contents reported by Tartèse et al. [57] are consistent with estimates made using the ‘missing component’ method from EPMA analyses by McCubbin et al. [65], who reported approximately 400–7300 ppm H2O (average= ∼3900±2800 ppm) for apatites in the paired meteorite LAP 02205. It is worth pointing out that apatites in both MIL 05035 and LAP 04841 display large variations in H2O contents with relatively restricted D/H variations, sharing similar characteristics with other Apollo 12 and 15 low-Ti mare basalts (figure 6a). Recently, Tartèse et al. [59] analysed the OH-δD systematics for apatites from the KREEP-rich basaltic meteorite NWA 773 and reported a range in H2O content for apatites between 520 and 2850 ppm H2O with associated δD values in the range of −273 to 184‰.
Overall, lunar apatites in mare basaltic rocks contain between 0 and approximately 8000 ppm H2O, this range of apatite H2O abundance being identical to the range measured in terrestrial apatites (figure 6b). The distribution of H2O contents in apatites from mare rocks is also similar to the distribution in terrestrial apatites from mafic rocks, with a majority of analyses yielding between approximately 0 and 2500 ppm H2O (figure 6b).
(e). Primitive lunar crust
The lunar highlands are generally considered synonymous with the primitive lunar crust. The material comprising the primitive lunar crust includes some of the earliest products of LMO crystallization such as the ferroan anorthosites (FANs) [25,67–72] as well as other LMO-derived rock types such as the magnesian suite (norites, troctolite, dunites) and alkali suite (granites, felsites, quartz monzodiorites (QMDs)) of rocks [25,68]. As in the case of mare basalts, much of the initial work relating to measurement of water content in lunar highlands samples has been focused on analyses of apatite which is present in these rocks albeit in trace amounts [65]. Where apatite is not present, other mineral phases have been analysed to ascertain water abundances in these rocks [73].
(i). Anorthosites
Currently the only study conducted on FAN samples is that of Hui et al. [73]. These authors used a Bruker Vertex 70 Fourier transform infrared (FTIR) spectrometer at the Astromaterial and Research Exploration Science (ARES) directorate of the NASA-Johnson Space Center, Houston, TX, USA, with additional data collected at the University of Michigan, USA. They collected spectral data for plagioclase grains from two Apollo FANs (15415 and 60015). The O–H bands (broadly approx. 3700 to approx. 3100 cm−1) observed in plagioclase crystals corresponded to up to 6.4 ppm H2O in the mineral structure [73].
(ii). Norites
McCubbin et al. [65] investigated a norite clast in an impact-melt breccia. They reported a paucity of apatite in all of the magnesian- and alkali-suite samples they studied. In a few apatites that they analysed no statistically significant ‘missing component’ corresponding to OH was found, while the F content ranged between 2.70 and 2.90 wt% and the Cl content ranged between 1.30 and 1.72 wt%. The only other study which has targeted the norites is that of Barnes et al. [64], who used a Cameca NanoSIMS 50L at the Open University to measure the OH content and D/H ratio in an apatite from samples 77215 and 78235. They showed that apatite in sample 77215 contained up to approximately 1800 ppm H2O with a corresponding weighted average δD of −281±49‰ and, similarly, apatite in sample 78235 contained up to 1600 ppm H2O, with the associated δD having a weighted average of approximately −27±98‰ (figure 7).
Figure 7.

H2O contents and H isotopic compositions measured for apatite grains in a range of highlands samples. Note the log scale for the x-axis. Data sources are from [54,64,74,75]. All data except for sample 77538 have been corrected for contribution of spallogenic H and D. Cosmic ray exposure ages used to correct data are 210±30 Ma for 76535 [76–78], 292±14 Ma for 78235 [79], 310 Ma for 14161 [80] and 27.6±1.5 Ma for 14305 [81]. Additional data sources: mare basalt, [52,54,57]; CI chondrites, [48]; and Earth's mantle range, [82] and references therein].
(iii). Troctolites
Troctolite 76535 is one of the better-studied highlands samples, probably because it is deemed pristine (i.e. low siderophile abundances) and un-shocked [83]. Sample 76535 has been described in the literature as having particularly good examples of apatite and merrillite [84]. McCubbin et al. [65] and Elardo et al. [85] investigated the volatile contents in apatites from melt inclusions and intercumulus apatite in troctolite sample 76535. Both studies used the EPMA technique and did not detect any ‘missing component’ (to within 0.08 apfu uncertainty) corresponding to structural OH. Apatite analyses showed the presence of 2.3–3.3 wt% F and 1.2–1.8 wt% Cl [65,85]. In addition to analysis of FANs, Hui et al. [73] performed FTIR analyses of olivine and plagioclase grains from 76535. These authors did not detect any H2O in the two olivine crystals they studied and only detected H2O in one of three plagioclase grains, recording up to 2.7 ppm H2O. More recently, this sample has been investigated for its OH-δD systematics. As highlighted by Saal et al. [46], care needs to be taken when interpreting the δD signatures associated with such low OH contents, where spallation processes can contribute significant amounts of D resulting in elevated δD values. All the values discussed below have been corrected for spallogenic-produced D and H (see caption of figure 7 for details). Barnes et al. [64] measured 28 ppm H2O in a large intercumulus apatite grain but these authors found that almost all of the D and H could have been produced by spallogenic reactions and thus could not assign an indigenous D/H ratio to their H2O data (figure 7). Robinson et al. [74] also analysed apatite in troctolite 76535 and measured D, H and 18O in situ using a Cameca 1280 ion microprobe at the University of Hawaii, USA. They reported three analyses of up to 85 ppm H2O with corresponding δD values between approximately 340 and 640‰ (figure 7). Finally, Boyce et al. [75] used a Cameca 7f-GEO SIMS instrument at Caltech to analyse an apatite grain in troctolite 76535 and measured approximately 100 ppm H2O and δD of approximately 40‰ (figure 7). These data are comparable to the range observed thus far for mare basalts and basaltic lunar meteorites (as discussed above).
(iv). Granites and felsites
McCubbin et al. [65] investigated the volatile contents of apatite grains in a felsite and, as in the case of 76535, did not observe any statistically significant ‘missing component’ (OH). The F content varied between 2.7 and 3.09 wt% and Cl content ranged from 1.06 to 1.56 wt% [65]. McCubbin et al. [56] also analysed apatite in an alkali-suite clast in an impact-melt breccia. They reported between 115 and 530 ppm H2O, 0.9–1.2 wt% Cl and 2.5–2.6 wt% F. Apatites in two felsites and two QMDs were investigated for their H2O contents and H isotopic compositions by Robinson et al. [74]. They reported a range of 175 to 190±55 ppm H2O with an associated δD of between 335 and 410±75‰ in the felsites, and a range of 160 to 190±55 ppm H2O and associated δD of between approximately 120 and 340±90‰ in the QMDs (figure 7). Barnes et al. [64] investigated apatite in a granite clast from the alkali suite which contained up to 1200 ppm H2O with an associated average δD of −105±130‰. Finally, Greenwood et al. [54] reported that an apatite grain in an alkali anorthosite clast contained approximately 80±20 ppm H2O and weighted average δD of approximately 280±120‰ (figure 7).
(v). Impact melts
Apatite grains in KREEPy impact-melt rocks (including a lunar meteorite) were analysed by McCubbin et al. [65]. They did not detect any significant ‘missing component’ OH, and the apatites contained 1.99–3.89 wt% F and 0.77–3.62 wt% Cl. In addition to analysing apatite, Robinson et al. [74] analysed some KREEPy glass in sample 15358,6 and reported 65 and 90±10 ppm H2O with an associated δD of between 610 and 830±85‰.
4. How well lunar apatites preserve the indigenous magmatic water and D/H characteristics
(a). Terrestrial contamination
Ever since lunar samples collected during Apollo and Luna missions were brought to Earth, they have been stored and curated in specially designed ultra-clean curatorial facilities in order to minimize any terrestrial contamination. Although it may not be possible to rule out some surface contamination, it is highly unlikely that terrestrial contamination would have been introduced inside the rock samples (and furthermore into the apatite lattice) that are curated under such strict inert conditions. However, lunar meteorites have resided on the Earth's surface or within ice sheets for various lengths of time, and were inevitably subjected to terrestrial weathering. Specifically, the residence of meteorites in the Antarctic ice may have caused cryptic alteration of apatite grains. However, the efficiency of hydrogen exchange between crystalline minerals and Antarctic ice significantly decreases due to the very low temperatures prevailing in Antarctica. The D/H versus H2O systematics of apatite allows an assessment of such potential exchanges. In the region from where basaltic meteorites MIL 05035 and LAP 04841 have been recovered, the δD of Antarctic ice is around −300‰ [86]. Water mixing between high δD apatite and a low δD infinite water reservoir would thus result in negative correlations between D/H ratios and H2O abundances converging towards the D/H ratio of the ice at high water/rock ratios. Such a trend is not observed in analyses of apatites from basaltic meteorites LAP 04841 and MIL 05035 studied by Tartèse et al. [57] (figure 6a), suggesting that alteration of indigenous OH contents and D/H ratios in apatite in these meteorites remained limited, if any. Cl isotopes also provide a good tool to assess terrestrial contamination. In terrestrial materials, ratios of isotopes 37Cl to 35Cl are almost constant and δ37Cl values do not vary greatly from approximately 0±2‰ [87]. Apatites in some lunar meteorites have among the highest δ37Cl values reported for lunar samples (approx. 20‰ in NWA 2977 and approx. 75‰ in Dhofar 458; [66,75]), consistent with Cl isotopic composition reported for apatites and bulk-rock samples from Apollo collections [88]. It thus appears highly unlikely that apatites in these lunar meteorites would have preserved the heaviest δ37Cl values known in the Solar System if they were significantly compromised by weathering on the Earth.
(b). Shock-induced effects
Impacts among planetary materials have been of universal occurrence throughout the history of the Solar System. Such impacts commonly trigger shock events at extreme pressures and temperatures that could potentially volatilize hydrogen from hydrous minerals and change their original D/H characteristics. In such a scenario, the lighter isotope, H, is much more susceptible to escape than the heavier isotope. Minitti et al. [89] performed experiments on kaersutite at a pressure of approximately 32 GPa and demonstrated that shock events increased kaersutite H2O contents by 2500–9000 ppm, together with an increase in δD values by 70–90‰. Minitti et al. [89] explained these observations by a two-step shock process: (i) shock-induced devolatilization inducing preferential loss of H over D and (ii) kaersutite reaction with the ambient atmosphere leading to an increase in its H2O contents. Hydrogen isotope analyses carried out on apatites in Martian meteorites [90–94] provide useful insights into the potential effects of impacts on the D/H systematics in apatites. Hydrogen implantation from the surrounding atmosphere during shock-induced volatilization [89] might indeed in part explain the very high δD values (approx. 1000–4000‰) recorded in several minerals in Martian meteorites, as the Martian atmosphere is heavily enriched in D (δD approx. 5000–7000‰; [95]). However, not all apatites from Martian meteorites display such elevated δD signatures as in the case of Nakhla [92]. Similarly, δD values reported for melt inclusions in the shergottite group of Martian meteorites also display terrestrial-like δD signatures [96]. Therefore, it is difficult to generalize the magnitude and effect (if any) of shock on D/H systematics of apatites and melt inclusions in extra-terrestrial samples. Besides, since the Moon has virtually no atmosphere, shock-induced implantation of hydrogen into apatites from the surrounding atmosphere is unlikely to have altered their OH contents and H isotopic compositions.
(c). Hydrogen from solar wind
Material residing on the lunar surface is exposed to SW and other cosmic/galactic radiations. As hypothesized, and indeed supported by actual measurements in lunar agglutinates [42], SW is a major source of H in the lunar regolith, associated with extremely negative δD signatures. A mixing between SW H and any indigenous H with elevated δD may result in a δD signature intermediate between those of the two components. Greenwood et al. [54] used such a scenario to account for the low δD values measured in apatites in sample 14053 through interaction with SW-implanted hydrogen in the lunar regolith during an impact-heating event. Rock 14053 is characterized by a unique petrologic history, as it is the most reduced rock from the Moon, as exemplified by mesostasis fayalite and spinels being extensively reduced in the exterior portions of this rock [58]. Most of the analyses of apatites in basaltic meteorites MIL 05035 and LAP 04841 yielded δD values of approximately 300–400‰ [57] (figure 6a), marginally lower than the majority of analyses carried out in apatites in Apollo mare basalts. However, numerous sub-samples of meteorites MIL 05035 and of the different paired LAP stones have been studied in detail [97–103] but textural observations indicative of a peculiar history similar to 14053 have never been reported. It is, therefore, unlikely that δD values in apatites in MIL 05035 and LAP 04841 were altered due to interaction with SW-implanted hydrogen in the lunar regolith during a metamorphic event. Nevertheless, if SW hydrogen was indeed involved in the petrogenesis of 14053 or other suspected samples, future studies of these samples for other SW-implanted elements and gases will be useful in further supporting or rejecting this hypothesis.
(d). Post-crystallization diffusion
Mare basalts are typically characterized by liquidus and solidus temperatures of approximately 1200–1300°C and approximately 900–1000°C, respectively, and generally crystallize over a temperature interval of approximately 300°C [104]. Cooling rate estimates for mare basalts range from 0.1 to 30°C per hour [104,105], implying that they would completely crystallize from liquidus to solidus temperatures in approximately 10 hours to approximately 125 days, assuming linear cooling rates. F–Cl–OH exchange in fluorapatite was investigated at 1 atm and 1 GPa by Brenan [106], who postulated that F, Cl and OH chemical diffusion was coupled due to the requirement of charge balance neutrality. These data showed that F and Cl diffusion coefficients are indeed similar. On the other hand, the low spatial resolution of the OH measurements prevented Brenan [106] from calculating an accurate OH diffusivity. Yet, profiles for Cl and OH contents in the 1 GPa hydrothermal experiment showed a simultaneous drop for both species at similar positions in the profiles, which would qualitatively indicate similar diffusion coefficients for Cl and OH. In detail, Brenan [106] also found that the diffusion rate of Cl slightly varied with the OH content of apatite cores in the 1 atm experiments, suggesting relatively rapid migration of H through the apatite lattice accompanied by the formation/destruction of an equivalent amount of oxyapatite component. However, without more robust constraints on OH diffusivity based on precise measurements, it is not possible to quantitatively assess this issue any further. Results obtained by Brenan [106] also showed that there is a strong anisotropy in diffusion; diffusion parallel to the c-axis being about three orders of magnitude faster than diffusion along the a-axis at 1 atm. Brenan [106] also investigated the effect of pressure at 1 GPa and the data showed that anion diffusion parallel to the c-axis is about two orders of magnitude higher than at 1 atm. Using the data from Brenan [106], and considering a specific set of temperatures, initial cooling rate and diffusion parameters, it is possible to explore the conditions under which apatite cores would be affected by re-equilibration of OH, F and Cl by diffusion, according to the following relationship:
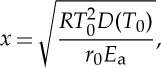 |
where x is the effective transport distance in m for a grain of spherical geometry, R is the gas constant, T0 is the initial temperature in kelvin, D(T0) is the diffusion coefficient at the initial temperature in m2 s−1, r0 is the initial cooling rate in kelvin per second and Ea is the activation energy [106]. Figure 8 shows the results of this calculation using the diffusion parameters determined parallel to the c-axis at 1 atm and 1 GPa and for different apatite radii of 10, 50 and 100 μm. If the initial temperature and the cooling rate are above the curve calculated for a specific combination of grain size and pressure, then apatite cores will retain their initial volatile contents. For cooling rates typical of mare basalts and apatite saturation temperatures around the solidus at 900–1000°C, figure 8 shows that, at 1 atm, diffusive exchange of volatiles reaching cores of apatite in mare basalts could only occur if (i) grain radius is around 10 μm and (ii) the magma experiences very slow cooling rates below 1°C h−1. Using the 1 GPa diffusivity parameters, it seems that the core of a 10 μm apatite is least likely to retain its primary volatile signature for typical mare basalt cooling rates, whereas volatile contents in cores of larger apatite grains would only be modified in the most slowly cooled mare basalts (figure 8). Overall, it thus seems that mare basalt emplaced on the lunar surface cooled down too quickly to allow significant diffusion of volatiles—OH, F and Cl in apatites—to have occurred, consistent with Brenan's [106] observation that, under conditions of rapid cooling (more than 1°C per 100 years at T>750°C), apatites would preserve zoning inherited at the time of crystallization, and that post-crystallization diffusion might not be an important process in altering halogen concentrations in volcanic apatites [107].
Figure 8.

Diagram displaying the log of the initial cooling rate (r0) as a function of the initial temperature of the magma. Each curve illustrates the maximum r0 below which cores of spherical apatite grains, with radius of 10, 50 and 100 μm, would not retain their initial volatile content. Calculations have been made using the diffusion parameters determined at 1 atm and 1 GPa by Brenan [106].
Based on detailed measurements of OH, F and Cl contents of apatite phenocrysts from the Cerro Galan ignimbrite (Argentina), Boyce & Hervig [108] have shown that diffusion of halogens in apatite is an important process to consider before estimating magmatic OH, F and Cl contents from apatite analysis. Indeed, they showed that prolonged residence of apatite at high temperature around 800°C induced diffusive mobility of volatiles in apatite. Emplacement of a new, hot, basaltic lava flow on top of an older, cold one would re-heat it, but would it be sufficiently heated, and for a sufficient period of time, to allow volatile diffusion in apatite grains? Simple one-dimensional numerical computation of cooling of a 10 m thick basaltic flow emplaced at 1200°C on top of a cold basaltic flow at 0°C (figure 9a) suggests (i) that it takes several years for a 10 m thick lava flow to completely cool down (figure 9b) and (ii) that some heat is indeed transferred from the hot basaltic flow to the cold basalt over a few metres, but that temperature in the host basalt does not exceed approximately 550°C (figure 9c), which would be insufficient to cause volatile diffusion in apatite. Results of this highly simplified one-dimensional numerical modelling are consistent with more thorough investigations such as those of Fagents et al. [109]. Once the parent magma has crystallized and reached sub-solidus temperatures (less than approx. 800°C), apatite appears to be pretty robust to volatile diffusion. This observation is consistent with a study by Stormer & Carmichael [110] about the F–OH exchange between biotite and apatite in the altered Leucite Hills lavas, in which they showed that while biotite exchanged F and OH with late-stage low-temperature aqueous fluids, apatite did not. Also, experiments carried out by Nadeau et al. [111] on several specimens of F-rich apatites showed that approximately 80–90% of the water was released from apatite grains only after prolonged heating of approximately 10–50 h at a very high temperature of 1500°C. The conditions required to devolatilize different apatite samples were correlated with grain size, suggesting that dehydration was controlled by a diffusive process, which thus appears to be very limited at low to moderate temperatures.
Figure 9.
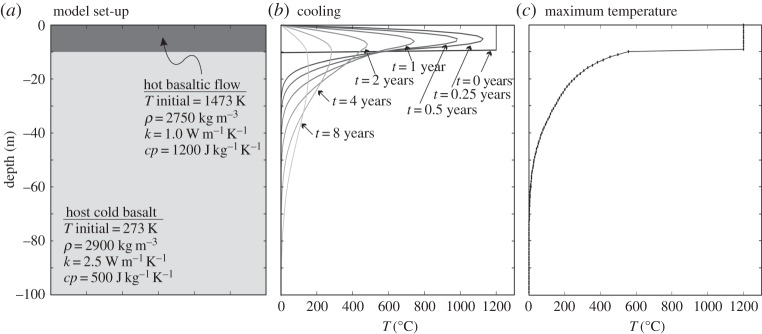
Simple one-dimensional modelling of cooling of a basaltic lava flow. (a) Model set-up, showing the initial geometry and giving the parameters used (T, temperature; ρ, volumetric mass; k, thermal conductivity; cp, heat capacity). (b) Plot of temperature versus depth showing the results of the simulation after different times t. (c) Maximum temperature reached for each point of the model.
5. Water in the lunar magma ocean and mare basalt source regions
Before delving into modelling of parental magma water contents and the origin(s) of lunar volatiles it is worthwhile to briefly revisit the LMO hypothesis for lunar differentiation following accretion. As originally proposed [69,70] the model starts with the Moon fully or partially molten (figure 10) and, through fractional crystallization, the first minerals to crystallize from LMO were higher density ultramafic minerals such as Mg-rich olivine and pyroxenes that sank towards the bottom of the LMO. The crystallization sequence progressed such that, by about 80% crystallization, less-dense feldspar-dominated mineral assemblages began to crystallize. The residual LMO liquid at this stage was very Fe–Ti-rich and, therefore, denser than the crystallizing feldspar, which could essentially float at or near the surface of the LMO, where it eventually became the main component of the primary lunar crust [69–72]. Not only was the residual LMO liquid dense, it was also highly enriched in incompatible elements such as potassium (K), rare earth elements (REEs) and phosphorus (P) (KREEP). Thus the final ‘layers’ to crystallize from the residual liquid were KREEP-rich (termed urKREEP; [112,113]) and Fe–Ti-rich. It is worth noting that the depth of the LMO is debated to be between 200 and 1000 km [71,114,115] or even whole Moon melting [116–118]. The dominant mode of LMO crystallization also remains contentious as to whether it was fractional, equilibrium or a mixture of both [70,119,120]. The estimated time for complete crystallization of the LMO varies between approximately 10 and 220 Ma [121,122]. In such a scenario having a dense Fe–Ti material on top of less-dense Mg-rich material is gravitationally unstable, such that the newly crystallized LMO cumulate pile (Mg-rich olivine and pyroxene layers towards the bottom while KREEP and Fe–Ti layers towards the top) re-organized itself via an ‘overturn event’ [72,114,122]. In a nutshell, the dense, KREEP-rich, Fe–Ti-rich material sank through the LMO cumulate pile and probably triggered the onset of Mg-rich basaltic magmatism (i.e. onset of Mg-suite and alkali-suite magmatism [123,124]). However, it is not known as to how much mixing of interior material was achieved by the overturn event.
Figure 10.
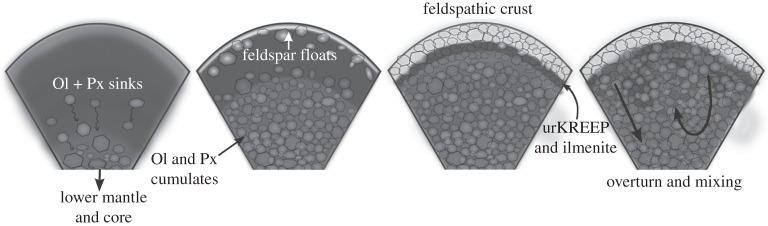
A cartoon showing a possible LMO scenario (depth of melting not specified) (after PSRD graphic, G. J. Taylor). The model proceeds through crystallization of Mg-rich minerals (Ol+Px) which sink, then of low-density Ca-rich minerals (feldspar) which float, and finally of KREEP and Fe-Ti rich (ilmenite) layers. Note that this diagram is not to scale.
(a). From the perspective of primitive lunar crust
To a first order, we now know that there are highlands samples in which nominally anhydrous minerals (NAMs) and apatites contain appreciable amounts of water [64,73]. We can use these water contents to estimate the H2O contents of the magmas from which these minerals crystallized. To do this, we need to know the timing and mode of crystallization of a particular mineral and the partitioning behaviour of volatiles into different mineral phases in a lunar context. However, these types of calculations are complicated by the lack of lunar-specific mineral–melt partition coefficients, both for NAMs [73] and for apatites (see §5c(ii)). Hui et al. [73] used the range of water contents obtained from FAN 60015 (0.8–6.4 ppm H2O) and estimates of plagioclase–melt partitioning to calculate that the LMO melt from which the primary anorthositic lunar crust crystallized was characterized by approximately 1600 ppm H2O. According to their calculations, the residual late-stage urKREEP portion of the LMO should then have been characterized by approximately 1.4 wt% H2O. Hui et al. [73] also postulated that the olivine and pyroxene cumulates which crystallized first from the LMO contained approximately 11 ppm H2O, which would suggest that the source regions for the primitive Mg-rich melts were hydrated. At the other end of the spectrum, the most hydrated highlands sample seems to be norite 78235 [64]. Using the measured H2O content of apatite in this norite and a conservative apatite–melt partition coefficient for water of 0.3 [125], and considering that apatite crystallized in equilibrium with the melt, this equates to approximately 700–5500 ppm H2O in the melt at the time of apatite crystallization. Assuming that the range of H2O contents measured in the apatite from norite 78235 can be explained by protracted apatite crystallization over the range between 96 and 99.5% crystallization (e.g. the ‘driest’ apatite in 78235 crystallized after 96% crystallization while the ‘wettest’ crystallized after 99.5%), we estimate a parental magma H2O content of approximately 30 ppm (using partition coefficients of 0.004 and 0.007 for plagioclase and pyroxene, respectively, as given in Hui et al. [73]). Considering that these melts formed through 3–20% partial melting, their mantle source regions contained approximately 1–6 ppm H2O. Despite many assumptions associated with these calculations, these estimates for the norite source regions are in the range of H2O content calculated by Hui et al. [73] for the mantle source cumulates that co-crystallized together with FANs, which are proposed to contain approximately 11 ppm H2O. This observation strongly suggests that the magmas involved in primary crust production on the Moon were indeed hydrated, and, by extension, the hydrated nature of LMO.
(b). From the perspective of mantle-derived volcanic products
In their study of the volatile contents of pyroclastic glasses, Saal et al. [43] carried out a traverse within a very-low-Ti glass bead and measured concentration profiles of H2O, F, S and Cl. These data showed that volatile contents decrease from core to rim, for example from approximately 30 ppm to approximately 14 ppm for H2O, indicating that indigenous volatiles were affected by degassing upon eruption. Based on modelling of diffusive degassing of the volatiles, Saal et al. [43] estimated the pre-degassing H2O content to be at least 260 ppm, the best fit being obtained for an initial H2O content of 745 ppm. The follow-on study of Hauri et al. [44] on melt inclusions trapped in olivine within these lunar volcanic glasses confirmed that magmas that formed pyroclastic glasses were indeed volatile-rich, melt inclusions containing approximately 270–1200 ppm H2O [44]. As the glasses quenched very soon after eruption, post-eruptive H diffusion out of the melt inclusions was probably minimal, and measured H2O contents therefore constituted a direct measurement of volatile contents of a primary magma. Hauri et al. [44] estimated that the mantle sources of these magmas might have contained approximately 80–400 ppm H2O, after taking into consideration the range of partial melting (5–30%) of their source regions to produce such magmas. These estimated H2O contents are consistent with estimates made for mantle sources of some terrestrial MORBs [45].
Estimates for magmas and their mantle source regions have also been made from apatite water contents, though these calculations are still hampered by the lack of adequate partition coefficients (see §5c(ii)) and an assumption of equilibrium crystallization for apatite, which may not always be the case. Boyce et al. [53] calculated that the melt from which apatite crystallized in 14053 contained approximately 4000 ppm H2O. These authors assumed that apatite crystallized after 95% crystallization of NAMs, implying that the primitive melt contained approximately 100–200 ppm H2O. Although Boyce et al. [53] did not calculate the water content of the mantle source for sample 14053, assuming 5–15% partial melting, this would correspond to approximately 6–30 ppm H2O. From apatite analysis in lunar meteorite NWA 2977, McCubbin et al. [56] calculated that melt from which apatite crystallized contained 7000–17 000 ppm H2O. Considering that apatite entered the phase assemblage in NWA 2977 after 95% crystallization of NAMs, this would imply that primitive melt contained 360–850 ppm H2O. Such values are very similar to those measured by Hauri et al. [44] in melt inclusions in pyroclastic glasses. McCubbin et al. [56] made a very conservative estimate that apatite crystallized after 99% crystallization, in which case the primitive magma contained 70–170 ppm H2O. To estimate the H2O content of the mantle source region of NWA 2977, McCubbin et al. [56] again used the most conservative estimate that NWA 2977 primitive melt formed by 3% partial melting, which implies that the mantle source region contained 2–5 ppm H2O. Based on H2O content measured in apatite grains in these two studies [53,56], one can therefore conclude that mare basalt mantle source region H2O contents were probably in the range of 1–30 ppm H2O, which is notably lower than estimates made by Hauri et al. [44] from melt inclusion H2O contents. However, these two studies [53,56] did not consider any loss of H2O through magma degassing, which could account for this apparent discrepancy. Tartèse et al. [57] also calculated mare basalt mantle source region H2O contents based on apatite H2O contents. Using a partition coefficient (D) of 0.3 for H2O between apatite and melt and assuming equilibrium crystallization of apatite, they calculated that H2O contents in the basaltic melts at the time of apatite crystallization ranged between 2860 and 5770 ppm for Apollo high-Ti basalts, from 690 to 12 070 ppm for Apollo low-Ti mare basalts, and from 6830 to 25 400 ppm for lunar basaltic meteorites. In addition to performing similar calculations to those of Boyce et al. [53] and McCubbin et al. [56], Tartèse et al. [57] also considered degassing of mare magmas while estimating the H2O contents of parental melts and subsequently that of their mantle source regions. For high-Ti mare basalts, Tartèse et al. [57] argued that apatite crystallization and degassing were concomitant, preventing any reliable back-calculation. In the case of low-Ti mare basalts (Apollo and meteorites), the observation of relatively restricted variations in D/H ratios for a large spread in apatite H2O contents has been interpreted as reflecting protracted apatite crystallization after magmatic degassing. The δD values measured in apatites in low-Ti mare basalts require that approximately 85–99% of H had been lost through degassing, depending on the sample [57]. Considering such a degree of degassing and that apatite started to crystallize after approximately 98% crystallization of NAMs, Tartèse et al. [57] calculated that the primitive low-Ti basaltic melts contained between approximately 670 and 5580 ppm H2O. As these low-Ti basalts are thought to be produced by 5–10% partial melting of their mantle source regions [126,127], this would imply that the source regions of Apollo 15 mare basalts contained approximately 9–360 ppm H2O, and the source region of Apollo 12 mare basalts contained approximately 270–585 ppm H2O, and the source regions of MIL 05035 and LAP 04841 contained approximately 45–180 ppm H2O. Overall, these ranges of mare basalt mantle source region H2O contents, calculated after taking into account magmatic degassing, are very consistent with the estimates made by Hauri et al. [44] of approximately 80–400 ppm H2O for the mantle source regions of pyroclastic glasses. Such H2O contents are also in good agreement with the range of approximately 60–350 ppm H2O estimated for the terrestrial mantle [45].
(c). Complexities associated with estimating the water content of the lunar interior
(i). Role of magmatic processes
Firstly, we need to consider the potential for fractionation of D and/or H during the partial melting of lunar magma source regions. Based on the work carried out by Bindeman et al. [128], Tartèse & Anand [129] proposed that small degrees of partial melting (1–15%) of a hypothetical source region for mare basalts characterized by approximately 25 ppm H and an initial δD value of approximately 100‰ did not significantly fractionate D over H. This model scenario should be equally suitable for the mantle source regions of the Mg-rich lunar magmas (i.e. pertaining to Mg-suite rocks from the lunar highlands). Secondly, during crystallization, cooling and transport of magma two processes can affect the concentration of total H-component (including H, OH and H2O) in the melt. The first is the progressive crystallization of NAMs such as pyroxene and plagioclase. As H behaves incompatibly in silicate melts [130], crystallization of NAMs will lead to enrichment of the total H-component in the remaining melt, and this process has restricted effect on fractionation of H and D isotopes; of the order of a few tens of permil [128]. The second magmatic process at play is degassing of H-bearing species from a magma, which would lead to a decrease in the total H-component. Whereas degassing of OH/H2O results in very limited D/H fractionations of the order of a few tens of permil [131,132], the degassing of H-bearing species such as H2, CH4 or HCl, for example, strongly fractionates H from D [57,133], leaving a D-enriched residual total H-component in the melt. Two important considerations arise when attempting to constrain the fractionation factor α, involved in degassing models: (i) temperature-dependent H isotope fractionation among gaseous molecules H2Ot and H2 and (ii) H isotopic fractionation due to pure kinetic degassing in a vacuum (i.e. Rayleigh fractionation). For the former, Richet et al. [133] have calculated α as a function of temperature (α=0.857–0.891 over the temperature range 900–1000°C). For kinetic degassing in a vacuum, α is given by the square root of the ratio of the light and heavy isotopologues H2 and HD (α=0.866) and is independent of the temperature. Nevertheless, in both cases, the values of α are comparable and therefore either scenario is suitable for present purposes. Ultimately, the OH contents measured in apatites in mare basalts are probably a net result of the complex interplay between crystallization and degassing processes. In the lunar case, degassing of 95–99% of H species (as H2) in mare magmas under lunar fO2 (approx. IW-1) raises the δD value of the remaining H in the melt by approximately 700–1000‰ [46,57,129,134]. Based on the large variations of D/H ratios at relatively constant H2O contents recorded in apatites in high-Ti basalts, Tartèse et al. [57] also proposed that degassing of H2 and apatite crystallization occurred simultaneously at relatively lower temperatures compared with apatite crystallization in low-Ti mare basalts, which probably occurred after degassing of H2 at higher temperatures.
(ii). Inadequate apatite–melt partition coefficients
In order to estimate the water content of lunar parental melts from which the apatites crystallized, appropriate apatite–melt partition coefficients are required corresponding to specific lunar magmatic conditions (e.g. fO2, melt composition). Unfortunately, such information is currently unavailable and, even for non-lunar cases, the partitioning data are somewhat limited. Therefore, any estimates of water content of the parental melts, and, by extension, of the mantle source regions, are subject to large uncertainties and at best should be taken as indicative figures only. The inadequacy in our present understanding of apatite–melt partition coefficients for OH, F and Cl is partly reflected in the large ranges of estimated water contents of lunar mantle source regions of mare basalts [56,57]. New experiments are being performed to better constrain partitioning behaviour of OH, F and Cl between apatite and melt corresponding to lunar magmatic conditions [135,136]. Recent experimental work has also highlighted the importance of treating partitioning data for volatiles between apatite and silicate melt in terms of exchange equilibria, considering such volatiles are essential structural constituents in apatite [136]. In this scenario, in order to calculate the water content of the silicate melt in equilibrium with an apatite (of known F, Cl and OH content), it is essential to know either the F or Cl concentration in the co-existing melt. Another additional complexity in ascertaining the volatile contents of parent melts using apatite measurements is related to their mode of crystallization. If the apatite grew from a parental melt through fractional crystallization, it may not be possible to use the measured volatile abundances in apatites to estimate the volatile contents of their parental melt [137]. Thus, availability of other petrological indicators for equilibrium or fractional crystallization of apatite will be extremely useful in this context.
As apatite primarily crystallizes towards the very late stages of melt evolution (more than 95% crystallization in most lunar basaltic melts) and is commonly associated with petrographically distinct portions of lunar basalts known as mesostasis perhaps it will be more informative to use the re-calculated bulk compositions of these mesostasis areas as starting compositions in apatite-melt partitioning studies. As previous investigations in terrestrial systems have indicated strong influence of silica activity [138,–140] in the melt on the partitioning behaviour of OH between apatite and melt, it will be instructive to compare the results of these types of experimental studies with those carried out using the bulk composition of whole rocks. In addition, experiments should be conducted on a range of starting melt compositions with varying OH, F and Cl contents to crystallize apatites with a range in OH, F and Cl contents corresponding to compositional ranges observed in apatites from natural samples.
(iii). Representativeness of the samples
On the basis of global coverage of the lunar surface achieved by a number of recent orbital missions and the lunar meteorites, it has been argued that the Apollo and Luna missions sampled a geochemically anomalous region of the Moon, not necessarily representative of the average lunar surface (see [26] for an excellent discussion on this topic). Therefore, it is important to bear this in mind while drawing inferences about certain bulk geochemical characteristics of the Moon based on data acquired on Apollo or Luna samples. Lunar meteorites have undoubtedly provided invaluable additional and wider sampling of the lunar surface compared with samples from Apollo and Luna collections. However, without knowing their exact provenance, it is not possible to fully integrate and interpret lunar meteorite data in our evolving hypothesis for the formation and evolution of the Moon. Besides restricted geographical sampling of the lunar surface, the vertical sampling of the lunar crust and mantle underneath remains even more incomplete. Except for a few drill cores collected during the Apollo missions, impact cratering has been the main process to provide some insights into vertical lithological variations by excavating rocks from crustal/sub-crustal levels and exposing them at the lunar surface. It has been argued that, in rare cases such as during the formation of the South Pole–Aitkin (SPA) basin, the impactor may have excavated the lunar mantle, which is now postulated to be exposed in rings of the SPA basin [141]. Notwithstanding this exciting possibility, all lunar samples in our collections (Apollo, Luna and meteorites) are derived from surface or near-surface lithologies representing the lunar crust. As yet, no genuine sample of lunar mantle has been reported or found. Therefore, our current understanding of the geochemical make-up of the lunar mantle is solely based on analysis of various rock types occurring at the surface of the Moon. With increasing resolution of lunar remote sensing dataset, due consideration should be given to locations that could potentially be rich in diverse geologic rock types, especially in lithologies suspected to be mantle material.
(d). View from other volatiles (e.g. F, Cl, S)
Not all recent laboratory investigations of lunar samples are in favour of water-rich lunar interior. For example, Sharp et al. [88] measured chlorine isotopic composition of a range of Apollo samples and reached the conclusion that the lunar interior had to be anhydrous. The δ37Cl values (δ37Cl=[(37Cl/35Cl)sample/(37Cl/35Cl)SMOC−1]×1000, where (37Cl/35Cl)SMOC is the 37Cl/35Cl ratio of the standard mean ocean chloride) they measured in a suite of lunar samples ranged from −1 to 24‰. In comparison, Cl isotopic composition of terrestrial and a vast majority of non-lunar extra-terrestrial materials is clustered around 0±2‰ [88]. Wang et al. [66] and Boyce et al. [75] have since confirmed the extreme enrichment in 37Cl of some lunar materials. Sharp et al. [88] interpreted the extreme enrichment in 37Cl as a result of different behaviour of the two Cl isotopes during near-surface magma degassing, the lighter isotope being preferentially partitioned into the vapour phase, the heavier isotope, bonded with metals as metal chloride, being either left behind in the melt or re-deposited from vapour phase on volcanic glasses, leading to large fractionation of Cl isotopes. Presence of any water in the system would have significantly influenced the behaviour of chlorine isotopes such that there would not have been any significant isotopic fractionation as seen in terrestrial systems. Sharp et al. [134] expanded their petrogenetic model and argued that the requirement for an anhydrous melt during Cl degassing did not preclude the possibility that significant H-bearing species existed in the melt before Cl loss, and was lost more quickly than Cl as also observed in experiments performed by Ustunisik et al. [142].
To investigate variations in abundances of other volatiles such as Cl and F among different lunar lithologies, McCubbin et al. [65] carried out a comprehensive study based on EPMA analysis of the volatile composition of apatites from mare basalts, the magnesian suite, the alkali suite, and KREEP-rich impact-melt rocks. Consistent with previous SIMS analyses of apatite water contents, McCubbin et al. [65] found that many of the apatites analysed from mare basalts had a significant ‘missing component’ that they attributed to OH, while apatites from the magnesian and alkali suites and from KREEP-rich impact melts had, on average, no detectable ‘missing component’. Most lunar apatite grains are F-rich. Apatite volatile chemistry indicates that, in the case of mare basalt, the late-stage residual melts from which apatites crystallized were enriched in F and H2O relative to Cl, which is consistent with the relative volatile abundances determined for the pyroclastic glasses [43]. As argued by McCubbin et al. [65], this common feature of Cl depletion relative to F and H2O is an inherent feature of the source regions of the mare volcanic products. By contrast, at the time of apatite crystallization, the magnesian suite, alkali suite and KREEP-rich impact melts were enriched in Cl relative to F and H2O. As these lithologies display some strong KREEP signatures, the volatile chemistry of apatites probably reflects the relative volatile abundances of urKREEP. If so, it appears that the Cl/F ratio in the mantle source regions of non-mare lithologies is drastically different from that of the mantle source regions of the mare volcanic products. Taken at face value, these results indicate that the distribution of magmatic volatiles within the Moon is heterogeneous, as the difference in relative magmatic volatile abundances among various lithologies cannot be reconciled by differential degassing of magmatic volatiles from a uniform initial source.
(e). The origin and source(s) of water in the Moon
Up to seven sources have been suggested for lunar volatiles including water: the Sun (SW reduction of lunar regolith), the Earth, the Moon (internal degassing), comets, asteroids, interplanetary dust, and giant interstellar molecular clouds [143]. Among these, the dominant sources are likely to be primordial water acquired during lunar accretion, water ice delivered to the Moon by cometary and asteroidal impacts, and water being produced by the interaction of SW hydrogen and oxygen-bearing mineral species in the lunar soil. It is unlikely that any one source could account for all of the lunar water and most probably a combination of these sources have contributed to the lunar water inventory during the different stages of lunar evolution. The polar ice deposits on the Moon are thought to be primarily cometary in origin although recent reports have indicated the possibility of migration of SW-derived water from lower latitudes to polar cold traps during the diurnal cycle of the sunlit side of the Moon [22,23]. However, no direct geochemical measurements of polar water ice deposits have yet been made which could help distinguish these competing hypotheses.
The first in situ D/H measurements carried out in lunar apatites revealed that most of the samples were characterized by elevated D/H ratios compared with terrestrial rocks, and that the highest D/H ratios were found in mare basalts [54]. D/H ratios in Oort cloud comets seemed to be a good match for those measured in lunar apatites by Greenwood et al. [54], with δD values around approximately 500–1500‰ [144]. As a result, Greenwood et al. [54] argued for the existence of a D-rich reservoir, originating from the delivery of cometary water, in the lunar interior. Based on the data published by Greenwood et al. [54], Tartèse & Anand [129] proposed an alternative mechanism which could explain the elevated D/H ratios measured in apatites in some mare basalts. In magmas evolving at the very low lunar fO2 (approx. IW-1), the total H-component dissolved in the basaltic melts is a mixture of molecular H2 and of the hydrous species H2O and OH (referred as H2Ot; [47,67,145]), probably largely dominated by the OH species [146]. At low pressures prevailing during mare basalt eruptions, H2Ot probably dominated the budget of dissolved H-bearing species [47,146]. On the other hand, the reduced species molecular H2 will dominate the H2O component in the vapour phase degassed from the magmas upon eruption and cooling (e.g. [147] and references therein). As a result, degassing of mare basalts had redox implications as it converted some dissolved H2Ot to H2 in the vapour phase. As argued by Sharp et al. [134], the presence of iron metal in mare basalts probably buffered the reduction of H2Ot to H2 and O2−, preventing the fO2 in the melt to rise and allowing continuous loss of H2. As described above in §5c(i), degassing of a H2-dominated vapour induces large fractionation of H isotopes. Degassing of 95–99% of H species initially present in mare basalts as H2 would have raised the δD value of the remaining H in the melt by approximately 700–1000‰ [57,129,134]. Therefore, elevated D/H ratios measured in apatites from mare basalts can be reconciled with magmas having an initial H isotope composition consistent with δD values of approximately 0±200‰, characteristics of terrestrial and carbonaceous chondrite-type materials [48,146]. The H isotopic composition of some melt inclusions trapped in olivine in high-Ti pyroclastic glasses measured by Saal et al. [46] is consistent with ‘Earth-like’ initial H isotopic composition for lunar magmas. As pointed out by these authors, the lowest δD value of approximately 190‰ they measured in the wettest melt inclusion is an upper limit for the primary D/H ratios of these magmas, as it is not possible to assess whether some degassing had already occurred when melt inclusions were trapped. From these data on melt inclusions, Saal et al. [46] reached the same conclusion that lunar indigenous water has an isotopic composition indistinguishable from that of carbonaceous chondrite materials. Finally, a small dataset is now available for samples from the lunar highlands, and especially interesting are the water content and H isotope composition of apatite in norites 77215 and 78235 and granite clast 14303 [64]. In these samples, apatite contains H2O contents between approximately 200 and 1800 ppm, with δD ranging from −384 to 203‰. All of these samples are thought to have crystallized deep in the lunar crust under plutonic conditions such that it is unlikely that vapour degassing occurred. In the case of the norite samples, the measured D/H ratio probably reflects directly the H isotopic composition of the norite mantle source regions, and, by extension, that of the primitive LMO [64].
Inferences and interpretations based on the wealth of new in situ data seem to converge towards a consensus view of water in the lunar interior having a similar H isotope composition to that of the Earth. It is interesting to revisit and reassess data from some pioneering studies involving bulk measurements for water in lunar samples that were made in the 1970s. At that time, the interpretation of stepwise heating analysis of volatiles trapped in lunar soils and regolith breccias was that H2 consisted of D-free hydrogen implanted on the lunar surface by the SW, whereas H2O extracted from these samples was terrestrial water that has contaminated the samples [35]. Yet, if indigenous lunar water is characterized by Earth-like H isotope composition, then it is impossible to distinguish between indigenous lunar water and terrestrial water. Also, the few bulk analyses of H2 and H2O abundance and isotopic compositions carried out on mare basalts yielded δD values ranging from approximately −100 and approximately 300‰ [37,39]. These authors interpreted the lower end of this δD range as largely resulting from implantation of SW H, while the higher end of the range was related to cosmic-ray spallation processes [37,39]. But could this relatively heavy hydrogen extracted from bulk mare basalts be genuine indigenous lunar hydrogen? Indeed, modal mixing of 90% of early-crystallized NAMs with δD of 100‰ with 10% of late-crystallized minerals such as apatite having δD of 900‰ would result in a bulk δD of 180‰. To test this scenario, it is indispensable to move towards a combined analysis of NAMs and apatite, together with melt inclusions they host.
6. Challenges and future prospects
(a). Analytical challenges
We have already witnessed that recent technological advances have enabled re-analysis of samples to a higher precision combined with low detection limits than was possible during the Apollo era. However, such high-precision measurements are extremely challenging and are constrained by analytical conditions. Recent studies [73,148] have highlighted the application of in situ analytical techniques for measurements of water content in NAMs. However, a major challenge in making accurate and precise measurements of H2O contents at such low abundances with an instrument such as SIMS is the quantification of background hydrogen. Methods used to reduce extraneous contribution of H during measurements include baking-out of the instrument, mounting samples and analytical standards in a hydrogen-free medium, and storing samples in a vacuum oven/desiccator prior to analysis [149,150]. As a result, indium metal is now routinely used as a mounting medium for the standards used to calibrate against unknowns [149,150]. However, typical polished sections of lunar materials that are commonly available for SIMS analysis are mounted using epoxy resins, and, therefore, for low-detection work, such as those on NAMs, individual chips have to be requested, and then polished and mounted in indium for SIMS measurements, which requires certain specific skills and expertise.
Another obstacle inherent to any precise SIMS measurements is the characterization of appropriate standards, which can be particularly tricky in the case of NAMs, for example, as one has to know not only the H2O content of the minerals but also their chemical homogeneity [149,151,152]. Unfortunately, there are currently only a handful of laboratories in the world that have such standards, not easily accessible to the wider lunar science community. However, advancements in this field may be aided by combining several techniques which have recently seen improvements in detection limits and sensitivities such as electron recoil detection analysis (ERDA) coupled with Rutherford back-scatter spectroscopy (RBS), which can measure simultaneously H content and major element abundances [153,154], with traditional FTIR and SIMS techniques.
(b). Wider sampling of the Moon and availability of other analogue materials
Probably one of the most challenging aspects to studies of extra-terrestrial materials is their availability, and this is also true for Apollo and Luna samples and lunar meteorites. As these samples are very precious and of limited quantity, they are very carefully curated and managed under strict protocols, and generally allocated sample sizes are very small. This poses limitations in terms of representativeness of a given sample. However, despite the limited amount of material available for study, lunar sample science is continually advancing by using and often reusing the samples that have been available since the 1970s.
As discussed in §5c(iii), wider and additional sampling of the Moon both laterally as well as vertically is highly desirable in order to better understand the geologic evolution of the Moon as well as to better inform the models proposed for the origin and evolution of the Moon. Such sampling should ideally be carried out either by an automated or, preferably, by human-assisted/human-led missions to areas of the Moon that appear to be rich in geologic diversity and potentially could provide key samples (e.g. genuine samples of lunar mantle) for geochemical investigations through which a number of existing models and hypotheses could be verified.
An understanding of the volatile inventory of the Moon can also be improved by investigating other Solar System materials that are considered analogues to Moon rocks such as the Howardite–Eucrite–Diogenite (HED) suite of meteorites [155]. HEDs are currently thought to have originated from asteroid 4Vesta under similar oxygen fugacity conditions as that of the lunar interior and thus could provide critical insights into the processes such as degassing affecting the H isotopic composition of apatites—an aspect not afforded by terrestrial apatites on account of higher oxygen fugacity in the Earth's mantle affecting the speciation of H in the basaltic melt. Similarly, there are other differentiated meteorites sharing similar petrogenetic histories to that of lunar samples that could be investigated for their abundances and isotopic composition of water and other associated volatile elements.
(c). Multi-proxy approach
Tremendous progress has been made recently both technologically as well as scientifically in measuring and understanding the inventory of water in lunar samples through laboratory investigations. While providing key insights into the sources and origin of water in the lunar interior, these new studies have also highlighted a number of issues that need to be resolved and all observations reconciled in order to develop as complete an understanding of the volatile history of the Moon as is permissible by the latest dataset. For example, there is an apparent disagreement regarding the volatile inventory of the lunar interior as indicated by Cl isotope measurements compared with other studies involving measurements of OH abundance and H isotopic measurements of lunar samples. However, no significant piece of research has been published which involved a multi-proxy approach carrying out both Cl and H isotope measurements (and other volatile element abundances) on the same samples/phases in the same laboratory using the same set of standards. Such an approach would eliminate any inter-laboratory biases and minimize uncertainties associated with analytical protocols, thereby allowing a more robust scientific interpretation of the resulting dataset in the context of lunar origin and evolution. In the case of apatites that are also amenable to U/Pb or Pb–Pb age dating, integration of the radiogenic and stable isotope dataset provides a powerful tool to investigate any secular evolution in the volatile inventory of the lunar interior.
7. Summary
Lunar science is presently going through a renaissance period as evidenced by a surge in lunar exploration by several space agencies as well as renewed interest in laboratory-based analysis of lunar samples for their volatile inventories using the latest advancements in analytical instrumentation and techniques. These efforts have already resulted in several new discoveries about the Moon, one of which has been an unambiguous detection and quantification of water in lunar samples. Furthermore, the H isotopic composition of this measured water in lunar samples points to a common origin for the water in the Earth–Moon system. Nevertheless, our understanding of complex magmatic processes influencing the volatile inventory of lunar samples is in its infancy. Samples of the primitive lunar crust seem to have preserved a record of water in the LMO, necessitating incorporation of water in models proposed for the lunar origin and solidification of the LMO. Ongoing experiments for determining partitioning behaviour of volatile elements corresponding to lunar conditions will further improve our ability to provide more robust estimates of the volatile inventory of the lunar interior through measurements made on returned lunar samples. Ultimately, future analytical work using a multi-proxy approach and targeting a range of lunar samples, minerals and other analogue materials will lead to new discoveries and better understanding of the abundance, distribution and sources of water in the Moon.
Acknowledgements
We thank David Stevenson and Alex Halliday for this invited contribution. We gratefully acknowledge critical insights provided by Francis McCubbin, Alberto Saal, Bernard Marty, Jeremy Boyce and Ian Franchi during our various discussions and deliberations on this topic. We also thank Erik Hauri and an anonymous reviewer for their constructive comments. Philippe Yamato is thanked for providing the numerical code used to perform one-dimensional simulation of lava flow cooling.
Funding statement
We also thank STFC for a PhD studentship to J.J.B. and PDRA funding to R.T. (via grant no. ST/I001298/1 to M.A.).
References
- 1.De Meijer RJ, Anisichkin VF, van Westrenen W. 2013. Forming the Moon from terrestrial silicate-rich material. Chem. Geol. 345, 40–49. ( 10.1016/j.chemgeo.2012.12.015) [DOI] [Google Scholar]
- 2.Reufer A, Meier MMM, Benz W, Wieler R. 2012. A hit-and-run giant impact scenario. Icarus 221, 296–299. ( 10.1016/j.icarus.2012.07.021) [DOI] [Google Scholar]
- 3.Canup RM. 2012. Forming a Moon with an Earth-like composition via a giant impact. Science 338, 1052–1055. ( 10.1126/science.1226073) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stewart ST Ćuk M. 2012. Making the Moon from a fast-spinning Earth: a giant impact followed by resonant despinning. Science 338, 1047–1052. ( 10.1126/science.1225542) [DOI] [PubMed] [Google Scholar]
- 5.Pahlevan K, Stevenson DJ. 2007. Equilibration in the aftermath of the lunar-forming giant impact. Earth Planet. Sci. Lett. 262, 438–449. ( 10.1016/j.epsl.2007.07.055) [DOI] [Google Scholar]
- 6.Watson K, Murray BC, Brown H. 1961. The behavior of volatiles on the lunar surface. J. Geophys. Res. 66, 3033–3045. ( 10.1029/JZ066i009p03033) [DOI] [Google Scholar]
- 7.Arnold JR. 1979. Ice in the lunar polar regions. J. Geophys. Res. 84, 5659–5668. ( 10.1029/JB084iB10p05659) [DOI] [Google Scholar]
- 8.Harmon JK, Slade MA. 1992. Radar mapping of Mercury: full-disk images and polar anomalies. Science 258, 640–643. ( 10.1126/science.258.5082.640) [DOI] [PubMed] [Google Scholar]
- 9.Feldman WC, Maurice S, Binder AB, Barraclough BL, Elphic RC, Lawrence DJ. 1998. Fluxes of fast and epithermal neutrons from Lunar Prospector: evidence for water ice at the lunar poles. Science 281, 1496–1500. ( 10.1126/science.281.5382.1496) [DOI] [PubMed] [Google Scholar]
- 10.Feldman WC, Lawrence DJ, Elphic RC, Barraclough BL, Maurice S, Genetay I, Binder AB. 2000. Polar hydrogen deposits on the Moon. J. Geophys. Res. 105, 4175–4195. ( 10.1029/1999JE001129) [DOI] [Google Scholar]
- 11.Nozette S, Lichtenberg CL, Spudis P, Bonner R, Ort W, Malaret E, Robinson M, Shoemaker EM. 1996. The Clementine bistatic radar experiment. Science 274, 1495–1498. ( 10.1126/science.274.5292.1495) [DOI] [PubMed] [Google Scholar]
- 12.Nozette S, Shoemaker EM, Spudis PD, Lichtenberg CL. 1997. The possibility of ice on the Moon. Science 278, 144–145. ( 10.1126/science.278.5335.144) [DOI] [Google Scholar]
- 13.Campbell DB, Campbell BA, Carter LM, Margot J-L, Stacy NJS. 2006. No evidence for thick deposits of ice at the lunar south pole. Nature 443, 835–837. ( 10.1038/nature05167) [DOI] [PubMed] [Google Scholar]
- 14.Colaprete A, et al. 2010. Detection of water in the LCROSS ejecta plume. Science 330, 463–468. ( 10.1126/science.1186986) [DOI] [PubMed] [Google Scholar]
- 15.Gladstone GR, et al. 2010. LRO-LAMP observations of the LCROSS impact plume. Science 330, 472–476. ( 10.1126/science.1186474) [DOI] [PubMed] [Google Scholar]
- 16.Hayne PO, Greenhagen BT, Foote MC, Siegler MA, Vasavada AR, Paige DA. 2010. Diviner Lunar radiometer observations of the LCROSS impact. Science 330, 477–479. ( 10.1126/science.1197135) [DOI] [PubMed] [Google Scholar]
- 17.Mitrofanov IG, et al. 2010. Hydrogen mapping of the Lunar South Pole using the LRO neutron detector experiment LEND. Science 330, 483–486. ( 10.1126/science.1185696) [DOI] [PubMed] [Google Scholar]
- 18.Paige DA, et al. 2010. Diviner Lunar radiometer observations of cold traps in the Moon's South Polar Region. Science 330, 479–482. ( 10.1126/science.1187726) [DOI] [PubMed] [Google Scholar]
- 19.Schultz PH, Hermalyn B, Colaprete A, Ennico K, Shirley M, Marshall WS. 2010. The LCROSS cratering experiment. Science 330, 468–472. ( 10.1126/science.1187454) [DOI] [PubMed] [Google Scholar]
- 20.Spudis PD, et al. 2010. Initial results for the North Pole of the Moon from Mini-SAR, Chandrayaan-1 mission. Geophys. Res. Lett. 37, L06204 ( 10.1029/2009GL042259) [DOI] [Google Scholar]
- 21.Clark RN. 2009. Detection of adsorbed water and hydroxyl on the Moon. Science 326, 562–564. ( 10.1126/science.1178105) [DOI] [PubMed] [Google Scholar]
- 22.Pieters CM, et al. 2009. Character and spatial distribution of OH/H2O on the surface of the Moon seen by M3 on Chandrayaan-1. Science 326, 568–572. ( 10.1126/science.1178658) [DOI] [PubMed] [Google Scholar]
- 23.Sunshine JM, Farnham TL, Feaga LM, Groussin O, Merlin F, Milliken RE, A'Hearn MF. 2009. Temporal and spatial variability of Lunar hydration as observed by the Deep Impact spacecraft. Science 326, 565–568. ( 10.1126/science.1179788) [DOI] [PubMed] [Google Scholar]
- 24.Klima R, Cahill J, Hagerty J, Lawrence D. 2013. Remote detection of magmatic water in Bullialdus Crater on the Moon. Nat. Geosci. 6, 737–741. ( 10.1038/ngeo1909) [DOI] [Google Scholar]
- 25.Shearer CK, et al. 2006. Thermal and magmatic evolution of the Moon. In New views of the Moon (eds Jolliff BL, Wieczorek MA, Shearer CK, Neal CR.), pp. 365–518. Reviews in Mineralogy and Geochemistry, vol. 60 Chantilly, VA: Mineralogical Society of America. [Google Scholar]
- 26.Korotev RL. 2005. Lunar geochemistry as told by lunar meteorites. Chemie der Erde 65, 297–346. ( 10.1016/j.chemer.2005.07.001) [DOI] [Google Scholar]
- 27.Joy KH, Arai T. 2013. Lunar meteorites: new insights into the geological history of the Moon. Astron. Geophys. 54, 4.28–4.32. [Google Scholar]
- 28.Papike JJ, Taylor LA, Simon S. 1991. Lunar minerals. In The lunar source book (eds Heiken GH, Vaniman DT, French BM.), pp. 121–182. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 29.El Goresy A, Ramdohr P, Pavićević M, Medenbach O, Müller O, Gentner W. 1973. Zinc, lead, chlorine and FeOOH-bearing assemblages in the Apollo 16 sample 66095: origin by impact of a comet or a carbonaceous chondrite?. Earth Planet. Sci. Lett. 18, 411–419. ( 10.1016/0012-821X(73)90097-6) [DOI] [Google Scholar]
- 30.Gibson EK, Jr, Moore GW. 1973. Volatile-rich lunar soil: evidence of possible cometary impact. Science 179, 69–71. ( 10.1126/science.179.4068.69) [DOI] [PubMed] [Google Scholar]
- 31.Taylor LA, Mao HK, Bell PM. 1974. Identification of the hydrated iron oxide mineral Akaganéite in Apollo 16 lunar rocks. Geology 2, 429–432. () [DOI] [Google Scholar]
- 32.Epstein S, Taylor HP. 1970. 18O/16O, 30Si/28Si, D/H, and 13C/12C studies of lunar rock and minerals. Science 167, 533–535. ( 10.1126/science.167.3918.533) [DOI] [PubMed] [Google Scholar]
- 33.Epstein S, Taylor HP. 1971. O18/O16, Si30/Si28, D/H, and C13/C12 ratios in lunar samples. In Proc. 2nd Lunar Sci. Conf. Houston, TX, 11–14 January 1971, pp. 1421–1441. Cambridge, MA: MIT Press. [Google Scholar]
- 34.Epstein S, Taylor HP. 1972. O18/O16, Si30/Si28, C13/C12, and D/H studies of Apollo 14 and 15 samples., pp. 1429–1454. Cambridge, MA: MIT Press. [Google Scholar]
- 35.Epstein S, Taylor HP. 1973. The isotopic composition and concentration of water, hydrogen, and carbon in some Apollo 15 and 16 soils and in the Apollo 17 orange soil. In Proc. 4th Lunar Sci. Conf. Houston, TX, 5–8 March 1973, pp. 1559–1575. Cambridge, MA: MIT Press. [Google Scholar]
- 36.Friedman I, O'Neill JR, Adami LH, Gleason JD, Hardcastle K. 1970. Water, hydrogen, deuterium, carbon, carbon-13, and oxygen-18 content of selected lunar material. Science 167, 533–535. ( 10.1126/science.167.3918.538) [DOI] [PubMed] [Google Scholar]
- 37.Friedman I, O'Neill JR, Gleason JD, Hardcastle K. 1971. The carbon and hydrogen content and isotopic composition of some Apollo 12 materials. In Proc. 2nd Lunar Sci. Conf. Houston, TX, 11–14 January 1971, pp. 1407–1415. Cambridge, MA: MIT Press. [Google Scholar]
- 38.Merlivat L, Nief G, Roth E. 1972. Deuterium content of lunar material., pp. 1473–1477. Cambridge, MA: MIT Press. [Google Scholar]
- 39.Merlivat L, Lelu M, Nief G, Roth E. 1974. Deuterium, hydrogen, and water content of lunar material. In Proc. 5th Lunar Sci. Conf. Houston, TX, 18–22 March 1974, pp. 1885–1895. Philadelphia, PA: Elsevier. [Google Scholar]
- 40.Merlivat L, Lelu M, Nief G, Roth E. 1976. Spallation deuterium in rock 70215., pp. 649–658. New York, NY: Pergamon Press. [Google Scholar]
- 41.Hashizume K, Chaussidon M, Marty B, Robert F. 2000. Solar wind record on the Moon: deciphering presolar from planetary nitrogen. Science 290, 1142–1145. ( 10.1126/science.290.5494.1142) [DOI] [PubMed] [Google Scholar]
- 42.Liu Y, Guan Y, Zhang Y, Rossman GR, Eiler JM, Taylor LA. 2012. Direct measurement of hydroxyl in the lunar regolith and the origin of lunar surface water. Nat. Geosci. 5, 779–782. ( 10.1038/ngeo1601) [DOI] [Google Scholar]
- 43.Saal AE, Hauri EH, Lo Cascio M, Van Orman JA, Rutherford MC, Cooper RF. 2008. Volatile content of lunar volcanic glasses and the presence of water in the Moon's interior. Nature 454, 192–196. ( 10.1038/nature07047) [DOI] [PubMed] [Google Scholar]
- 44.Hauri EH, Weinreich T, Saal AE, Rutherford MC, Van Orman JA. 2011. High pre-eruptive water contents preserved in lunar melt inclusions. Science 333, 213–215. ( 10.1126/science.1204626) [DOI] [PubMed] [Google Scholar]
- 45.Saal AE, Hauri EH, Langmuir CH, Perfit MR. 2002. Vapour undersaturation in primitive mid-ocean-ridge basalt and the volatile content of Earth's upper mantle. Nature 419, 451–455. ( 10.1038/nature01073) [DOI] [PubMed] [Google Scholar]
- 46.Saal AE, Hauri EH, Van Orman JA, Rutherford MJ. 2013. Hydrogen isotopes in lunar volcanic glasses and melt inclusions reveal a carbonaceous chondrite heritage. Science 340, 1317–1320. ( 10.1126/science.1235142) [DOI] [PubMed] [Google Scholar]
- 47.Hirschmann MM, Withers AC, Ardia P, Foley NT. 2012. Solubility of molecular hydrogen in silicate melts and consequences for volatile evolution of terrestrial planets. Earth Planet. Sci. Lett. 345–348, 38–48. ( 10.1016/j.epsl.2012.06.031) [DOI] [Google Scholar]
- 48.Alexander CMO'D, Bowden R, Fogel ML, Howard KT, Herd CDK, Nittler LR. 2012. The provenances of asteroids, and their contributions to the volatile inventories of the terrestrial planets. Science 337, 721–723. ( 10.1126/science.1223474) [DOI] [PubMed] [Google Scholar]
- 49.Sha LK. 2000. Whitlockite solubility in silicate melts: some insights into lunar and planetary evolution. Geochim. Cosmochim. Acta 64, 3217–3236. ( 10.1016/S0016-7037(00)00420-8) [DOI] [Google Scholar]
- 50.McCubbin FM, Nekvasil H, Lindsley DH. 2007. Is there evidence for water in lunar magmatic minerals?: A crystal chemical investigation. In Proc. 38th Lunar Planet. Sci. Conf., League City, TX, 12–16 March 2007, Abstract 1354. Houston, TX: Lunar and Planetary Institute. [Google Scholar]
- 51.McCubbin FM, Nekvasil H, Jolliff BL, Carpenter PK, Zeigler RA. A survey of lunar apatite volatile contents for determining bulk lunar water: how dry is ‘bone dry’? In Proc. 39th Lunar Planet. Sci. Conf., League City, TX, 10–14 March 2008,, Abstract 1788 Houston, TX: Lunar and Planetary Institute. [Google Scholar]
- 52.Barnes JJ, Franchi IA, Anand M, Tartèse R, Starkey NA, Koike M, Sano Y, Russell SS. 2013. Accurate and precise measurements of the D/H ratio and hydroxyl content in lunar apatites using NanoSIMS. Chem. Geol. 337–338, 48–55. ( 10.1016/j.chemgeo.2012.11.015) [DOI] [Google Scholar]
- 53.Boyce JW, Liu Y, Rossman GR, Guan Y, Eiler JM, Stolper EM, Taylor LA. 2010. Lunar apatite with terrestrial volatile abundances. Nature 466, 466–469. ( 10.1038/nature09274) [DOI] [PubMed] [Google Scholar]
- 54.Greenwood JP, Itoh S, Sakamoto N, Warren P, Taylor L, Yurimoto H. 2011. Hydrogen isotope ratios in lunar rocks indicate delivery of cometary water to the Moon. Nat. Geosci. 4, 79–82. ( 10.1038/ngeo1050) [DOI] [Google Scholar]
- 55.McCubbin FM, Steele A, Nekvasil H, Schnieders A, Rose T, Fries M, Carpenter PK, Jolliff BL. 2010. Detection of structurally bound hydroxyl in fluorapatite from Apollo mare basalt 15058,128 using TOF-SIMS. Am. Mineral. 95, 1141–1150. ( 10.2138/am.2010.3448) [DOI] [Google Scholar]
- 56.McCubbin FM, Steele A, Hauri EH, Nekvasil H, Yamashita S, Hemley R. 2010. Nominally hydrous magmatism on the Moon. Proc. Natl Acad. Sci. USA 27, 11223–11228. ( 10.1073/pnas.1006677107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tartèse R, Anand M, Barnes JJ, Starkey NA, Franchi IA, Sano Y. 2013. The abundance, distribution, and isotopic composition of hydrogen in the Moon as revealed by basaltic lunar samples: implications for the volatile inventory of the Moon. Geochim. Cosmochim. Acta 122, 58–74. ( 10.1016/j.gca.2013.08.014) [DOI] [Google Scholar]
- 58.Taylor LA, Patchen A, Mayne RG, Taylor DH. 2005. The most reduced rock from the moon, Apollo 14 basalt 14053: its unique features and their origin. Am. Mineral. 89, 1617–1624. [Google Scholar]
- 59.Tartèse R, Anand M, McCubbin FM, Elardo SM, Shearer CK, Franchi IA. 2014. Apatites in lunar KREEP basalts: the missing link to understanding the H isotope systematics of the Moon. Geology 42, 363–366. ( 10.1130/G35288.1) [DOI] [Google Scholar]
- 60.Boudreau A, Somon A. 2007. Crystallization and degassing in the Basement Sill, McMurdo Dry Valleys, Antarctica. J. Petrol. 48, 1369–1386. ( 10.1093/petrology/egm022) [DOI] [Google Scholar]
- 61.Chu MF, Wang KL, Griffin WL, Chung SL, O'Reilly SY, Pearson NJ, Iizuka Y. 2009. Apatite composition: tracing petrogenetic processes in Transhimalayan granitoids. J. Petrol. 50, 1829–1855. ( 10.1093/petrology/egp054) [DOI] [Google Scholar]
- 62.Drinkwater JL, Czamanske GK, Ford AB. 1990. Apatite of the Dufek intrusion: distribution, paragenesis and chemistry. Can. Mineral. 28, 835–854. [Google Scholar]
- 63.Zhang C, Holtz F, Ma C, Wolff PE, Li X. 2012. Tracing the evolution and distribution of F and Cl in plutonic systems from volatile-bearing minerals: a case study from the Liujiawa pluton (Dabie orogen, China). Contrib. Mineral. Petrol. 154, 869–879. [Google Scholar]
- 64.Barnes JJ, Tartèse R, Anand M, McCubbin FM, Franchi IA, Starkey NA, Russell SS. 2014. The origin of water in the primitive Moon as revealed by the lunar highlands samples. Earth Planet. Sci. Lett. 390, 244–252. ( 10.1016/j.epsl.2014.01.015) [DOI] [Google Scholar]
- 65.McCubbin FM, Jolliff BJ, Nekvasil H, Carpenter PK, Zeigler RA, Steele A, Elardo SM, Lindsley DH. 2011. Fluorine and chlorine abundances in lunar apatite: implications for heterogeneous distributions of magmatic volatiles in the lunar interior. Geochim. Cosmochim. Acta 75, 5073–5093. ( 10.1016/j.gca.2011.06.017) [DOI] [Google Scholar]
- 66.Wang Y, Guan Y, Hsu W, Eiler JM. 2012. Water content, chlorine and hydrogen isotope compositions of lunar apatite. In Proc. 75th Met. Soc. Annual Meeting, Cairns, Australia, 12–17 August 2012, Abstract 5170. Houston, TX: Lunar and Planetary Institute. [Google Scholar]
- 67.Elkins-Tanton LT, Grove TL. 2011. Water (hydrogen) in the lunar mantle: results from petrology and magma ocean modeling. Earth Planet. Sci. Lett. 307, 173–179. ( 10.1016/j.epsl.2011.04.027) [DOI] [Google Scholar]
- 68.Shearer CK, Papike JJ. 1999. Magmatic evolution of the Moon. Am. Mineral. 84, 1469–1494. [Google Scholar]
- 69.Smith JV, Anderson AT, Newton RC, Olsen EJ, Wyllie PJ. 1970. Petrologic history of the moon inferred from petrography, mineralogy and petrogenesis of Apollo 11 rocks. In Proc. Apollo 11 Lunar Science Conf., Houston, TX, 5–8 January 1970, pp. 897–925. New York, NY: Pergamon Press. [Google Scholar]
- 70.Wood JA, Dickey JS, Jr, Marvin UB, Powell BN. 1970. Lunar anorthosites and a geophysical model of the moon., pp. 965–988. New York, NY: Pergamon Press. [DOI] [PubMed] [Google Scholar]
- 71.Taylor SR, Jjakes P. 1974. The geochemical evolution of the moon. In Proc. 5th Lunar Sci. Conf. Houston, TX, 18–22 March 1974, pp. 1287–1305. Houston, TX: Lunar and Planetary Institute. [Google Scholar]
- 72.Ringwood AE, Kesson SE. 1976. A dynamic model for mare basalt petrogenesis., pp. 1697–1722. Houston, TX: Lunar and Planetary Institute. [Google Scholar]
- 73.Hui H, Peslier AH, Zhang Y, Neal CR. 2013. Water in lunar anorthosites and evidence for a wet early Moon. Nat. Geosci. 6, 177–180. ( 10.1038/ngeo1735) [DOI] [Google Scholar]
- 74.Robinson KL, Nagashima K, Taylor GJ. 2013. D/H of intrusive moon rocks: implications for lunar origin. In Proc. 44th Lunar Planet. Sci. Conf., The Woodlands, TX, 18–22 March 2013, Abstract 1327 Houston, TX: Lunar and Planetary Institute. [Google Scholar]
- 75.Boyce JW, Guan Y, Treiman AH, Greenwood JP, Eiler JM, Ma C. 2013. Volatile components in the moon: abundances and isotope ratios of Cl and H in lunar apatites. In Proc. 44th Lunar Planet. Sci. Conf., The Woodlands, TX, 18–22 March 2013, Abstract 2851. Houston, TX: Lunar and Planetary Institute. [Google Scholar]
- 76.Bogard DD, Nyquist LE, Bansal BM, Wiesmann H, Shih CY. 1975. 76535: an old lunar rock. Earth Planet. Sci. Lett. 26, 69–80. ( 10.1016/0012-821X(75)90178-8) [DOI] [Google Scholar]
- 77.Crozaz G, Drozd R, Hohenberg C, Morgan C, Ralston C, Walker R, Yuhas D. 1974. Lunar surface dynamics: some general conclusions and new results from Apollo 16 and 17. In Proc. 5th Lunar Sci. Conf. Houston, TX, 18–22 March 1974, pp. 2475–2499. Philadelphia, PA: Elsevier. [Google Scholar]
- 78.Lugmair GW, Marti K, Kurtz JP, Scheinin NB. 1976. History and genesis of lunar troctolite 76535 or: how old is old?, pp. 2009–2033. New York, NY: Pergamon Press. [Google Scholar]
- 79.Drozd RJ, Hohenberg CM, Morgan CJ, Podosek FA, Wroge ML. 1977. Cosmic-ray exposure history at Taurus-Littrow. In Proc. 8th Lunar Sci. Conf. Houston, TX, 14–18 March 1977, pp. 3027–3043. New York, NY: Pergamon Press. [Google Scholar]
- 80.Kirsten T, Duebner J, Horn P, Kaneoka I, Kiko J, Schaeffer OA, Thio SK. 1972. The rare gas record of Apollo 14 and 15 samples., pp. 1865–1889. Cambridge, MA: The MIT Press. [Google Scholar]
- 81.Eugster O, Eberhardt P, Geiss J, Grogler N, Jungck M, Meier F, Morgell M, Niederer F. 1984. Cosmic ray exposure histories of Apollo 14, Apollo 15, and Apollo 16 rocks. Proc. Lunar Planet. Sci. Conf. 14, 498 [Google Scholar]
- 82.Lécuyer C, Gillet P, Robert F. 1998. The hydrogen isotope composition of seawater and the global water cycle. Chem. Geol. 145, 249–261. ( 10.1016/S0009-2541(97)00146-0) [DOI] [Google Scholar]
- 83.Warren PH. 1993. A concise compilation of petrologic information on possibly pristine nonmare Moon rocks. Am. Mineral. 78, 360–376. [Google Scholar]
- 84.McCallum IS, Swartz JM. 2001. Lunar Mg suite: thermobarometry and petrogenesis of parental magmas. J. Geophys. Res. 106, 27969–27983. ( 10.1029/2000JE001397) [DOI] [Google Scholar]
- 85.Elardo SM, McCubbin FM, Shearer CK., Jr 2012. Chromite symplectites in Mg-suite troctolite 76535 as evidence for infiltration metasomatism of a lunar layered intrusion. Geochim. Cosmochim. Acta 87, 154–177. ( 10.1016/j.gca.2012.03.030) [DOI] [Google Scholar]
- 86.Masson-Delmotte V, et al. 2008. A review of Antarctic surface snow isotopic composition: observations, atmospheric circulation, and isotopic modeling. J. Climate 21, 3359–3387. ( 10.1175/2007JCLI2139.1) [DOI] [Google Scholar]
- 87.Sharp ZD, Mercer JA, Jones JH, Brearley AJ, Selverstone J, Bekker A, Stachel T. 2013. The chlorine isotope composition of chondrites and Earth. Geochim. Cosmochim. Acta 107, 189–204. ( 10.1016/j.gca.2013.01.003) [DOI] [Google Scholar]
- 88.Sharp ZD, Shearer CK, McKeegan KD, Barnes JD, Wang YQ. 2010. The chlorine isotope composition of the Moon and implications for an anhydrous mantle. Science 329, 1050–1053. ( 10.1126/science.1192606) [DOI] [PubMed] [Google Scholar]
- 89.Minitti ME, Leshin LA, Darby Dyar M, Ahrens TJ, Guan Y, Luo SN. 2008. Assessment of shock effects on amphibole water contents and hydrogen isotope compositions: 2. Kaersutitic amphibole experiments. Earth Planet. Sci. Lett. 266, 288–302. ( 10.1016/j.epsl.2007.11.012) [DOI] [Google Scholar]
- 90.Boctor NZ, Alexander CMO'D, Wang J, Hauri EH. 2003. The sources of water in martian meteorites: clues from hydrogen isotopes. Geochim. Cosmochim. Acta 67, 3971–3989. ( 10.1016/S0016-7037(03)00234-5) [DOI] [Google Scholar]
- 91.Greenwood JP, Itoh S, Sakamoto N, Vicenzi EP, Yurimoto H. 2008. Hydrogen isotope evidence for loss of water from Mars through time. Geophys. Res. Lett. 35, L05203 ( 10.1029/2007GL032721) [DOI] [Google Scholar]
- 92.Hallis LJ, Taylor GJ, Nagashima K, Huss GR. 2012. Magmatic water in the martian meteorite Nakhla. Earth Planet. Sci. Lett. 359–360, 84–92. ( 10.1016/j.epsl.2012.09.049) [DOI] [Google Scholar]
- 93.Leshin LA. 2000. Insights into martian water reservoirs from analyses of martian meteorite QUE 94201. Geophys. Res. Lett. 27, 2017–2020. ( 10.1029/1999GL008455) [DOI] [Google Scholar]
- 94.Watson LL, Hutcheon ID, Epstein S, Stolper EM. 1994. Water on Mars: clues from D/H and water contents of hydrous phases in SNC meteorites. Science 265, 85–90. ( 10.1126/science.265.5168.86) [DOI] [PubMed] [Google Scholar]
- 95.Webster CR, et al. 2013. Mars atmospheric escape recorded by H, C and O isotope ratios in carbon dioxide and water measured by the SAM tunable laser spectrometer on the Curiosity rover. In Proc. 44th Lunar Planet. Sci. Conf., The Woodlands, TX, 18–22 March 2013, Abstract 1365. Houston, TX: Lunar and Planetary Institute. [Google Scholar]
- 96.Usui T, Alexander CMO'D, Wang J, Simon JI, Jones JH. 2012. Origin of water and mantle-crust interactions on Mars inferred from hydrogen isotopes and volatile element abundances of olivine-hosted melt inclusions of primitive shergottites. Earth Planet. Sci. Lett. 357–358, 119–129. ( 10.1016/j.epsl.2012.09.008) [DOI] [Google Scholar]
- 97.Anand M, Taylor LA, Floss C, Neal CR, Terada K, Tanikawa S. 2006. Petrology and geochemistry of LaPaz Icefield 02205: a new unique low-Ti mare-basalt meteorite. Geochim. Cosmochim. Acta 70, 246–264. ( 10.1016/j.gca.2005.08.018) [DOI] [Google Scholar]
- 98.Day JMD, Taylor LA, Floss C, Patchen AD, Schnare DW, Pearson DG. 2006. Comparative petrology, geochemistry and petrogenesis of evolved, low-Ti lunar mare basalt meteorites from the LaPaz icefield, Antarctica. Geochim. Cosmochim. Acta 70, 1581–1600. ( 10.1016/j.gca.2005.11.015) [DOI] [Google Scholar]
- 99.Hill E, Taylor LA, Floss C, Liu Y. 2009. Lunar meteorite LaPaz Icefield 04841: petrology, texture, and impact-shock effects of a low-Ti mare basalt. Meteorit. Planet. Sci. 44, 87–94. ( 10.1111/j.1945-5100.2009.tb00719.x) [DOI] [Google Scholar]
- 100.Joy KH, Crawford IA, Downes H, Russell SS, Kearsley AT. 2006. A petrological, mineralogical, and chemical analysis of the lunar mare basalt meteorite LaPaz Icefield 02205, 02224, and 02226. Meteorit. Planet. Sci. 41, 1003–1025. ( 10.1111/j.1945-5100.2006.tb00500.x) [DOI] [Google Scholar]
- 101.Joy KH, Crawford IA, Anand M, Greenwood RC, Franchi IA, Russell SS. 2008. The petrology and geochemistry of Miller Range 05035: a new lunar gabbroic meteorite. Geochim. Cosmochim. Acta 72, 3822–3844. ( 10.1016/j.gca.2008.04.032) [DOI] [Google Scholar]
- 102.Liu Y, Floss C, Day JMD, Hill E, Taylor LA. 2009. Petrogenesis of lunar mare basalt meteorite Miller Range 05035. Meteorit. Planet. Sci. 44, 261–284. ( 10.1111/j.1945-5100.2009.tb00733.x) [DOI] [Google Scholar]
- 103.Righter K, Collins SJ, Brandon AD. 2005. Mineralogy and petrology of the LaPaz Icefield lunar mare basaltic meteorites. Meteorit. Planet. Sci. 40, 1703–1722. ( 10.1111/j.1945-5100.2005.tb00139.x) [DOI] [Google Scholar]
- 104.Taylor GJ, Warren P, Ryder G, Delano J, Pieters C, Lofgren G. 1991. Lunar rocks. In The lunar source book (eds Heiken GH, Vaniman DT, French BM.), pp. 183–284. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 105.Holness MB, Richardson C, Anand M. 2012. A new proxy for dolerite crystallisation times in planetary samples. In Proc. 43rd Lunar Planet. Sci. Conf., The Woodlands, TX, 19–23 March 2012, Abstract 1589. Houston, TX: Lunar and Planetary Institute. [Google Scholar]
- 106.Brenan J. 1994. Kinetics of fluorine, chlorine and hydroxyl exchange in fluorapatite. Chem. Geol. 110, 195–210. ( 10.1016/0009-2541(93)90254-G) [DOI] [Google Scholar]
- 107.Piccoli PM, Candela PA. 2002. Apatite in igneous systems. Rev. Mineral. Petrol. 48, 255–292. [Google Scholar]
- 108.Boyce JW, Hervig RL. 2008. Magmatic degassing histories from apatite volatile stratigraphy. Geology 36, 63–66. ( 10.1130/G24184A.1) [DOI] [Google Scholar]
- 109.Fagents SA, Rumpf ME, Crawford IA, Joy KH. 2010. Preservation potential of implanted solar wind volatiles in lunar palaeoregolith deposits buried by lava flows. Icarus 207, 595–604. ( 10.1016/j.icarus.2009.11.033) [DOI] [Google Scholar]
- 110.Stormer JC, Carmichael ISE. 1971. Fluorine-hydroxyl exchange in apatite and biotite: a potential geothermometer. Contr. Mineral. Petrol. 31, 127–131. ( 10.1007/BF00373455) [DOI] [Google Scholar]
- 111.Nadeau SL, Epstein S, Stolper E. 1999. Hydrogen and carbon abundances and isotopic ratios in apatite from alkaline intrusive complexes, with a focus on carbonatites. Geochim. Cosmochim. Acta 63, 1837–1851. ( 10.1016/S0016-7037(99)00057-5) [DOI] [Google Scholar]
- 112.Warren PH. KREEP: major-element diversity, trace-element uniformity (almost). In Workshop on Moon in transition: Apollo 14, KREEP, and evolved lunar rocks (eds Taylor GJ, Warren PH.), pp. 149–153. LPI Technical Report Number 89-03 Houston, TX: Lunar and Planetary Institute. [Google Scholar]
- 113.Warren PH, Wasson JT. 1979. The origin of KREEP. Rev. Geophys. Space Phys. 17, 73–88. ( 10.1029/RG017i001p00073) [DOI] [Google Scholar]
- 114.Hess PC, Parmentier EM. 1995. A model for the thermal and chemical evolution of the Moon's interior: implications for the onset of mare volcanism. Earth Planet. Sci. Lett. 134, 501–514. ( 10.1016/0012-821X(95)00138-3) [DOI] [Google Scholar]
- 115.Nakamura Y, Lammlein D, Latham G, Ewing M, Dorman J, Press F, Toksöz N. 1973. New seismic data on the state of the deep lunar interior. Science 181, 49–51. ( 10.1126/science.181.4094.49) [DOI] [PubMed] [Google Scholar]
- 116.Mueller S, Taylor GJ, Phillips RJ. 1988. Lunar composition: a geophysical and petrological synthesis. J. Geophys. Res. 93, 6338–6352. ( 10.1029/JB093iB06p06338) [DOI] [Google Scholar]
- 117.Canup RM, Asphaug E. 2001. Origin of the Moon in a giant impact near the end of the Earth's formation. Nature 412, 708–712. ( 10.1038/35089010) [DOI] [PubMed] [Google Scholar]
- 118.Longhi J. 2006. Petrogenesis of picritic mare magmas: constraints on the extent of early lunar differentation. Geochim. Cosmochim. Acta 70, 5919–5934. ( 10.1016/j.gca.2006.09.023) [DOI] [Google Scholar]
- 119.Elkins-Tanton LT, Burgess S, Yin Q-Z. 2011. The lunar magma ocean: reconciling the solidification process with lunar petrology and geochronology. Earth Planet. Sci. Lett. 304, 326–336. ( 10.1016/j.epsl.2011.02.004) [DOI] [Google Scholar]
- 120.Snyder GA, Neal CR, Taylor LA, Halliday AN. 1995. Processes involved in the formation of magnesian-suite plutonic rocks from the highlands of the Earth's Moon. J. Geophys. Res. 100, 9365–9388. ( 10.1029/95JE00575) [DOI] [Google Scholar]
- 121.Meyer J, Elkins-Tanton LT, Wisdom J. 2010. Coupled thermal-orbital evolution of the early Moon. Icarus 208, 1–10. ( 10.1016/j.icarus.2010.01.029) [DOI] [Google Scholar]
- 122.Elkins-Tanton LT, Van Orman JA, Hager BH, Grove TL. 2002. Re-examination of the lunar magma ocean cumulate overturn hypothesis: melting or mixing is required. Earth Planet. Sci. Lett. 196, 239–249. ( 10.1016/S0012-821X(01)00613-6) [DOI] [Google Scholar]
- 123.Shearer CK, Papike JJ. 2005. Early crustal building processes on the Moon: models for the petrogenesis of the magnesian suite. Geochim. Cosmochim. Acta 69, 3445–3461. ( 10.1016/j.gca.2005.02.025) [DOI] [Google Scholar]
- 124.Elardo SM, Draper DS, Shearer CK. 2011. Lunar magma ocean crystallization revisited: bulk composition, early cumulate mineralogy, and the source regions of the highlands Mg-suite. Geochim. Cosmochim. Acta 75, 3024–3045. ( 10.1016/j.gca.2011.02.033) [DOI] [Google Scholar]
- 125.Vander Kaaden KE, McCubbin FM, Whitson ES, Hauri EH, Wang J. 2012. Partitioning of F, Cl, and H2O between apatite and a synthetic shergottite liquid (QUE 94201) at 1.0 GPa and 990–1000°C. In Proc. 43rd Lunar Planet. Sci. Conf., The Woodlands, TX, 19–23 March 2012, Abstract 1247. Houston, TX: Lunar and Planetary Institute. [Google Scholar]
- 126.Shearer CK, Papike JJ. 1993. Basaltic magmatism on the Moon: a perspective from volcanic picritic glass beads. Geochim. Cosmochim. Acta 57, 4785–4812. ( 10.1016/0016-7037(93)90200-G) [DOI] [Google Scholar]
- 127.Hallis LJ, Anand M, Strekopytov S. 2014. Trace-element modeling of mare basalt source regions: implications for a heterogeneous lunar mantle. Geochim. Cosmochim. Acta 134, 289–316 doi.org/10.1016/j.gca.2014.01.012 [Google Scholar]
- 128.Bindeman IN, Kamenetsky VS, Palandri J, Vennemann T. 2012. Hydrogen and oxygen isotope behaviors during variable degrees of upper mantle melting: example from the basaltic glasses from Macquarie Island. Chem. Geol. 310–311, 126–136. ( 10.1016/j.chemgeo.2012.03.031) [DOI] [Google Scholar]
- 129.Tartèse R, Anand M. 2013. Late delivery of chondritic hydrogen into the lunar mantle: insights from mare basalts. Earth Planet Sci. Lett. 361, 480–486. ( 10.1016/j.epsl.2012.11.015) [DOI] [Google Scholar]
- 130.Aubaud C, Hauri EH, Hirschmann MM. 2004. Hydrogen partition coefficients between nominally anhydrous minerals and basaltic melts. Geophys. Res. Lett. 31, L20611 ( 10.1029/2004GL021341) [DOI] [Google Scholar]
- 131.Kyser TK, O'Neill JR. 1984. Hydrogen isotope systematics of submarine basalts. Geochim. Cosmochim. Acta 48, 2123–2133. ( 10.1016/0016-7037(84)90392-2) [DOI] [Google Scholar]
- 132.Taylor BE, Eichelberger JC, Westrich HR. 1983. Hydrogen isotopic evidence of rhyolitic magma degassing during shallow intrusion and eruption. Nature 306, 541–545. ( 10.1038/306541a0) [DOI] [Google Scholar]
- 133.Richet P, Bottinga Y, Javoy M. 1977. A review of hydrogen, carbon, nitrogen, oxygen, sulphur, and chlorine stable isotope fractionation among gaseous molecules. Annu. Rev. Earth Planet. Sci. 5, 65–110. ( 10.1146/annurev.ea.05.050177.000433) [DOI] [Google Scholar]
- 134.Sharp ZD, McCubbin FM, Shearer CK. 2013. A hydrogen-based oxidation mechanism relevant to planetary formation. Earth Planet. Sci. Lett. 380, 88–97. ( 10.1016/j.epsl.2013.08.015) [DOI] [Google Scholar]
- 135.Potts NJ, Anand M, Van Westrenen W, Tartèse R, Franchi IA. 2013. Using lunar apatite to assess the volatile inventory of the lunar interior. NLSI Lunar Volatiles Workshop, NASA Lunar Science Institute (NLSI), AMES Research Centre, Mountain View, CA, USA. [Google Scholar]
- 136.McCubbin FM, et al. 2014. Apatite-melt partitioning in basaltic magmas: the importance of exchange equilibria and the incompatibility of the OH component in halogen-rich apatite. In Proc. 45th Lunar Planet. Sci. Conf., The Woodlands, TX, 17–21 March 2014, Abstract 2741. Houston, TX: Lunar and Planetary Institute. [Google Scholar]
- 137.Boyce JW, Tomlinson SM, McCubbin FM, Greenwood JP, Treiman AH. 2014. Equilibrium exchange apatite hygrometry and a solution to the lunar apatite paradox. In Proc. 45th Lunar Planet. Sci. Conf., The Woodlands, TX, 17–21 March 2014, Abstract 2096. Houston, TX: Lunar and Planetary Institute; [DOI] [PubMed] [Google Scholar]
- 138.Watson EB. 1979. Apatite saturation in basic to intermediate magmas. Geophys. Res. Lett. 6, 937–940. ( 10.1029/GL006i012p00937) [DOI] [Google Scholar]
- 139.Harrison TM, Watson EB. 1984. The behavior of apatite during crustal anatexis: equilibrium and kinetic considerations. Geochim. Cosmochim. Acta 48, 1467–1477. ( 10.1016/0016-7037(84)90403-4) [DOI] [Google Scholar]
- 140.Tollari N, Toplis MJ, Barnes S-J. 2006. Predicting phosphate saturation in silicate magmas: an experimental study of the effects of melt composition and temperature. Geochim. Cosmochim. Acta 70, 1518–1536. ( 10.1016/j.gca.2005.11.024) [DOI] [Google Scholar]
- 141.Yamamoto S, et al. 2010. Possible mantle origin of olivine around lunar impact basins detected by SELENE. Nat. Geosci. 3, 533–536. ( 10.1038/ngeo897) [DOI] [Google Scholar]
- 142.Ustunisik G, Nekvasil H, Lindsley DH. 2011. Differential degassing of H2O, Cl, F, and S: potential effects on lunar apatite. Am. Mineral. 96, 1650–1653. ( 10.2138/am.2011.3851) [DOI] [Google Scholar]
- 143.Lucey PG. 2009. The poles of the Moon. Elements 5, 41–46. ( 10.2113/gselements.5.1.41) [DOI] [Google Scholar]
- 144.Bockelée-Morvan D, et al. 2012. Herschel measurements of the D/H and 16O18O ratios in water in the Oort-cloud comet C/2009 P1 (Garradd). Astron. Astrophys. 544, L15 ( 10.1051/0004-6361/201219744) [DOI] [Google Scholar]
- 145.Zhang Y. 2012. Water in lunar basalts: the role of molecular hydrogen (H2), especially in the diffusion of the H component. In Proc. 43rd Lunar Planet. Sci. Conf., The Woodlands, TX, 19–23 March 2012, Abstract 1957. Houston, TX: Lunar and Planetary Institute. [Google Scholar]
- 146.Wetzel DT, Rutherford MJ, Jacobsen SD, Hauri EH, Saal AE. 2013. Degassing of reduced carbon from planetary basalts. 110, 8010–8013. ( 10.1073/pnas.1219266110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhang Y, Ni H. 2010. Diffusion of H, C, and O components in silicate melts. Rev. Mineral. Geochem. 72, 171–225. ( 10.2138/rmg.2010.72.5) [DOI] [Google Scholar]
- 148.Liu Y, Mosenfelder JL, Guan Y, Rossman GR, Eiler JM, Taylor LA. 2012. SIMS analyses of water abundance in nominally anhydrous minerals in lunar basalts. In Proc. 43rd Lunar Planet. Sci. Conf., The Woodlands, TX, 19–23 March 2012, Abstract 1866. Houston, TX: Lunar and Planetary Institute. [Google Scholar]
- 149.Hauri EH, Wang J, Dixon JE, King PL, Mandeville C, Newman S. 2002. SIMS analysis of volatiles in silicate glasses: 1. Calibration, matrix effects and comparisons with FTIR. Chem. Geol. 183, 99–114. ( 10.1016/S0009-2541(01)00375-8) [DOI] [Google Scholar]
- 150.Koga K, Hauri EH, Hirschmann MM, Bell DR. 2003. Hydrogen concentration analyses using SIMS and FTIR: comparison and calibration for nominally anhydrous minerals. Geochem. Geophys. Geosyst. 4 ( 10.1029/2002GC000378) [DOI] [Google Scholar]
- 151.Aubaud C, Withers AC, Hirschmann MM, Guan Y, Leshin LA, Mackwell SJ, Bell DR. 2007. Intercalibration of FTIR and SIMS for hydrogen measurements in glasses and nominally anhydrous minerals. Am. Mineral. 92, 811–828. ( 10.2138/am.2007.2248) [DOI] [Google Scholar]
- 152.Mosenfelder JL, Le Voyer M, Rossman GR, Guan Y, Bell DR, Asimow PD, Eiler JM. 2011. Analysis of hydrogen in olivine by SIMS: evaluation of standards and protocol. Am. Mineral. 96, 1725–1741. ( 10.2138/am.2011.3810) [DOI] [Google Scholar]
- 153.Raepsaet C, Bureau H, Khodja H, Aubaud C, Carraro A. 2008. μ-Erda developments in order to improve the water content determination in hydrous and nominally anhydrous mantle phases. Nucl. Instrum. Method B 266, 1333–1337. ( 10.1016/j.nimb.2008.01.028) [DOI] [Google Scholar]
- 154.Withers AC, Bureau H, Raepsaet C, Hirschmann MM. 2012. Calibration of infrared spectroscopy by elastic recoil detection analysis of H in synthetic olivine. Chem. Geol. 334, 92–98. ( 10.1016/j.chemgeo.2012.10.002) [DOI] [Google Scholar]
- 155.Sarafian AR, Roden MF, Patiño Douce AE. 2013. The volatile content of Vesta: clues from apatite in eucrites. Meteorit. Planet. Sci. 48, 2135–2154. ( 10.1111/maps.12124) [DOI] [Google Scholar]