

Abstract
The Moon is depleted in volatile elements relative to the Earth and Mars. Low abundances of volatile elements, fractionated stable isotope ratios of S, Cl, K and Zn, high μ (238U/204Pb) and long-term Rb/Sr depletion are distinguishing features of the Moon, relative to the Earth. These geochemical characteristics indicate both inheritance of volatile-depleted materials that formed the Moon and planets and subsequent evaporative loss of volatile elements that occurred during lunar formation and differentiation. Models of volatile loss through localized eruptive degassing are not consistent with the available S, Cl, Zn and K isotopes and abundance data for the Moon. The most probable cause of volatile depletion is global-scale evaporation resulting from a giant impact or a magma ocean phase where inefficient volatile loss during magmatic convection led to the present distribution of volatile elements within mantle and crustal reservoirs. Problems exist for models of planetary volatile depletion following giant impact. Most critically, in this model, the volatile loss requires preferential delivery and retention of late-accreted volatiles to the Earth compared with the Moon. Different proportions of late-accreted mass are computed to explain present-day distributions of volatile and moderately volatile elements (e.g. Pb, Zn; 5 to >10%) relative to highly siderophile elements (approx. 0.5%) for the Earth. Models of early magma ocean phases may be more effective in explaining the volatile loss. Basaltic materials (e.g. eucrites and angrites) from highly differentiated airless asteroids are volatile-depleted, like the Moon, whereas the Earth and Mars have proportionally greater volatile contents. Parent-body size and the existence of early atmospheres are therefore likely to represent fundamental controls on planetary volatile retention or loss.
Keywords: volatile elements, Moon, the Earth, Mars, isotopic fractionation, zinc
1. Introduction
The origin of the Moon and the chemical and physical evolution of the Earth–Moon system remain unresolved, fundamental science problems. Dynamical models of planet formation and giant impact provide a broad range of possible formation scenarios, some of which appear to overcome previous angular momentum constraints [1–3]. Chronological evidence for the timing of formation of the Moon remains controversial [4–6], with age uncertainties resulting from lack of resolution of parent/daughter ratios, limitations in precision of measured isotopic ratios and restrictions in accurate dating of the oldest available crustal samples that have ubiquitously suffered later impact processes at the lunar surface. Some of the strongest available evidence for a co-genetic origin for the Moon and the Earth comes from the identical stable isotope compositions of oxygen [7–9], titanium [10] and silicon [11,12]. These results implicate the formation of the Moon from a silicate reservoir that was isotopically identical to the bulk silicate Earth for these elements. Whether this isotopic homogeneity reflects mixing during a giant impact [1,2], isotopic homogenization between the proto-Earth and a silicate melt–vapour disc [13], or if the terrestrial planet formation region was already isotopically well mixed prior to the formation of the Earth, the Moon and Venus from previously differentiated planetary embryos, remains unresolved.
The Moon is depleted with respect to the bulk silicate Earth in elements that are cosmochemically volatile or moderately volatile (figure 1a; e.g. [19–21]). Volatile and moderately volatile elements are defined as those elements for which 50% condensation temperatures at cosmochemical gas pressures (less than 10−4 atm) are less than 665 and 1135 K, respectively [14]. These properties extend to volatile species such as H2O (or OH). Hydroxyl has been detected in some lunar pyroclastic glasses [22], as well as apatites formed during the final stages of fractional crystallization within mare basalts [23], but water appears to be more depleted in the Moon than in the Earth. For moderately volatile elements, such as S, K, Cl, Zn, Rb, Cd and Pb, the degree of depletion in the Moon is similar to other volatile-depleted and differentiated planetesimal bodies in the Solar System, such as the parent bodies of eucrite or angrite meteorites (figure 1b).
Figure 1.
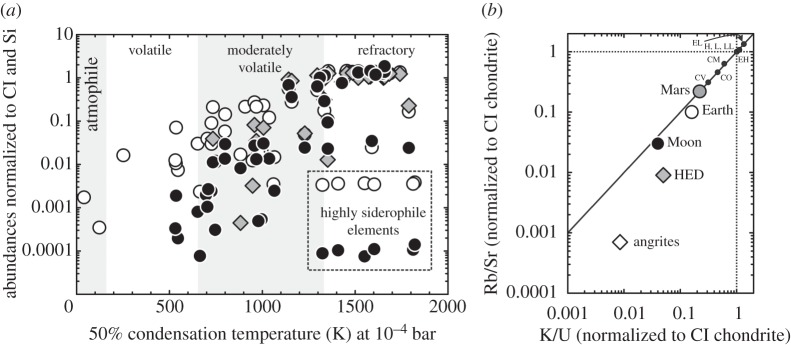
(a) Estimated abundances of the elements classified according to their 50% condensation temperatures at 10−4 bar (from [14]) in the mantles of the Earth (unfilled circles), Moon (filled circles) and the HEDPB (grey diamonds). (b) Carbonaceous Ivuna (CI) chondrite normalized Rb/Sr versus K/U ratios in bulk meteorites and planets. Data in (a) from Dreibus & Wanke [15], O'Neill [16], McDonough & Sun [17], with highly siderophile element abundance data for the Moon from Day et al. [18]. The dashed line in (a) denotes the region in which the highly siderophile elements are located for the Earth and the Moon. Panel (b) is modified from the data compilation of Davis [19].
Volatile and moderately volatile element depletions in the Moon are not recent effects, nor are their depletions solely related to removal in early, hot nebular gas [24]. The currently known chronology of volatile depletion for the Moon suggests both inheritance of volatile-depleted materials to form the Moon (and the Earth), as well as later volatile depletion. The ubiquitously low measured 87Sr/86Sr ratios (less than 0.7; initial 87Sr/86Sr=0.69906; e.g. [5,25]) and extremely high μ (238U/204Pb) values for lunar mare basalts (approx. 300) and lunar pyroclastic glass beads (19–55; for comparison, the lowest terrestrial 87Sr/86Sr ratio comes from 3450 Ma barite, 0.70053, e.g. [26]; compared with terrestrial 238U/204Pb ratios estimated at 6–8 [27]), indicate that Rb and Pb depletion occurred early, either prior to or during formation of the Moon. The timing of loss of moderately volatile elements is not well constrained, but is within current estimates of the timing of the Moon-forming event [4–6]. Equally, marginally more radiogenic measured 87Sr/86Sr ratios in lunar samples, compared with angrites and eucrites, indicate early loss of Rb on some differentiated asteroidal bodies (within the first 2–3 Ma after Solar System formation for angrites and eucrites; e.g. [28–30]).
Given the evidence for depletion of abundances of volatile and moderately volatile elements for the Moon relative to the Earth, an increasing number of studies are examining evaporative fractionation effects on stable isotopes of moderately volatile elements [31–34]. Their aim is to constrain the nature of volatile element depletion and so potentially to reveal how and when such depletion occurred and whether it relates to nebula (i.e. prior to the formation of planets and planetesimals), large-scale global (e.g. giant impact, lunar magma ocean) or localized (e.g. lava flow or fire fountain degassing) evaporation. Here, we review the underlying theory, seeking evidence for evaporative fractionation of stable isotopes of moderately volatile elements in planets and, using the currently available S, Cl, K and Zn data, place new constraints on how and when such processes occurred in the Moon. Our prime motivation is to explain the evidence for lower Zn abundances and high 66Zn/64Zn ratios measured in lunar mare basalts relative to martian or terrestrial basalts or ordinary and carbonaceous chondrites (figure 2). These differences have previously been interpreted to reflect a global-scale evaporation event associated with lunar formation [34]. In assessing evidence from fractionations observed in stable isotopes of S, Cl, K and Zn, we find the data to be most consistent with global-scale evaporative fractionation of moderately volatile and volatile elements immediately in the aftermath of the Earth–Moon forming event.
Figure 2.
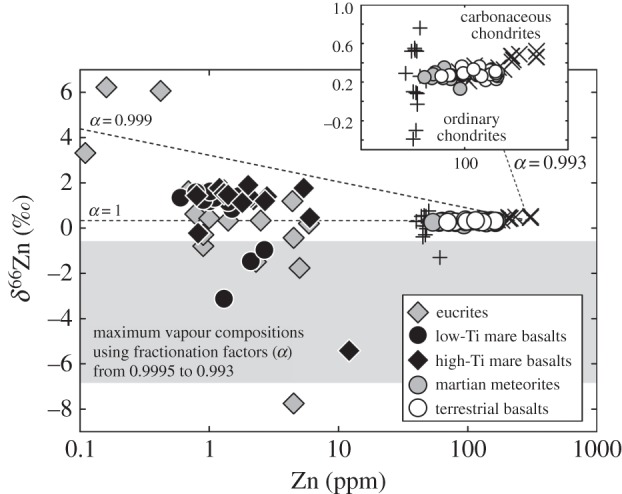
Zinc isotope compositions and abundances of planetary materials versus ordinary (plus symbols) and carbonaceous chondrites (cross symbols). Rayleigh distillation curves (dashed lines) for fractionation factors (α) of 1, 0.999 and 0.993 (upper limit of ideal fractionation behaviour for ZnCl) are shown for melts with a carbonaceous chondrite initial composition (δ66Zn=0.33‰; 300 ppm Zn). The corresponding field for maximum calculated vapour compositions for limited (0.9995) to ideal fractionation behaviour for ZnCl is shown for comparison. Inset shows expanded view of chondrite–Earth–Mars data. Data are from [34–37].
2. Isotopic fractionation of moderately volatile elements in cosmochemistry
Element volatility is a fundamental chemical property occurring in response to temperature variations experienced by a chemical system, the associated condensation temperatures of the elements, and the two processes of evaporation and condensation that define volatility. Under solar nebula conditions (i.e. gas pressures of less than 10−4 atm; [14]) all meteorite classes, and terrestrial and lunar rocks are depleted in the volatile elements compared with carbonaceous Ivuna-type (CI) chondrites (figure 1). CI chondrites are generally regarded as being representative of the bulk Solar System elemental composition (with the exception of abundances of highly volatile elements (H, C, N, O, noble gases) and elements that are reactive in stellar environments (Li, Be, B)), because of the similarity of their elemental distribution with those determined spectroscopically in the solar photosphere [14,38–40]. Volatile depletions for various types of undifferentiated planetary materials, relative to CI chondrites, differ, but for the most part have the common characteristic of being reasonably smooth functions of volatility (expressed as 50% condensation temperatures). The volatile element patterns of differentiated planetary rocks are typically less smooth owing to the effects of igneous processes. Some incompatible elements, however, are not significantly decoupled by igneous differentiation, and ratios of volatile to refractory incompatible elements can be used to assess the level of volatile depletion in planets and parent bodies [19,39,41].
A logical system for defining cosmochemical volatility has been adopted that divides the elements into different groups based on their physical and chemical properties [42]. The primary cosmochemical classification of the elements is based on volatility, with the elements divided into those that are refractory (having equilibrium condensation temperatures higher than those of the most abundant rock-forming elements, magnesium, silicon and iron), moderately volatile (having condensation temperatures lower than those of the refractory elements, but higher than that of FeS), and highly volatile (having condensation temperatures below that of FeS). For a gas of solar composition at a total pressure of 10−4 atm, refractory elements have 50% condensation temperatures above 1335 K, moderately volatile elements between 1335 and 665 K, and volatile elements below 665 K [14]. A fourth group, the atmophile elements (hydrogen, carbon, nitrogen, oxygen, and the noble gases), have condensation temperatures of 180 K or lower and are depleted in CI chondrites, relative to the Sun. The other important classification of elements is based on which mineral phase, or phases, elements concentrate into: siderophile elements concentrate into iron–nickel metal, chalcophile elements into sulfides, and lithophile elements into oxides and silicates. An example of how this concentration behaviour affects elemental distributions is oxygen; although it is an atmophile element, it behaves as somewhat refractory owing to its early incorporation into oxides and silicates. Clearly, the timing of volatile depletion, coupled with the incorporation of elements into phases based on their concentration behaviour, is profoundly important in the current distribution of elements in the Solar System.
Volatile element depletion can be simplified into two processes: partial condensation from a cooling gas; and partial evaporation from condensed dust or melt. For elements with more than one isotope, a physical kinetic or fractional equilibrium stable isotope effect during evaporation then provides a measure of the extent of partial evaporation. The kinetic isotope effect can be best understood using the moderately volatile elements such as S, Cl, K and Zn, for which there is no evidence for large fractionations of their isotopes during planetary differentiation and which have well-determined abundance depletions. In classical theory, natural mass-dependent stable isotopic fractionation of an element occurs during any exchange reaction and is due to the difference in vibrational energy of the bonds formed by the different isotopes at equilibrium or under kinetic conditions [43,44]. The general rule behind isotopic fractionation is that heavy isotopes of an element will have a tendency to be enriched in the compounds where that element forms the stiffest (usually the shortest and strongest) bonds [45]. A natural implication of this effect is that, during evaporation/condensation processes, the lightest isotopes will have the tendency to be enriched in the vapour phase while the heaviest isotopes will be enriched in the condensed phase. The amplitude of these effects is proportional to the mass difference between the isotopes and therefore it is most notable for light elements (e.g. H, Li, O, S). The development of a new generation of multi-collector inductively coupled plasma mass spectrometers (MC-ICP-MS) in the late 1990s has allowed high-precision measurements of the isotopic ratios of heavier elements (e.g. Fe, Zn, Cu) and small but resolvable isotopic variations are now measurable [46]. For geochemical, cosmochemical and lunar applications, much of the theoretical form of work has been reviewed previously [19,45,47] and the reader is referred to these articles for further details.
In order to choose stable isotopes that can effectively demonstrate isotopic fractionation during moderately volatile element evaporation, some of the same rules apply as for ‘standard’ stable isotope systems, such as oxygen. Classic stable isotopic theory states that elements that show stable isotopic variations in nature generally have: (i) low atomic mass; (ii) large relative mass differences between stable isotopes; (iii) a tendency to form highly covalent bonds; (iv) multiple oxidation states or other chemical variability; and (v) two isotopes or more with sufficient abundance to make measurements feasible (cf. [45,48]). While these rules still apply, development of clean chemistry and precise and accurate MC-ICP-MS methods has expanded the ability to measure higher atomic masses at smaller relative mass differences (e.g. Zn, U). Additionally, stable isotope systems such as Li, Mg, Si, Ca and Zn have a single predominant oxidation state that can be advantageous for simplifying interpretation of stable isotope data. For some volatile and moderately volatile elements, the determining factor in stable isotope ratio measurement is absolute abundance in natural materials. For example, low abundances of Hg, Cd and Sn in lunar rocks makes measurement of their isotopic ratios difficult given the available sample masses of lunar and asteroidal materials for study from the Apollo missions and from meteorite falls and finds. Zinc, on the other hand, while depleted in absolute abundances in lunar rocks, occurs in sufficient quantity (more than 1 μg g−1) that its isotopic ratios can be measured precisely.
3. The S, Cl, K and Zn compositions of lunar, terrestrial and martian rocks
In the following section, we review available stable isotopic and associated elemental abundance data for cosmochemically moderately volatile elements (S, Cl, K, Zn) for which there are comparable data for lunar, terrestrial and martian basalts, as well as data for eucrite meteorite samples. We note that there are numerous moderately volatile or volatile elements that have two or more isotopes and can potentially provide information on evaporation and condensation processes operating during the evolution of the Moon, applying the caveats described in §2 (e.g. 11B/10B, 908 K, moderately volatile; 65Cu/63Cu, 1037 K, moderately volatile siderophile (MVS); 71Ga/69Ga, 968 K, MVS; 74,73,72,70Ge, 883 K, MVS; 78,77,76Se, 697 K, highly volatile chalcophile (HVC); 81Br/79Br, 546 K, highly volatile; 87Rb/85Rb, 800 K, moderately volatile; Cd isotopes, 652 K, HVC; Sn isotopes, 704 K, HVC; Hg isotopes, 252 K, HVC). For the rest of this paper, isotopic ratios are reported as delta (parts per one thousand or per mil (‰)) notation, where
 |
3.1 |
where X is the heavier mass isotope (34S, 37Cl, 41K, 66Zn) and Y is the lighter mass isotope (32S, 35Cl, 39K, 64Zn).
(a). Sulfur
Sulfur has four stable isotopes, 32S (95.02%), 33S (0.75%), 34S (4.21%) and 36S (0.02%), with the typical mass-dependent stable isotope value reported as 34S/32S (δ34S) relative to Canyon Diablo Troilite (CDT; 34S/32S=0.045). Sulfur has the lowest 50% condensation temperature (664 K) of the elements discussed here and, as well as being highly volatile under cosmochemical conditions, is also chalcophile [14], making its behaviour distinct from those of Cl, K or Zn. Early studies of lunar samples revealed a relatively large spread in δ34S values for mare basalts (−1.6 to +0.9‰; [49]), and significantly 34S-enriched components in lunar regolith and soil materials (+6.1 to +13.5‰ and 320–790 ppm; [50]), interpreted to reflect the loss of 20–30% of the abundance of S (and K) from the lunar regolith by ion sputtering [51]. A recent study has revealed a restricted range in δ34S for mare basalts of +0.57±0.09‰ [52], while work to identify mass-independent S fractionation in shergottites indicates an average δ34S for martian basalts of −0.05±0.3‰ [53].
High-precision 34S/32S data have been obtained for terrestrial mid-ocean ridge basalts (MORB), giving an average δ34S of −0.98±0.51‰ [54,55]. Correlations with Sr and Nd isotope ratios in MORB suggest mixing with a low-δ34S mantle and a high-δ34S crustal component, in turn indicating that the depleted mantle has low δ34S (−1.28±0.33‰) when compared to lunar rocks (+0.57±0.09‰), potentially indicating a core fractionation effect [55] and/or a loss of the lighter S isotopes from the Moon. The range of S concentrations in lunar basalts (730 to >1500 ppm; [52]), martian shergottites (300 to >1500 ppm; [53]) and terrestrial MORB (728–1221 ppm; [54]) are similar, but lunar mare basalts have isotopically heavier δ34S values (figure 3a). The similarities in the range of S concentrations for lunar, terrestrial and martian rocks may at first appear an impediment for volatile loss. However, S is typically exhausted after more than 20% partial melting, with most mantle-derived partial melts from the Earth or the Moon being S-undersaturated [60,61], indicating that enrichment of S is expected in mantle melt products.
Figure 3.

Comparisons of (a) S, (b) Cl, (c) K and (d) Zn isotope ratios (reported as per mil values) measured in lunar mare basalts compared with lunar regolith/soil, terrestrial lavas and martian shergottites. All data are for bulk rock samples. Inset panel in (d) shows pyroclastic glass beads and mare basalts with isotopically light Zn inherited from post-crystallization condensation. Data for (a) from [53,54,56], for (b) from [32,57–59], for (c) from [31] and for (d) from [34,37].
(b). Chlorine
Chlorine has two stable isotopes, 35Cl (75.78%) and 37Cl (24.22%), and the delta notation (δ37Cl) is typically expressed relative to standard mean ocean chloride (SMOC). Chlorine has a 50% condensation temperature estimated as 948 K and is highly volatile [14]. It is also volatile at magmatic temperatures, incompatible during silicate melting and water-soluble, and it has consequently been identified as a powerful potential tracer of interactions between the terrestrial hydrosphere, crust and mantle [57,59]. Lack of significant Cl isotope fractionation in the terrestrial mantle, crust and carbonaceous chondrites led Sharp et al. [57] to conclude lack of nebular fractionation of Cl isotopic compositions, or of late accretion Cl additions to the Earth. Compilations of terrestrial MORB data give an average Cl content of 205 ppm (range=42–1400 ppm), with δ37Cl of −0.7±0.6‰ [57,59,62]. The large range in Cl contents and elevated δ37Cl relative to estimated mantle composition (−1.6‰) indicates ubiquitous alteration of terrestrial volcanic rocks by seawater, brines or hydrothermally altered surface rocks. Since terrestrial magmas and lavas show a limited range of δ37Cl, it has been argued that there is always an excess of H relative to Cl, and that the dominant volatile chloride species is therefore HCl gas with a limited isotopic fraction between basaltic melts [32]. However, more recent work by Sharp et al. [33] provided evidence for significant isotopic fractionation of Cl isotopes in HCl. Limited data for martian shergottites also indicate a limited range in Cl isotope compositions (δ37Cl=−0.4±0.4‰) and a range in Cl contents from 24 to 115 ppm [58].
Chlorine isotopes have been strongly fractionated in some lunar materials (−1 to +24‰), leading Sharp et al. [32] to suggest a mechanism of volatilization of Cl as metal halides (e.g. NaCl, ZnCl2, FeCl2), loss of Cl during basalt eruption on the lunar surface and, consequently, an essentially anhydrous lunar interior. Volatilization of Cl on the Moon is possible because of the low calculated H/Cl ratio for lunar basalts and from the presence of metal halides on the surface coatings of lunar pyroclastic glass beads [63]. Sharp et al. [32] presented Cl isotope and abundance data for bulk rock analyses of lunar mare basalts, regolith materials and lunar pyroclastic glass beads by gas-source mass spectrometry, as well as Cl isotope analyses of apatites performed by secondary ionization mass spectrometry. Lunar regolith components (+5.6 to +16‰ and 8–117 ppm Cl) are enriched in 37Cl relative to mare basalts (0 to +4.2‰ and 5–25 ppm Cl) and apatites within mare basalts and KREEP-rich (potassium–rare earth elements–phosphorus) rocks (lunar sample no. 72275) are also enriched in 37Cl (+13.1 to +24.5‰).
Sharp et al. [32] reported apatite in sample 12040 with δ37Cl of +17.2 and 0.83 wt% Cl and reported a bulk composition for the same mare basalt of δ37Cl=0‰ and 11 ppm Cl. Assuming 0.1–0.2 modal per cent phosphate in 12040—a reasonable approximation for a primitive low-Ti mare basalt [64–66]—and using the Cl concentration measured by Sharp et al. [32] indicates that approximately 9 ppm (out of the 11 ppm measured [32]; i.e. approx. 80% of the total Cl) is hosted within apatite minerals in mare basalts and implies either that the isotopic composition of the bulk should be much heavier than that reported by Sharp et al. [32], or that the approximately 20% of Cl not hosted within apatites should be extremely (and improbably) isotopically light (approx. −70‰). Variable δ37Cl values occur for apatite in individual lunar samples, and δ37Cl in excess of +25‰ has been measured in apatites with more than 5000 ppm chlorine and with relatively high OH contents [67]. Therefore, values provided by individual apatites can be variable within a single rock and may reflect late-stage, secondary volatile-loss effects during extreme fractional crystallization. Given uncertainties in how to interpret the apatite Cl isotope and OH contents, the possibility for heterogeneous distribution of δ37Cl within individual apatites and the late-stage fractionation of apatite within lunar mare basalts, we only report bulk rock gas-source mass spectrometry analyses of Cl for mare basalts in figure 3b. Bulk rock data indicate that martian and terrestrial basalts have similar δ37Cl, whereas mare basalts are characterized by 37Cl-enriched compositions and Cl contents that are typically 2–40 times lower than basalts from the Earth or Mars.
(c). Potassium
Pioneering work in the 1990s [31,68] revealed limited isotopic fractionation of the two stable isotopes of potassium, 39K (93.3%) and 41K (6.7%; the long-lived radioisotope 40K makes up 0.012% of the natural abundance), in planetary materials. The work of Humayun and others reports 41K/39K ratios as δ41K relative to Suprapur KNO3 [68,69]. Limited work on K has been done in the past 20 years, although a study of K in terrestrial tektites revealed no discernible isotopic fractionation [69]. Given that potassium is moderately volatile and has a 50% condensation temperature of 1006 K, a lack of isotopic fraction led Humayun & Clayton [68] to conclude that K and other moderately volatile alkalis (e.g. Na, Rb, Cs) must have been depleted prior to chondrule, chondrite or planet formation, probably from a hot dust stage in the early solar nebula.
Despite lack of 41K/39K isotopic fractionation between terrestrial basalts (δ41K=+0.28±0.21‰), martian shergottites (+0.5‰), eucrites (+0.9 to +2.7‰) and mare basalts (+0.1 to +1.5‰), Humayun & Clayton [68] found isotopically light δ41K in a ferroan anorthosite (60015, −3.9‰), and isotopically heavy δ41K in lunar soils (+5 to +12.7‰) [31] (figure 3c). Additionally, these authors confirmed previous studies [51] that had observed higher K contents in terrestrial basalts and martian shergottites (typically greater than 1000 ppm), relative to mare basalts (300–750 ppm) or eucrites (160–300 ppm). The lack of isotopic fractionation of 41K/39K in basalts formed by high-temperature igneous processes, or in tektites formed by dry melting during hypervelocity impacts, indicates lack of K fractionation, either during melting, or by partial evaporation [69]. Recasting the K isotopic data suggests variable and slight heavy isotopic enrichment in mare basalts relative to terrestrial or martian igneous rocks (figure 3c).
(d). Zinc
Numerous studies have been performed to characterize the zinc isotopic composition of planetary materials [34–37,70,71]. Zinc is composed of five stable isotopes, 64Zn (48.6%), 66Zn (27.9%), 67Zn (4.1%), 68Zn (18.8%) and 70Zn (0.6%), but, because of its low natural abundance, 70Zn is rarely reported in high-precision isotopic measurement studies. The only recent study reporting high-precision 70Zn/64Zn ratio showed that this ratio is mass-dependently fractionated within chondritic material [72]. Along with Cl, Zn is classified as highly volatile, with a 50% condensation temperature of 726 K [14]. Zinc isotope ratios are reported as 66Zn/64Zn (δ66Zn), 67Zn/64Zn (δ67Zn) or 68Zn/66Zn (δ68Zn), typically relative to the ‘Lyon’ Zn standard JMC 3–0749 L [46].
All the Zn isotopic data measured in Solar System materials to date fall on a slope 2 line on a δ68Zn versus δ66Zn plot, conforming to the expected mass-dependent mass-fractionation law. The same is true for 67Zn/64Zn and 70Zn/64Zn [72]. Terrestrial, martian and lunar rocks all lie on the same mass-dependent mass-fractionation line, along with all classes of chondritic meteorites [34,35,73], indicating that Solar System Zn evolved from a single, isotopically homogeneous reservoir. All reported isotopic variations are therefore due to mass-dependent fractionations. Mass-dependent fractionation of Zn isotopes also rules out secondary neutron capture effects, which would lead to non-mass-dependent behaviour.
Zinc is more volatile than both K and Cl under solar nebula conditions [14] and exhibits limited isotopic fractionation during igneous processes [37]. On the Earth, and in primitive meteorites such as chondrites, variations of δ66Zn larger than 1‰ have only been observed associated with evaporation events [70,73,74]. Any Zn isotopic fractionation, combined with differences in Zn concentrations between planetary igneous rocks from differentiated bodies such as the Earth, the Moon and Mars, can reveal important differences in volatile depletion and replenishment events for these bodies. Extensive studies of terrestrial basalts have revealed a limited variation in Zn isotopic compositions and a precise bulk silicate Earth δ66Zn value of +0.28 ± 0.05‰ (2σ), with terrestrial basalts typically having between 60 and more than 160 ppm Zn, the concentration of which broadly correlates with indices of differentiation [37]. Martian meteorites span a limited range in δ66Zn (+0.13 to +0.35‰), with an average δ66Zn of +0.25±0.03‰ [34]. Zinc concentrations in shergottite meteorites are similar to those in terrestrial basalts at between 41 and 130 ppm, and Zn concentrations in nakhlite meteorites lie within the range of shergottite meteorites at between 61.5 and 89 ppm [34].
Lunar samples exhibit the largest range of δ66Zn from any differentiated planetary body in the Solar System. However, systematic variations are observed between: (i) lunar pyroclastic glass beads, which have negative δ66Zn values (−2.9 to −4.2‰) and high Zn concentrations (129–231 ppm); (ii) pristine low-Ti mare basalts, which have an average δ66Zn value of +1.31±0.13‰ and are isotopically identical to high-Ti mare basalts (δ66Zn=+1.39±0.31‰), and have Zn concentrations (0.6–12 ppm) at least a factor of 10 lower than in lunar pyroclastic glass beads; and (iii) lunar regolith, soils and anorthositic regolith breccia meteorites (MAC 88105), which have isotopically heavy δ66Zn (+2.6 to +5.6‰) (figure 3d) [34,70,71]. The high δ66Zn of the regolith and soil samples reflect both sputtering effects and impact gardening of the lunar regolith [70,71] and do not represent primary magmatic compositions. There is a class of mare basalts with isotopically light δ66Zn (figure 3d, inset) that result in a similar range in lunar δ66Zn compared with eucrite meteorites (−7.8 to +6.2‰) [36] and have been interpreted as the consequence of condensation of isotopically light zinc after mare basalt crystallization [34,75].
4. Evidence for a non-ideal Rayleigh regime during isotopic fractionation of Zn
An important aspect of the Zn isotope data for the Moon is that the isotopic fractionation does not conform to ideal isotopic fractionation by distillation of ZnCl2or ZnS, which are both possible (although not the only) species involved in lunar degassing (figure 2). For ideal evaporation of a liquid into a vacuum, the Rayleigh distillation equation is
| 4.1 |
where F is the fraction of 64Zn remaining in the melt and α is the fractionation factor between vapour and melt:
| 4.2 |
The fractionation factor (α) can be estimated as an upper limit for ideal behaviour using the square-root of the light isotopologue over the heavy isotopologue of the vaporizing species in the case of kinetic fractionation [76],
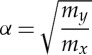 |
4.3 |
where my and mx are the masses of the light and heavy isotopologue, respectively. For ZnCl2 the α value is 0.993. Extreme fractionations of Zn isotopes in the melt and associated vapour phase are computed for reducing Zn concentrations in the melt fraction. Fractionations greater than those measured can be calculated, suggesting either incomplete evaporation, or the competing effect of diffusion rates being insufficient to maintain isotopic homogeneity in lunar melts (effect exemplified as lower α value of 0.999, for reference; figure 2). Fractionations less than theoretical calculations are observed experimentally [56,77] and would be expected if diffusion rates are insufficient to maintain isotopic homogeneity of the melt.
Potential analogues to explain inefficient and partial evaporation of Zn and, by extension, stable isotope fractionations of other volatile elements, are tektites. Tektites are terrestrial natural glasses that are produced during hypervelocity impacts or airbursts of a projectile into terrestrial target rocks or soils [78,79]. Some tektites are extremely water-poor (less than 0.02 wt% H2O) compared with precursor rocks (sediments with more than 1 wt% H2O), making them some of the most anhydrous naturally occurring terrestrial materials. Although volatilization at high temperature is a probable cause of this effect, K and Cd isotope studies to seek evidence for evaporative stable isotope fractionations have yielded equivocal results [69,80,81]. In contrast, some tektites are enriched in the heavy isotopes of Zn compared to upper continental crust [74], and Zn abundance is negatively correlated with δ66Zn, indicating that isotopic fractionation occurred during evaporation under the high-temperature conditions of tektite formation (above 2273 K; figure 4). A similar isotopic effect has been observed for Cu isotopes [82]. Tektites do not follow the calculated Rayleigh distillation curves for Zn isotope fractionation, which has been interpreted as evaporative isotopic fractionation in a diffusion-limited regime in which the molten tektite is advecting by viscous coupling with air [74]. The tektite is not re-homogenized instantly after evaporation, as it is in ideal Rayleigh distillation, and the magnitude of the isotopic fractionation decreases. Therefore, both natural [74] and experimental [77] evidence points to inefficient evaporative fractionation behaviour of moderately volatile elements. By analogy, the restricted and consistent Zn isotope fractionations observed for lunar mare basalts (approx. +1.3‰), compared with ideal Rayleigh behaviour for the Zn concentrations observed (δ66Zn at least more than 5‰; figure 2) provides an important constraint on the type of process responsible for the approximately 1‰ difference in δ66Zn between the Earth and the Moon.
Figure 4.
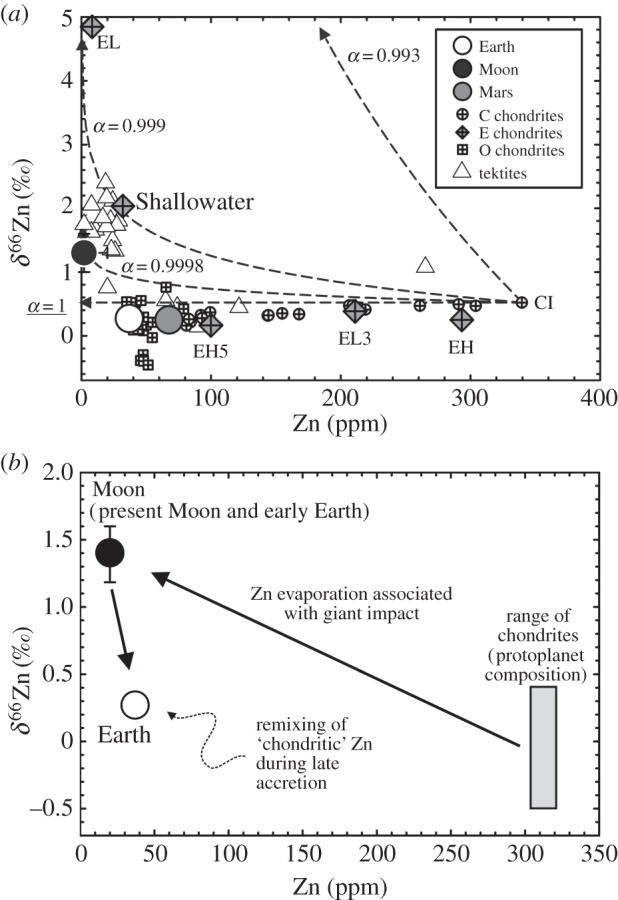
(a) Zinc isotope and abundance systematics in chondrites [35,73] and terrestrial tektites [74] versus estimates of lunar, terrestrial and martian mantle composition (from [34]). Enstatite chondrites (EH, EL3, EH5, EL and Shallowater) are indicated on the figure. Rayleigh distillation curves of α=1, 0.9998, 0.999 and 0.993 are shown for comparison. (b) A possible scenario of volatile element depletion associated with the giant impact, from a chondritic precursor, followed by stochastic late accretion in higher proportions to the Earth than to the Moon. See text for details.
5. Mechanisms for generating moderately volatile element depletion in the Moon
(a). Nebula versus planetary volatile loss
Extreme volatile depletion of the Moon, as implied from the low concentration and isotopically heavy Zn isotope enrichment seen in lunar mare basalts, contrasts with more elevated Zn abundance and broadly chondritic Zn isotopic compositions for the Earth and Mars [34]. Comparison of volatile and moderately volatile element abundance and isotopic compositions indicate that mare basalts are systematically volatile-depleted and are enriched in the heavy isotopes of moderately volatile elements compared with terrestrial or martian basalts (e.g. figures 1 and 3). Taken as a whole, the data indicate that moderately volatile element depletion is ubiquitous among mare basalts, through a large- rather than a small-scale process, or by hydrodynamic escape [83]. Given the differences in moderately volatile element abundances between the Moon and the Earth, it is not likely that this variation solely reflects depletion prior to planet formation [24,31]. A depletion process prior to the formation of chondrules, chondrites and planet formation in the hot, dusty solar nebula can explain the systematic depletion of volatile elements in chondrites [19], which are proxy materials for the building blocks of planets, but other depletion and/or replenishment events are also required to explain the differing volatile budgets among differentiated planetary bodies.
At least two phases of volatile and moderately volatile depletion must have occurred during the formation of the Solar System. The first is at the nebula scale, prior to or during the formation of the first solids (cf. calcium–aluminium inclusions, chondrules). The second volatile depletion event, or series of events, is associated with planetesimal and planet formation and appears to relate with escape velocity (figure 5) and whether the body could retain a long-term atmosphere (e.g. the Earth, Mars, Venus) or not (e.g. Moon, Mercury, angrite or howardite–eucrite–diogenite parent bodies (HEDPB)), both during and after their differentiation. We refer to the first event as nebula volatile depletion and to the second as planetary volatile depletion. It is planetary volatile depletion that is considered in detail here, and that is applied to understanding the Moon's origin.
Figure 5.
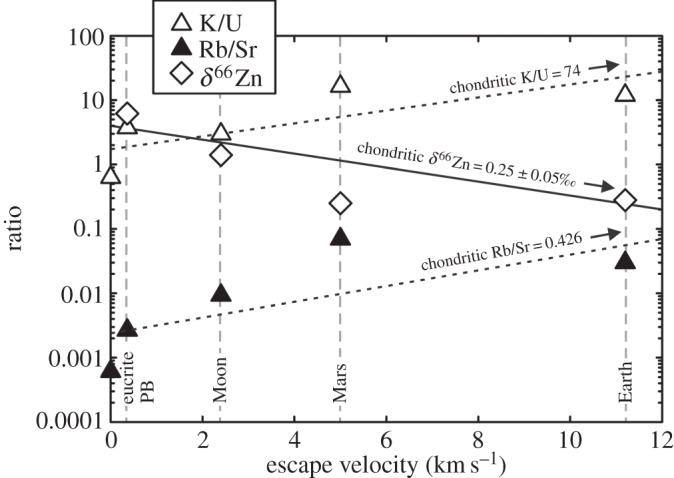
Escape velocity versus abundance ratios of K/U and Rb/Sr, and 66Zn/64Zn ratios (as δ66Zn in per mil) for the Earth, Mars, Moon, the eucrite parent body (eucrite PB) and the angrite parent body (on the y-axis; data reported in table 1). Exponential regressions are shown for δ66Zn (R2=0.66; solid line), Rb/Sr (R2=0.55; dashed line) and K/U (R2=0.54; dashed line). Ratios of Rb/Sr and K/U decrease, and δ66Zn shows heavy-isotope enrichment, with lower escape speeds. Chondritic values for Rb/Sr, K/U and 66Zn/64Zn are reported along the exponential regression lines.
We suggest two process-driven mechanisms for planetary volatile depletion for the Moon and the Earth after the initial phase of nebula volatile depletion.
First, evaporation of volatile and moderately volatile elements could have occurred during the high-energy Earth–Moon forming event [34,84], which was then followed by later replenishment of volatile elements to the Earth and Mars by proportionally larger amounts of late accretion than to the Moon [85]. This process would be unique to the Earth–Moon system and irrelevant to Mars or small planetesimals, such as the angrite or eucrite parent bodies, which did not obviously suffer such a high-energy and late-stage (probably more than 100 Ma after Solar System formation; [4–6]) event.
Second, planetary differentiation processes may have led to volatile and moderately volatile elements being lost on smaller (cf. lower mass, lower escape velocity) airless bodies, such as the Moon, more than on the Earth or Mars. These latter planets are considered to have formed nascent atmospheres early in their formation histories which may subsequently have been blown off, resulting in complex volatile retention trends [86]. An inevitable consequence of differentiation and incompatible volatile element loss on smaller bodies would also have been the formation of nascent atmospheres during outgassing. The likelihood of loss of atmospheres, either through smaller parent-body size or from impact-induced atmospheric blow-off, may have been a critical ingredient in the loss of volatile and moderately volatile elements in planets. There are two possible locations where volatile and moderately volatile elements could have been lost during planetary differentiation: (i) either during a large-scale magmatic event on the Moon, such as a magma ocean; or (ii) from local eruption and evaporation processes acting during basaltic eruption. Below, we review the evidence for these potential processes, in turn. The evidence suggests that giant impact and late accretion—or localized eruption processes—require special circumstances in order to explain the moderately volatile element compositions of lunar mare basalts. Volatile outgassing during a magma ocean event offers much promise for understanding planetary volatile depletion, if mechanisms for evaporative vapour loss can be effectively identified.
(b). Volatile loss during the Earth–Moon forming giant impact?
Since the mid-1970s, the concept of a giant impact has become the favoured theory for the formation of the Earth and the Moon [87], and remains consistent with the physical, geochemical and age constraints [1,5,7,8]. Such a large-scale and high-energy event can theoretically explain planetary volatile depletion in the Earth–Moon system owing to the potential for large thermal and chemical gradients in the associated transient protoplanetary disc (figure 6). Owing to the differences in moderately volatile elements retained between the Earth and the Moon, this scenario would require either: (i) more effective retention of volatiles in the Earth, perhaps due to shielding effects from a proto-atmosphere, a stronger gravity field than on the Moon, or differences in mantle oxidation state; or (ii) replenishment of volatiles and moderately volatile elements to the Earth after formation of the Moon (§5c).
Figure 6.
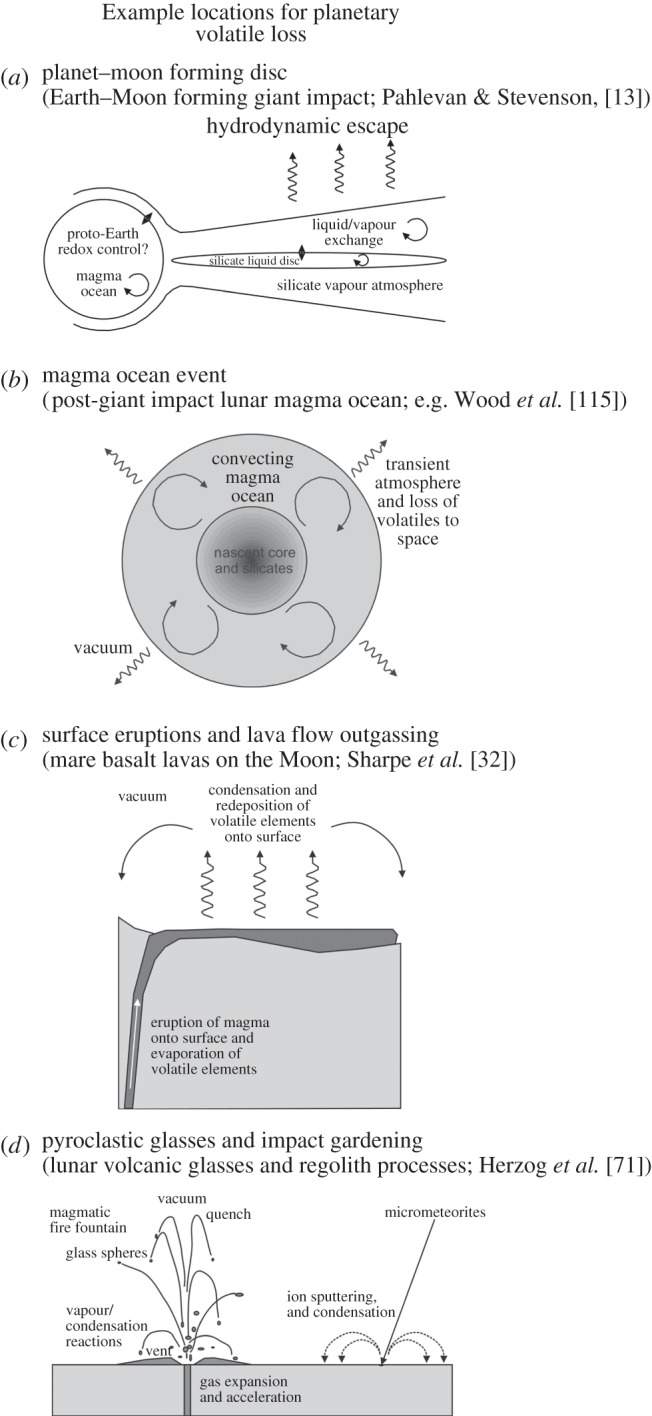
Illustrations of some of the possible conditions under which evaporation/condensation of moderately volatile elements can occur during planet formation and differentiation processes, with examples from the Moon: (a) a giant impact or high-temperature Earth–Moon forming event; (b) a lunar magma ocean event and prior to extensive anorthositic crust formation; (c) localized degassing of mare basalts on the lunar surface more than 1 Ga after initial lunar differentiation; (d) during evaporation in a magmatic fire fountain (cf. pyroclastic glass beads) and from impact gardening processes subsequent to lunar differentiation. Condensation of isotopically light vapours is expected as a consequence of events in (c) and (d), and possibly (b).
The current lack of resolution in models for planetary oxidation events restricts quantitative constraints [88], but oxidation state could effectively control the stability of mineral species that retain moderately volatile elements. For example, a difference in oxidation state between the proto-Earth and silicate liquid disc could explain the higher FeO contents in the present-day Moon relative to the terrestrial mantle [16]. For Zn, there is evidence from chondrite meteorites for the control of volatility through oxidation state. Enstatite chondrites, which are more reduced than ordinary or carbonaceous chondrites, show a trend of increasing δ66Zn with decreasing Zn abundance, and are consistent with evaporative fractionation [73]. With more reducing conditions from carbonaceous to enstatite chondrites, Zn can be preferentially partitioned into sulfides (or chlorides) relative to silicates [35,73]. On reduced bodies, such as the Moon, desulfidation and dechlorination will also lead to pronounced Zn loss and Zn isotopic fractionation. Depending on speciation, this mechanism may also explain why some moderately volatile elements, like K, do not appear to show strong isotopic fractionation between terrestrial and lunar materials. Oxidation of the Earth's mantle could have been triggered by sequestration of the impactor's core into the terrestrial core, or through the sudden increase in size of the Earth from accretion of the Moon-forming impactor, increasing generation of Mg-rich perovskite at the base of a terrestrial magma ocean, forcing disproportionation of ferrous iron into ferric iron plus metal [88,89]. On the other hand, new high-pressure elemental partitioning experiments have shown that the Earth's core could have been formed under rather oxidizing conditions [62].
There are some problems with a model of the Earth retaining volatile elements preferentially to the Moon during a giant impact event. Mars and differentiated asteroidal parent bodies (howardite–eucrite–diogenites (HED) or angrite meteorites) do not appear to have suffered giant impact events, yet the latter are more volatile-depleted than the Earth, while the former is not. It is also unclear how effective retention or loss of volatiles would be between a proto-Earth and silicate liquid disc. Pahlevan & Stevenson [13], in their analysis of this type of system, estimated equilibration times for oxygen on time scales of hundreds to thousands of years; moderately volatile element equilibration would presumably have been even more rapid [84]. It therefore seems likely that moderately volatile elements could not be effectively fractionated between different portions of the system following a giant impact, and that preferential replenishment of volatile and moderately volatile elements would be required in order to have a volatile-poor Moon relative to the Earth. However, it is also important to note that no studies have considered the subsequent volatile evolution of a planetary disc, so volatile depletion in giant impact disc environments remains an open question.
(c). A role for stochastic late accretion?
The elevated volatile contents of the Earth relative to the Moon can potentially be explained by replenishment of these elements from late accretion after the giant impact, a model that has been used to explain the inventory of volatiles in the present-day Earth [16,90–92]. A schematic illustration of how this would work for Zn is shown in figure 4. Depletion of volatile elements, such as Zn, occurs through evaporation during the giant impact and, subsequently, volatile-element-enriched ‘chondritic’ materials are accreted preferentially to the Earth, relative to the Moon, by post-core formation late accretion. This scenario is consistent with observations of chondrite-relative highly siderophile element (HSE: Os, Ir, Rh, Rh, Pt, Pd, Re, Au) abundances in the mantles and mantle products of the Earth, the Moon and Mars [61,93]. In this scenario, the approximately 20–40 times lower HSE abundances of the lunar mantle [18], compared with the Earth [94], can be explained by stochastic delivery of proportionally massive impactors to the Earth and the Moon [85].
Using estimated lunar (δ66Zn=+1.4‰; 2 ppm Zn) and terrestrial (δ66Zn=+0.27‰; 37 ppm Zn) bulk Zn compositions (table 1), approximately 12% late accretion addition of Zn is required to explain the differences observed between the Earth and the Moon by late accretion. These calculations, using concentrations estimated partly from basaltic melt products from the Earth and the Moon, assume identical partitioning of Zn from the mantle into the melt. Employing higher Zn contents in the bulk Moon proportionally reduces the amount of late accretion mass required (approx. 5.5% at 20 ppm Zn in the Moon), but cannot replicate the terrestrial Zn isotope composition. The estimated amount of mass of impactor material from Zn (approx. 12%) contrasts with that required for Pb (approx. 5%) [92] and HSE (0.5% or more) [93] abundances in the Earth's mantle and at first sight suggests that late accretion cannot be responsible for volatile distributions between the Earth and the Moon.
Table 1.
Escape velocities, K/U, Rb/Sr and Zn isotope and mantle abundance estimates for some planets and asteroids.
| planet/asteroid | escape velocity (km s−1) [95] | (K/U)/1000 | Rb/Sr | δ66Zn(‰) | ±2σ | Zn estimated mantle conc (ppm) | sources |
|---|---|---|---|---|---|---|---|
| Earth | 11.19 | 11.8 | 0.30 | 0.28 | 0.05 | 37 | [17,40] |
| Venus | 10.36 | ? | ? | ? | — | ? | |
| Mars | 5.03 | 16.4 | 0.070 | 0.25 | 0.03 | 66 | [34] |
| Mercury | 4.25 | ? | ? | ? | — | ? | |
| Moon | 2.38 | 3.0 | 0.009 | 1.4 | 0.1 | 2 | [16,34,75] |
| EPB | 0.36 | 3.7 | 0.003 | 6.2 | — | — | [36] |
| APB | <0.1 | 0.6 | 0.001 | — | — | — | estimated from [96] |
| CI chondrite | — | 74.3 | 0.426 | 0.25 | 0.05 | 310 | [17,35] |
Albarède et al. [92] have investigated a physical mechanism to explain the different apparent proportions of elements such as Zn, Pb and the HSE added to the Earth from late accretion. Models show that massive impactors involved in late accretion would have experienced partial vaporization [97], leading to a physical process of separation of elements according to phases that they concentrate into. For example, partial vaporization of a large impactor would lead to all of the lithophile and volatile elements being retained in the silicate mantle or resulting vapour cloud. In contrast, the HSE and Fe would mostly pool and sequester into the Earth's core, with some fraction of the HSE also being retained in the vapour cloud that would subsequently rain down on the Earth's surface, imparting a ‘late accretion’ signature (figure 7). Albarède et al. [92] argued that, during post-core formation accretion, partial vaporization could lead to the proportionally larger amounts of late accretionary mass required for volatiles, moderately volatile elements and Pb relative to the HSE. The current inventory of the HSE (approx. 0.5% late accretion addition) relative to Pb (approx. 5%) or Zn (approx. 12%) constrains the hypothetical vaporization ratios at 1:10 to 1:25. Furthermore, Albarède et al. [92] argued that this form of model would not violate W isotope constraints, because much of the impactor W would be segregated into the terrestrial core, along with the HSE. Models of addition of CI chondrite material to a present-day Moon composition for Zn show that simple mixing does not yield a present-day terrestrial composition (figure 7b). However, a 1:10 mixing model, incorporating accretion and partial vaporization of massive impactors, models the present-day composition of the terrestrial mantle quite well.
Figure 7.
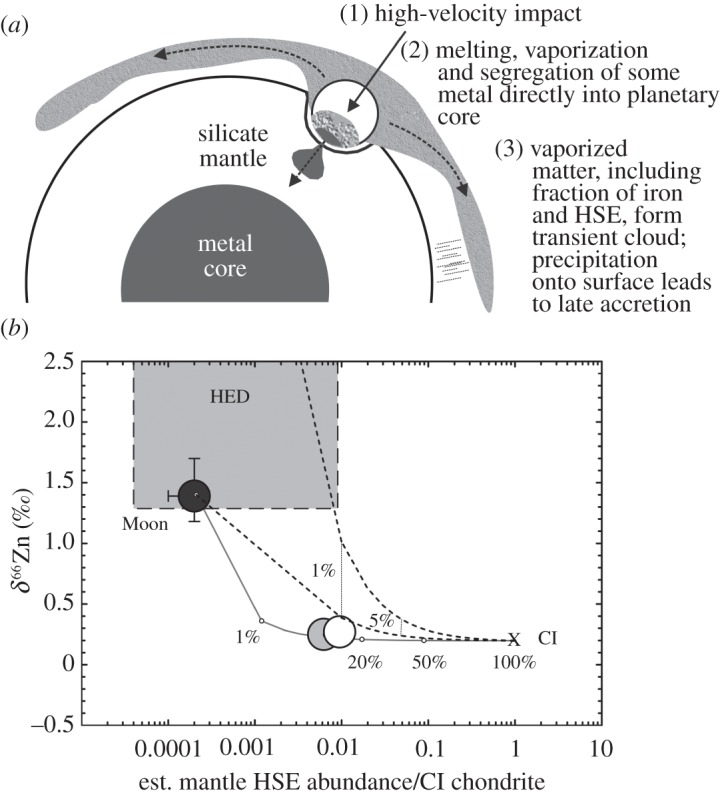
Impact vaporization as a process to explain differential proportions of volatile and siderophile late accretion. (a) Schematic diagram of an impactor striking a planet, partially vaporizing and forming a globally shrouding cloud enriched in all elements with concomitant differentiation of the melted fraction of the impactor into a metal fraction (removing the HSE) and a silicate melt fraction (containing lithophile elements). A similar model has been proposed to explain the different proportions of volatiles and siderophiles apparently added to the Earth after differentiation and metal–silicate equilibration [92]. (b) Zinc isotope compositions versus total HSE abundances for the Moon, the Earth, Mars, CI chondrites (data from [34,61]) and HED meteorites (data from [36,98] and J.M.D.D, 2010, unpublished data). Dashed mixing lines are for a eucrite composition (δ66Zn=+6‰; HSE abundance=0.0001×CI chondrite) and a lunar composition (δ66Zn=+1.4‰; HSE abundance=0.0001×CI chondrite) with CI chondrite. The solid grey line is for mixing with a lunar composition with CI chondrite, but assuming partitioning of HSE from volatiles like Zn using an impact vaporization scenario as depicted in (a) and assuming a 1:10 ratio of vaporized (step 3) to segregated (step 2) HSE.
A model of volatile element loss during giant impact, followed by stochastic late accretion of volatiles to the Earth and the Moon, has significant deficiencies. It is not clear how reasonable 5 to 10% or more late accretion for Pb and volatile elements is after the giant impact, given that the Earth–Moon forming event is widely considered the last major accretion event [2]. Nor is it known if the giant impact was a global ‘clearing house’ event for the HSE in the Earth. Understanding if both the Earth and the Moon were systematically depleted in volatile and highly siderophile elements during their formation is a crucial constraint for assessing whether a late accretion model for volatile enrichment can work. Scenarios that use a high ice/rock ratio (e.g. comets) could help to reduce the mass balance problem, but cometary material does not seem consistent with volatile element isotopic data [91]. The similar estimated HSE abundances of the Earth and Mars [61] also require explanation, given the large differences in timing of differentiation of the two bodies [99]. Recent work to constrain the timing of late accretion to the HED parent body(ies) has shown rapid addition, within 2–3 Ma, and that late accretion is simply the result of cessation of core formation on differentiated planetary bodies [98]. Thus, why Mars, whose core probably formed and ceased growth more rapidly than for the Earth [5,99], would have similar estimated HSE abundances in its mantle to the Earth, remains an outstanding science question. Given the discussion above, a giant impact and/or late accretion scenario to explain moderately volatile element distributions in the Earth and the Moon remains unconstrained, and alternative explanations need to be sought.
(d). Local eruption processes?
The large range in δ37Cl ratios measured for lunar rocks possibly indicates local eruption processes causing evaporation and volatile loss [32] (figure 6c,d). Independent evidence also exists that local eruption processes can lead to evaporation of moderately volatile elements and so volatile depletion in the Moon could reflect late-stage magmatic processes. For example, elevated volatile and siderophile abundances are surface-correlated in some lunar pyroclastic glass beads [63,100–106]. These observations can be reconciled by deposition of volatiles (e.g. Zn, S, Cl, F, Pb) occurring upon quenching of the beads within the associated vapour jet that expelled them, reduction of FeO to Fe-rich metal phases at the surface of the glass beads [107] and fractionation of Zn isotopes [71].
Localized eruption processes and impact gardening are important for partial condensation of isotopically light S, Cl, K and Zn on the lunar surface. For example, some mare basalts have δ66Zn<0.5‰ (e.g. 10017 and 12018) [34,75], and have higher Zn abundances (average=4.2 ppm) than the majority (80%) of mare basalts (average=1.7 ppm). Previously, light Zn isotope enrichment in pyroclastic glass beads has been attributed to Zn evaporation during fire fountaining, with condensation of isotopically light Zn on the surfaces of the ejected beads [70,75], as also proposed for 32S enrichments [104], and consistent with elevated Zn concentrations on their exteriors [63,71]. Vapour loss should increase the δ66Zn value of the residual magma, and this process can be modelled assuming exhalative evaporation of Zn as ZnCl2 on the Moon [63], versus for the Earth, where Zn is evaporated as Zn2+ [108], with essentially no fractionation between vapour and melt (α=1). Paniello et al. [34] showed that Rayleigh fractionation calculations can explain isotopically light vapour Zn isotope compositions conforming to those measured in 12018, 10017 and pyroclastic glass beads through evaporation and re-deposition of ZnCl2 from lunar melts (e.g. figure 2). A similar process would take place during impact gardening, with sputtering and re-deposition of isotopically light Zn on the lunar surface. We therefore exclude mare basalts with isotopically light Zn values because they are likely to reflect post-crystallization contamination at the lunar surface.
Both high- and low-Ti mare basalts with low Zn concentrations have δ66Zn of 1.4±0.3‰ [34,36,75]. The identical Zn isotopic compositions of low- and high-Ti mare basalts indicate identical processes acting on both basalt types, despite conspicuously differing crystallization and cooling histories and lunar mantle sources. Cooling and crystallization rates for mare basalts vary dramatically [109–111], with evidence for quenched and coarse-grained basalts at individual Apollo sites (see fig. 2 in [65], for an example at the Apollo 15 site). Additionally, low-Ti mare basalts are the predominant form of basalt exposed on the lunar surface (approx. 90%; [112]), and their prevalence over high-Ti mare basalts is consistent with magma ocean crystallization models that indicate that more than 78% of the lunar mantle would be dominated by olivine and orthopyroxene, whereas ilmenite-rich cumulates formed after 95% crystallization [113]. If localized eruption processes were the cause of Zn isotopic variations in lunar mare basalts, correlations might be expected between crystallization history or mantle source composition, yet the δ66Zn values seem invariant to these parameters. Furthermore, our reanalysis of published S, Cl, K and Zn isotope data (cf. figure 3) suggests that a large-scale global differentiation process, rather than local eruption processes, is a far more likely cause of the systematic depletion of these elements in the sources of mare basalts.
(e). Lunar magma ocean differentiation
The distinct geochemical traits of mare basalts and other lunar rocks underpin the concept of a lunar magma ocean to account for the plagioclase (Eu)-enriched crust overlying a plagioclase (Eu)-depleted mare basalt source region [114,115]. This form of ‘planet-scale’ melting and differentiation is probably an inevitable consequence of planetary differentiation, even on small bodies such as angrite or eucrite meteorites [116]. The chronology of events is poorly constrained [6], but the composition of mare basalts is consistent with derivation from mantle sources with different proportions of ilmenite [113]. The lunar magma ocean hypothesis predicts mineralogical variations in lunar cumulate reservoirs and in the source regions of low- and high-Ti mare basalts. Such features can also explain the significant stable O–Mg–Fe isotope variations measured for lunar basalts [8,117,118]. These studies have shown that, from existing knowledge of high-temperature mineral–melt partition coefficients, magmas generated from sources with variable modal mineralogies and/or mineral-source compositions would have fractionated stable isotope compositions.
Magma ocean differentiation offers an alternative explanation for the volatile element distribution and isotopic variations observed for the Moon relative to the Earth, Mars and other differentiated bodies (figure 6b). Outgassing of a magma ocean would lead to evaporation of volatile elements and the systematic and progressive heavy-isotope enrichment in the condensate relative to the vapour. An important aspect of outgassing of a magma ocean is that perfect volatility behaviour would not be expected, for three principal reasons. First, the duration of a lunar magma ocean phase is important [116], prior to generation of a permanent crust, which would presumably impede volatile element loss. It is possible that the shorter duration of a lunar magma ocean prior to crust formation, relative to more prolonged crustal growth above a magma ocean on smaller planetesimals, can partly explain the greater enrichment of δ66Zn in some unbrecciated eucrites [36]. Second, for loss of volatiles, atoms would be required to exceed the escape velocity, or would need to be lost during a process such as atmospheric blow-off, and this process may inhibit ideal Rayleigh distillation of moderately volatile stable isotopes. On small bodies (less than 500 km diameter), such as differentiated planetesimals, this would be a trivial issue, but the escape velocity of the present-day Moon is non-trivial (table 1) and an additional mechanism (e.g. vapour partial pressure above the magma ocean and formation and subsequent erosion of a transient atmosphere) would become critical. Third, evaporation of volatile elements would be expected to follow non-ideal Rayleigh fractionation. This is because only the upper portions of a magma ocean would be exposed to vacuum, and convection in the magma ocean would be a rate-limiting step in the efficiency of volatile loss in a lunar magma ocean.
There are a number of consequences of non-ideal Rayleigh fractionation and volatile loss in a lunar magma ocean that appear to fit most closely with the available volatile element data. First, ideal Rayleigh distillation behaviour would not be predicted, and this is certainly not observed (figure 2). Second, early-formed cumulates would be expected to record less volatile loss than later-formed cumulates, at least prior to the formation of the lunar anorthositic crust. Some deep, early-formed cumulates could retain a higher volatile inventory and more chondritic volatile stable isotope ratios and such sources could potentially fit with the deep mode of origin of lunar pyroclastic glass beads [119], their lower μ-values compared with mare basalts [27] and their high apparent water contents [22]. A conceptual model of magma ocean outgassing and volatile loss can explain some of the observed values for moderately volatile element abundances and stable isotope ratios (figure 8). Further work is required, however, to test the model (e.g. Zn isotope analyses of high-Mg suite rocks, which should be isotopically heavy) and to assess mechanisms that would explain the mass balance of light isotope depletion of moderately volatile elements in the Moon (e.g. transient atmospheric erosion or blow-off; storage of light isotopes of volatile elements in the lunar crust or regolith).
Figure 8.

Conceptual model of volatile outgassing during a lunar magma ocean, and the evolution of Zn isotopes. (a) Immediately after lunar accretion, an interior forms that does not witness interaction with the lunar surface, leading to a volatile-rich (Earth-like?) primitive composition. Convection and inefficient outgassing of the overlying magma ocean lead to formation of less (b) and more (c) volatile-depleted lower and upper lunar magma ocean cumulates (LLMOC, ULMOC), respectively. Outgassing and volatile depletion are eventually terminated by formation of the plagioclase-rich flotation crust. Continued fractional crystallization of the lunar magma ocean leads to formation of ilmenite and a KREEP-rich layers with the most volatile-depleted signatures. (d) Cumulate overturn [119] leads to formation of high-Ti mare basalt and high-Ti lunar pyroclastic glass (LPG) source regions. Partial melting of volatile-depleted (LLMOC and ULMOC) cumulates to form mare basalts and of primitive lunar interior to form LPG can explain the volatile abundance differences estimated for their source regions [22,34]. If high-Mg suite rocks form from LLMOC, a testable hypothesis is that they should have isotopically heavier δ66Zn. Conversely, ferroan anorthosites should have δ66Zn ranging from positive to negative values in response to volatile loss in their source region and later condensation (cf. figure 6d).
6. Conclusion and outlook
Available volatile element stable isotope variations indicate that a global-scale evaporation event was the most likely cause of volatile depletion in the Moon, and by extension in other fully differentiated and airless bodies. Despite this evidence, a number of fundamental science problems remain to be resolved in order to improve understanding of volatile element depletion:
(1) Additional high-quality S, Cl, K and Zn isotope data are needed for the Moon, the Earth and other planetary bodies, while elements with different volatilities, atomic masses or behaviours (e.g. Cu, Rb, Ga, Cd) would place additional constraints on some of the proposed models for planetary volatile element depletion.
(2) Analysis of both bulk rock samples and mineral phases will be important for understanding volatile element depletion and the siting of these elements within mantle-derived magmatic rocks from differentiated planetary bodies. Comparison of volatile and moderately volatile isotopic data from mineral phases with bulk rocks needs to be approached with caution, particularly if the mineral phase is a late-stage crystallization product (e.g. Cl in apatite).
(3) Further study of surface-correlated effects in the pyroclastic glass beads and careful assessment of surface coatings on the exteriors of lunar rocks are required in order to more fully understand localized eruptive processes. Apollo samples were all collected from the lunar regolith, so isotopically light condensation of moderately volatile elements from eruption and impact gardening could hamper resolution of magmatic and mantle signatures.
(4) The relationship between planetary escape velocity and volatile elements may be important in assessing degassing and volatile loss in a magma ocean. Current evidence suggests that the lunar pyroclastic glass eruptions led to condensation of isotopically light Zn [71], implying that lunar escape velocities were not effectively exceeded by this process. Similarly, if escape velocities were not exceeded during the earliest magma ocean phases, then an ephemeral atmosphere in a low-gravity environment followed by erosion of this atmosphere would be required. Rapid erosion of an atmosphere rich in isotopically light vapour may offer an explanation for the loss of volatiles from a lunar magma ocean.
(5) Refined experimental data will aid in understanding diffusion-limited regimes relevant to volatile loss [77]. Controversy surrounds the expected behaviour of volatiles into the hard vacuum at the lunar surface [120], but current evidence suggests inefficient isotopic fractionation of volatile stable isotopes. Further experimental and theoretical work is also required to understand the behaviour of moderately volatile elements with varying speciation. This form of approach is crucial for understanding the role of putative vapour–liquid Moon-forming discs after a giant impact scenario [84].
(6) More direct comparison of moderately volatile and highly volatile elements and associated species, such as H2O and OH, are desirable, given the competing evidence for a relatively ‘wet’ [22,23] or ‘dry’ Moon [32,34]. Did portions of the Moon retain a higher volatile element inventory in a magma ocean (figure 8), or were the source materials that formed the Moon ubiquitously volatile-depleted during a giant impact? If so, can heterogeneous addition of late accretion materials account for ‘wet’ regions of the Moon as sampled by lunar pyroclastic glasses?
Volatile depletion in planets remains a major challenge for understanding planet formation processes and the cosmochemistry of the earliest Solar System. Mare basalts from the Moon come from mantle source regions that were volatile-depleted relative to the source mantle of terrestrial or martian basalts and require a planetary volatile depletion separate from nebular volatile depletions observed in chondrites. It remains to be resolved what the exact process(es) of volatile depletion was (were) for the Moon, but the challenges associated with addressing this problem remain some of the most important in planetary science.
Acknowledgements
The authors thank the organizers of the Royal Society Origin of the Moon conference, D. Stevenson and A. Halliday, for the invitation to participate. We thank R. W. Carlson and T. Elliott for constructive review comments, A. Halliday for editorial handling, and F. Albarède, J. Dhaliwal, B. Fegley, T. Grove, S. Mukhopadhyay, Z. Sharp and J. Wasson for helpful suggestions.
Funding statement
This work was supported by NASA LASER (NNX11AG34G) and Cosmochemistry (NNX12AH75G) grants to J.M.D.D. and by a Chair of Excellence from the Sorbonne Paris Cité and NASA Cosmochemistry and Exobiology grants to F.M.
References
- 1.Canup RM. 2012. Forming a Moon with an Earth-like composition via a giant impact. Science 338, 1052–1055. ( 10.1126/science.1226073) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cuk M, Stewart ST. 2012. Making the Moon from a fast-spinning Earth: a giant impact followed by resonant despinning. Science 338, 1047–1052. ( 10.1126/science.1225542) [DOI] [PubMed] [Google Scholar]
- 3.Reufer A, Meier MMM, Benz W, Wieler R. 2012. A hit-and-run giant impact scenario. Icarus 221, 296–299. ( 10.1016/j.icarus.2012.07.021) [DOI] [Google Scholar]
- 4.Touboul M, Kleine T, Bourdon B, Palme H, Wieler R. 2007. Late formation and prolonged differentiation of the Moon inferred from W isotopes in lunar metals. Nature 450, 1206–1209. ( 10.1038/nature06428) [DOI] [PubMed] [Google Scholar]
- 5.Halliday AN. 2008. A young Moon-forming giant impact at 70–110 million years accompanied by late-stage mixing, core formation and degassing of the Earth. Phil. Trans. R. Soc. A 366, 4163–4181. ( 10.1098/rsta.2008.0209) [DOI] [PubMed] [Google Scholar]
- 6.Borg LE, Connelly JN, Boyet M, Carlson RW. 2011. Chronological evidence that the Moon is either young or did not have a global magma ocean. Nature 477, 70–72. ( 10.1038/nature10328) [DOI] [PubMed] [Google Scholar]
- 7.Wiechert U, Halliday AN, Lee DC, Snyder GA, Taylor LA, Rumble D. 2001. Oxygen isotopes and the Moon-forming giant impact. Science 294, 345–348. ( 10.1126/science.1063037) [DOI] [PubMed] [Google Scholar]
- 8.Spicuzza MJ, Day JMD, Taylor LA, Valley JW. 2007. Oxygen isotope constraints on the origin and differentiation of the Moon. Earth Planet. Sci. Lett. 253, 254–265. ( 10.1016/j.epsl.2006.10.030) [DOI] [Google Scholar]
- 9.Hallis LJ, Anand M, Greenwood RC, Miller MF, Franchi IA, Russell SS. 2010. The oxygen isotope composition, petrology and geochemistry of mare basalts: evidence for large-scale compositional variation in the lunar mantle. Geochim. Cosmochim. Acta 74, 6885–6899. ( 10.1016/j.gca.2010.09.023) [DOI] [Google Scholar]
- 10.Zhang J, Dauphas N, Davis AM, Leya I, Fedkin A. 2012. The proto-Earth as a significant source of lunar material. Nat. Geosci. 5, 251–255. ( 10.1038/ngeo1429) [DOI] [Google Scholar]
- 11.Armytage RMG, Georg RB, Williams HM, Halliday AN. 2012. Silicon isotopes in lunar rocks: implications for the Moon's formation and the early history of the Earth. Geochim. Cosmochim. Acta 77, 504–514. ( 10.1016/j.gca.2011.10.032) [DOI] [Google Scholar]
- 12.Fitoussi C, Bourdon B. 2012. Silicon isotope evidence against an enstatite chondrite Earth. Science 335, 1477–1480. ( 10.1126/science.1219509) [DOI] [PubMed] [Google Scholar]
- 13.Pahlevan K, Stevenson DJ. 2007. Equilibration in the aftermath of the lunar-forming giant impact. Earth Planet. Sci. Lett. 262, 438–449. ( 10.1016/j.epsl.2007.07.055) [DOI] [Google Scholar]
- 14.Lodders K. 2003. Solar System abundances and condensation temperatures of the elements. Astrophys. J. 591, 1220–1247. ( 10.1086/375492) [DOI] [Google Scholar]
- 15.Dreibus G, Wanke H. 1980. The bulk composition of the eucrite parent asteroid and its bearing on planetary evolution. Z. Naturforsch. 35a, 204–216 See http://zfn.mpdl.mpg.de/data/Reihe_A/35/ZNA-1980-35a-0204.pdf [Google Scholar]
- 16.O'Neill HStC. 1991. The origin of the Moon and the early history of the Earth—a chemical model. Part 1: The Moon. Geochim. Cosmochim. Acta 55, 1135–1157. ( 10.1016/0016-7037(91)90168-5) [DOI] [Google Scholar]
- 17.McDonough WF, Sun S-S. 1995. The composition of the Earth. Chem. Geol. 120, 223–254. ( 10.1016/0009-2541(94)00140-4) [DOI] [Google Scholar]
- 18.Day JMD, Pearson DG, Taylor LA. 2007. Highly siderophile element constraints on accretion and differentiation of the Earth–Moon system. Science 315, 217–219. ( 10.1126/science.1133355) [DOI] [PubMed] [Google Scholar]
- 19.Davis A. 2006. Volatile evolution and loss. In Meteorites and the early solar system II (eds Lauretta DS, McSween HY.), pp. 295–307. Tucson, AZ:University of Arizona Press [Google Scholar]
- 20.Wolf R, Anders E. 1980. Moon and Earth: compositional differences inferred from siderophiles, volatiles, and alkalis in basalts. Geochim. Cosmochim. Acta 44, 2111–2124. ( 10.1016/0016-7037(80)90208-2) [DOI] [Google Scholar]
- 21.Jones JH, Palme H. 2000. Geochemical constraints on the origin of the Earth and Moon. In Origin of the Earth and Moon (eds Canup RM, Righter K.), pp. 197–216. Tucson, AZ:University of Arizona Press [Google Scholar]
- 22.Hauri EH, Weinreich T, Saal A, Rutherford M, Van Orman JA. 2011. High pre-eruptive water contents preserved in lunar melt inclusions. Science 333, 213–215. ( 10.1126/science.1204626) [DOI] [PubMed] [Google Scholar]
- 23.McCubbin FM, Steele A, Hauri EH, Nekvasil H, Yamashita S, Hemley RJ. 2010. Nominally hydrous magmatism on the Moon. Proc. Natl Acad. Sci. USA 107, 11223–11228. ( 10.1073/pnas.1006677107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Halliday AN, Porcelli D. 2001. In search of lost planets—the paleocosmochemistry of the inner Solar System. Earth Planet. Sci. Lett. 192, 545–559. ( 10.1016/S0012-821X(01)00479-4) [DOI] [Google Scholar]
- 25.Carlson RW, Lugmair GW. 1988. The age of ferroan anorthosite 60025: oldest crust on a young Moon. Earth Planet. Sci. Lett. 90, 119–130. ( 10.1016/0012-821X(88)90095-7) [DOI] [Google Scholar]
- 26.McCulloch MT. 1994. Primitive 87Sr/86Sr from an Archean barite and conjecture on the Earth's age and origin. Earth Planet. Sci. Lett. 126, 1–13. ( 10.1016/0012-821X(94)90238-0) [DOI] [Google Scholar]
- 27.Tatsumoto M, Premo WR, Unruh DM. 1987. Origin of lead from green glass of Apollo 15426: a search from primitive lunar lead. J. Geophys. Res. 92, E361–E371. ( 10.1029/JB092iB04p0E361) [DOI] [Google Scholar]
- 28.Papanastassiou DA, Wasserburg GJ. 1978. Strontium isotopic anomalies in the Allende meteorite. Geophys. Res. Lett. 5, 595–598. ( 10.1029/GL005i007p00595) [DOI] [Google Scholar]
- 29.Moynier F, Day JMD, Okui W, Yokoyama T, Bouvier A, Walker RJ, Podosek FA. 2012. Planetary-scale strontium isotopic heterogeneity and the age of volatile depletion of early Solar System materials. Astrophys. J. 758, 45 ( 10.1088/0004-637X/758/1/45) [DOI] [Google Scholar]
- 30.Hans U, Kleine T, Bourdon B. 2013. Rb–Sr chronology of volatile depletion in differentiated protoplanets: BABI, ADOR and ALL revisited. Earth Planet. Sci. Lett. 374, 204–214. ( 10.1016/j.epsl.2013.05.029) [DOI] [Google Scholar]
- 31.Humayun M, Clayton RN. 1995. Precise determination of the isotopic composition of potassium: application to terrestrial rocks and lunar soils. Geochim. Cosmochim. Acta 59, 2115–2130. ( 10.1016/0016-7037(95)00131-X) [DOI] [Google Scholar]
- 32.Sharp ZD, Shearer CK, McKeegan KD, Barnes JD, Wang YQ. 2010. The chlorine isotope composition of the Moon and implications for an anhydrous mantle. Science 329, 1050–1053. ( 10.1126/science.1192606) [DOI] [PubMed] [Google Scholar]
- 33.Sharp ZD, Barnes JD, Fischer TP, Halick M. 2010. An experimental determination of chlorine isotope fractionation in acid systems and application to volcanic fumeroles. Geochim. Cosmochim. Acta 74, 264–273. ( 10.1016/j.gca.2009.09.032) [DOI] [Google Scholar]
- 34.Paniello RC, Day JMD, Moynier F. 2012. Zinc isotopic evidence for the origin of the Moon. Nature 490, 376–379. ( 10.1038/nature11507) [DOI] [PubMed] [Google Scholar]
- 35.Luck J-M, Ben Othman D, Albarède F. 2005. Zn and Cu isotopic variations in chondrites and iron meteorites: early solar nebula reservoirs and parent-body processes. Geochim. Cosmochim. Acta 69, 5351–5363. ( 10.1016/j.gca.2005.06.018) [DOI] [Google Scholar]
- 36.Paniello RC, Moynier F, Beck P, Barrat J-A, Podosek F, Pichat S. 2012. Zinc isotopes in HEDs: clues to the formation of 4-Vesta and the unique composition of Pecora Escarpment 82502. Geochim. Cosmochim. Acta 86, 76–87. ( 10.1016/j.gca.2012.01.045) [DOI] [Google Scholar]
- 37.Chen H, Savage PS, Teng F-Z, Helz RT, Moynier F. 2013. Zinc isotopic fractionation during magmatic differentiation and the isotopic composition of the bulk Earth. Earth Planet. Sci. Lett. 369–370, 34–42. ( 10.1016/j.epsl.2013.02.037) [DOI] [Google Scholar]
- 38.Anders E, Grevesse N. 1989. Abundances of the elements: meteoritic and solar. Geochim. Cosmochim. Acta 53, 197–214. ( 10.1016/0016-7037(89)90286-X) [DOI] [Google Scholar]
- 39.Palme H, Jones A. 2003. Solar system abundances of the elements. In Treatise on geochemistry (eds Holland HD, Turekian KK.), vol. 1, Meteorites, comets, and planets (ed. Davis AM.), pp.41–61. Amsterdam, The Netherlands: Elsevier; (doi:1016/-043751-6/01060-4) [Google Scholar]
- 40.Barrat JA, Zanda B, Moynier F, Bollinger C, Liorzou C, Bayon G. 2012. Geochemistry of CI chondrites: major and trace elements, and Cu and Zn isotopes. Geochim. Cosmochim. Acta 85, 79–92. ( 10.1016/j.gca.2011.12.011) [DOI] [Google Scholar]
- 41.Palme H, Larimer JW, Lipschutz ME. 1988. Moderately volatile elements. In Meteorites and the early Solar System (eds Kerridge JF, Matthews MS.), pp. 436–460. Tucson, AZ:University of Arizona Press [Google Scholar]
- 42.Larimer JW. 1988. The cosmochemical classification of the elements. In Meteorites and the early Solar System (eds Kerridge JF, Matthews MS.), pp. 375–389. Tucson, AZ:University of Arizona Press [Google Scholar]
- 43.Urey HC. 1947. The thermodynamic properties of isotopic substances. J. Chem. Soc. (London) 1947, 562–581. ( 10.1039/JR9470000562) [DOI] [PubMed] [Google Scholar]
- 44.Bigeleisen J, Mayer MG. 1947. Calculation of equilibrium constants for isotopic exchange reactions. J. Chem. Phys. 15, 261–267. ( 10.1063/1.1746492) [DOI] [Google Scholar]
- 45.Schauble EA. 2004. Applying stable isotope fractionation theory to new systems. Rev. Mineral. Geochem. 55, 65–111. ( 10.2138/gsrmg.55.1.65) [DOI] [Google Scholar]
- 46.Maréchal C, Telouk P, Albarède F. 1999. Precise analysis of copper and zinc isotopic compositions by plasma-source mass spectrometry. Chem. Geol. 156, 251–273. ( 10.1016/S0009-2541(98)00191-0) [DOI] [Google Scholar]
- 47.Humayun M, Cassen P. 2000. Processes determining the volatile abundances of the meteorites and terrestrial planets. In Origin of the Earth and Moon (eds Canup RM, Righter K.), pp. 3–23. Tucson, AZ:University of Arizona Press [Google Scholar]
- 48.O'Neil JR. 1986. Theoretical and experimental aspects of isotopic fractionation. Rev. Mineral. 16, 1–40. [Google Scholar]
- 49.Gibson EK, Chang S, Lennon K, Moore GW, Pearce GW. 1975. Sulfur abundances and distributions in mare basalts and their source magmas. In Proc. 6th Lunar Science Conf., Houston, TX, 17–21 March, pp. 1287–1301 New York, NY: Pergamon. [Google Scholar]
- 50.Kerridge JF, Kaplan IR, Petrowski C. 1975. Evidence for meteoritic sulfur in the lunar regolith. In Proc. 6th Lunar Science Conf., Houston, TX, 17–21 March, pp. 2151–2162 New York, NY: Pergamon. [Google Scholar]
- 51.Clayton RN, Mayeda TK, Hurd JM. 1974. Loss of oxygen, silicon, sulfur, and potassium from the lunar regolith. In Proc. 5th Lunar Science Conf., Houston, TX, 18–22 March, pp. 1801–1809 New York, NY: Pergamon. [Google Scholar]
- 52.Wing B, Farquhar J. In press Sulfur isotope homogeneity of lunar mare basalts. Geochim. Cosmochim. Acta. [Google Scholar]
- 53.Franz HB, et al. 2014. Isotopic links between atmospheric chemistry and the deep sulphur cycle on Mars. Nature 508, 364–368. ( 10.1038/nature13175) [DOI] [PubMed] [Google Scholar]
- 54.Labidi J, Cartigny P, Birck J-L, Assayag N, Bourrand JJ. 2012. Determination of multiple sulfur isotopes in glasses: a reappraisal of the MORB δ34S. Chem. Geol. 334, 189–198. ( 10.1016/j.chemgeo.2012.10.028) [DOI] [Google Scholar]
- 55.Labidi J, Cartigny P, Moriera M. 2013. Non-chondritic sulphur isotope composition of the terrestrial mantle. Nature 501, 208–211. ( 10.1038/nature12490) [DOI] [PubMed] [Google Scholar]
- 56.Richter FM, Janney PE, Mendybaev RA, Davis AM, Wadhwa M. 2007. Elemental and isotopic fractionation of type B CAI-like liquids by evaporation. Geochim. Cosmochim. Acta 71, 5544–5564. ( 10.1016/j.gca.2007.09.005) [DOI] [Google Scholar]
- 57.Sharp ZD, Barnes JD, Brearley AJ, Chaussidon M, Fisher TP, Kamenetsky VS. 2007. Chlorine isotope homogeneity of the mantle, crust and carbonaceous chondrites. Nature 446, 1062–1065. ( 10.1038/nature05748) [DOI] [PubMed] [Google Scholar]
- 58.Sharp ZD, Shearer CK, Agee CB, McKeegan KD. 2011. The chlorine isotope composition of Mars. In Proc. 42nd Lunar and Planetary Science Conf., The Woodlands, TX, 7–11 March, Abstract 2534. See http:www.lpi.usra.edu/meetings/lpsc2011/pdf/2534.pdf [Google Scholar]
- 59.Bonifacie M, Jendrzejewski N, Agrinier P, Humler E, Coleman M, Javoy M. 2008. The chlorine isotope composition of Earth's mantle. Science 319, 1518–1520. ( 10.1126/science.1150988) [DOI] [PubMed] [Google Scholar]
- 60.Bombardieri DJ, Norman MD, Kamenetsky VS, Danyushevsky LV. 2005. Major element and primary sulfur concentrations in Apollo 12 mare basalts: the view from melt inclusions. Meteorit. Planet. Sci. 40, 679–693. ( 10.1111/j.1945-5100.2005.tb00973.x) [DOI] [Google Scholar]
- 61.Day JMD. 2013. Hotspot volcanism and highly siderophile elements. Chem. Geol. 341, 50–74. ( 10.1016/j.chemgeo.2012.12.010) [DOI] [Google Scholar]
- 62.Siebert J, Badro J, Antonangeli D, Ryerson FJ. 2013. Terrestrial accretion under oxidizing conditions. Science 339, 1194–1197. ( 10.1126/science.1227923) [DOI] [PubMed] [Google Scholar]
- 63.Chou C-L, Boynton WV, Sundberg LL, Wasson JT. 1975. Volatiles on the surface of Apollo 15 green glass and trace-element distributions among Apollo 15 soils. In Proc. 6th Lunar Science Conf., Houston, TX, 17–21 March, pp. 1701–1727 New York, NY: Pergamon. [Google Scholar]
- 64.Day JMD, Taylor LA, Floss C, Patchen AD, Schnare DW, Pearson DG. 2006. Comparative petrology, geochemistry and petrogenesis of evolved, low-Ti lunar mare basalt meteorites from the La Paz icefield, Antarctica. Geochim. Cosmochim. Acta 70, 1581–1600. ( 10.1016/j.gca.2005.11.015) [DOI] [Google Scholar]
- 65.Schnare DW, Day JMD, Norman MD, Liu Y, Taylor LA. 2008. Laser-ablation ICP-MS study of Apollo 15 low-titanium olivine-normative and quartz-normative mare basalts. Geochim. Cosmochim. Acta 72, 2556–2572. ( 10.1016/j.gca.2008.02.021) [DOI] [Google Scholar]
- 66.Liu Y, Floss C, Day JMD, Hill E, Taylor LA. 2009. Petrogenesis of lunar mare basalt meteorite MIL 05035. Meteorit. Planet. Sci. 44, 261–284. ( 10.1111/j.1945-5100.2009.tb00733.x) [DOI] [Google Scholar]
- 67.Boyce JW, Guan Y, Treiman AH, Greenwood JP, Eiler JM, Ma C. 2013. Volatile components in the Moon: abundances and isotope ratios of Cl and H in lunar apatites. In Proc. 44th Lunar and Planetary Science Conf., The Woodlands, TX, 18–22 March, Abstract 2851. See http://www.lpi.usra.edu/meetings/lpsc2013/pdf/2851.pdf [Google Scholar]
- 68.Humayun M, Clayton RN. 1995. Potassium isotope geochemistry: genetic implications of volatile element depletion. Geochim. Cosmochim. Acta 59, 2115–2130. ( 10.1016/0016-7037(95)00131-X) [DOI] [Google Scholar]
- 69.Humayun M, Koeberl C. 2004. Potassium isotopic composition of Australasian tektites. Meteorit. Planet. Sci. 39, 1509–1516. ( 10.1111/j.1945-5100.2004.tb00125.x) [DOI] [Google Scholar]
- 70.Moynier F, Albarède F, Herzog G. 2006. Isotopic composition of zinc, copper, and iron in lunar samples. Geochim. Cosmochim. Acta 70, 6103–6117. ( 10.1016/j.gca.2006.02.030) [DOI] [Google Scholar]
- 71.Herzog GF, Moynier F, Albarède F, Berezhnoy AA. 2009. Isotopic and elemental abundances of copper and zinc in lunar samples, Zagami, Pele's hairs, and a terrestrial basalt. Geochim. Cosmochim. Acta 73, 5884–5904. ( 10.1016/j.gca.2009.05.067) [DOI] [Google Scholar]
- 72.Moynier F, Dauphas N, Podosek FA. 2009. Search for 70Zn anomalies in meteorites. Astrophys. J. Lett. 700, L92–L95. ( 10.1088/0004-637X/700/2/L92) [DOI] [Google Scholar]
- 73.Moynier F, Paniello RC, Gounelle M, Albarède F, Beck P, Podosek F, Zanda B. 2011. Nature of volatile depletion and genetic relationships in enstatite chondrites and aubrites inferred from Zn isotopes. Geochim. Cosmochim. Acta 75, 297–307. ( 10.1016/j.gca.2010.09.022) [DOI] [Google Scholar]
- 74.Moynier F, Beck P, Jourdan F, Yin Q-Z, Reimold U, Koeberl C. 2009. Isotopic fractionation of zinc in tektites. Earth Planet. Sci. Lett. 277, 482–489. ( 10.1016/j.epsl.2008.11.020) [DOI] [Google Scholar]
- 75.Kato C, Valdes M, Dhaliwal J, Day JMD, Moynier F. 2013. Origin of isotopically light Zn in lunar samples through vaporization and the Zn isotope composition of the lunar mantle. In American Geophysical Union Fall Meeting 2013 Abstract V33D-2788 [Google Scholar]
- 76.Esat TM. 1988. Physicochemical isotope anomalies. Geochim. Cosmochim. Acta 52, 1409–1424. ( 10.1016/0016-7037(88)90211-6) [DOI] [Google Scholar]
- 77.Xue S, Yu Y, Hewins RH, Hall GS, Herzog GF. 1996. Zinc losses from and zinc isotopic abundances in residues formed by heating of Zn, ZnO, ZeS, and Zn-doped silicate glass. In Proc. 27th Lunar and Planetary Science Conf., Houston, TX, 18–22 March, pp. 1461–1462 New York, NY: Pergamon. [Google Scholar]
- 78.Koeberl C. 1994. Tektite origin by hypervelocity asteroidal or cometary impact: target rocks, source craters, and mechanisms. In Large meteorite impacts and planetary evolution (eds Dressler BO, Grieve RAF, Sharpton VL.), Geological Society of America Special Paper, 293, pp. 133–152 [Google Scholar]
- 79.Wasson JT. 2003. Large aerial bursts: an important class of terrestrial accretionary events. Astrobiology 3, 163–179. ( 10.1089/153110703321632499) [DOI] [PubMed] [Google Scholar]
- 80.Herzog GF, Alexander CMO'D, Glass BP, Berger EL, Delaney JS. 2005. Potassium isotope fractionation in Australasian microtektites: evidence for evaporation and recondensation in a vapor plume. In Proc. 36th Lunar and Planetary Science Conf., League City, TX, 14–18 March, Abstract 1167. See http://www.lpi.usra.edu/meetings/lpsc2005/pdf/1167.pdf [Google Scholar]
- 81.Wombacher F, Rehkamper M, Mezger K, Munker C. 2003. Stable isotope compositions of cadmium in geological materials and meteorites determined by multiple-collector ICPMS. Geochim. Cosmochim. Acta 67, 4639–4654. ( 10.1016/S0016-7037(03)00389-2) [DOI] [Google Scholar]
- 82.Moynier F, Koeberl C, Beck P, Jourdan F, Telouk P. 2010. Isotopic fractionation of Cu in tektites. Geochim. Cosmochim. Acta 74, 799–807. ( 10.1016/j.gca.2009.10.012) [DOI] [Google Scholar]
- 83.Albarède F. 2004. The stable isotope geochemistry of copper and zinc. Rev. Mineral. Geochem. 55, 409–427. ( 10.2138/gsrmg.55.1.409) [DOI] [Google Scholar]
- 84.Visscher C, Fegley B. 2013. Chemistry of impact-generated silicate melt–vapor debris disks. Astrophys. J. 767, 12 ( 10.1088/2041-8205/767/1/L12) [DOI] [Google Scholar]
- 85.Bottke WF, Walker RJ, Day JMD, Nesvorny D, Elkins-Tanton L. 2010. Stochastic late accretion to Earth, the Moon, and Mars. Science 330, 1527–1530. ( 10.1126/science.1196874) [DOI] [PubMed] [Google Scholar]
- 86.Genda H, Abe Y. 2005. Enhanced atmospheric loss on protoplanets at the giant impact phase in the presence of oceans. Nature 433, 842–844. ( 10.1038/nature03360) [DOI] [PubMed] [Google Scholar]
- 87.Hartmann W, Davis D. 1975. Satellite-sized planetesimals and lunar origin. Icarus 24, 504–514. ( 10.1016/0019-1035(75)90070-6) [DOI] [Google Scholar]
- 88.Wade J, Wood BJ. 2005. Core formation and the oxidation state of the Earth. Earth Planet. Sci. Lett. 236, 78–95. ( 10.1016/j.epsl.2005.05.017) [DOI] [Google Scholar]
- 89.Frost DJ, Liebske C, Langenhorst F, McCammon CA, Trønnes RG, Rubie DC. 2014. Experimental evidence for the existence of iron-rich metal in the Earth's lower mantle. Nature 428, 409–412. ( 10.1038/nature02413) [DOI] [PubMed] [Google Scholar]
- 90.Albarède F. 2009. Volatile accretion history of the terrestrial planets and dynamic implications. Nature 461, 1227–1233. ( 10.1038/nature08477) [DOI] [PubMed] [Google Scholar]
- 91.Marty B. 2012. The origins and concentrations of water, carbon, nitrogen and noble gases on Earth. Earth Planet. Sci. Lett. 313–314, 56–66. ( 10.1016/j.epsl.2011.10.040) [DOI] [Google Scholar]
- 92.Albarède F, Ballhaus C, Blichert-Toft J, Lee C-T, Marty B, Moynier F, Yin Q-Z. 2013. Asteroidal impacts and the origin of terrestrial and lunar volatiles. Icarus 222, 44–52. ( 10.1016/j.icarus.2012.10.026) [DOI] [Google Scholar]
- 93.Walker RJ. 2009. Highly siderophile elements in the Earth, Moon and Mars: update and implications for planetary accretion and differentiation. Chem. Erde 69, 101–125. ( 10.1016/j.chemer.2008.10.001) [DOI] [Google Scholar]
- 94.Becker H, Horan MF, Walker RJ, Gao S, Lorand J-P, Rudnick RL. 2006. Highly siderophile element composition of the Earth's primitive upper mantle: constraints from new data on peridotite massifs and xenoliths. Geochim. Cosmochim. Acta 70, 4528–4550. ( 10.1016/j.gca.2006.06.004) [DOI] [Google Scholar]
- 95.Lodders K, Fegley B. 1998. The planetary scientists companion. Oxford, UK: Oxford University Press. [Google Scholar]
- 96.Riches AJV, Day JMD, Walker RJ, Simonetti A, Liu Y, Neal CR, Taylor LA. 2012. Rhenium–osmium isotope and highly-siderophile-element abundance systematics of angrite meteorites. Earth Planet. Sci. Lett. 353/354, 208–218. ( 10.1016/j.epsl.2012.08.006) [DOI] [Google Scholar]
- 97.O'Keefe JD, Ahrens TJ. 1977. Impact-induced energy partitioning, melting, and vapourization on terrestrial planets. In Proc. 8th Lunar Science Conf., Houston, TX, 14–18 March, pp. 3357–3374 New York, NY: Pergamon. [Google Scholar]
- 98.Day JMD, Walker RJ, Qin L, Rumble D. 2012. Late accretion as a natural consequence of planetary growth. Nat. Geosci. 5, 614–617. ( 10.1038/ngeo1527) [DOI] [Google Scholar]
- 99.Dauphas N, Pourmand A. 2011. Hf–W–Th evidence for rapid growth of Mars and its status as a planetary embryo. Nature 473, 489–492. ( 10.1038/nature10077) [DOI] [PubMed] [Google Scholar]
- 100.Chou C-L, Baedecker PA, Bild RW, Wasson JT. 1974. Volatile-element systematics and green glass in Apollo 15 lunar soils. In Proc. 6th Lunar Science Conf., Houston, TX March, pp. 1645–1657. New York, NY:Pergamon. [Google Scholar]
- 101.Meyer C, McKay DS, Anderson DH, Butler P. 1975. The sources of subliminates on the Apollo 15 green and Apollo 17 orange glass samples. In Proc. 6th Lunar Science Conf., Houston, TX, 17–21 March, pp. 1673–1699 New York, NY: Pergamon. [Google Scholar]
- 102.Cirlin EH, Housley RM. 1979. Scanning Auger microprobe and atomic absorption studies of lunar volcanic volatiles. In Proc. 10th Lunar and Planetary Science Conf., Houston, TX, 19–23 March, pp. 341–354 New York, NY: Pergamon. [Google Scholar]
- 103.Krähenbühl U. 1980. Distribution of volatile and non-volatile elements in grain-size fractions of Apollo 17 drive tube 74001/2. In Proc. 11th Lunar and Planetary Science Conf., Houston, TX, 17–21 March, pp. 1551–1564 New York, NY: Pergamon. [Google Scholar]
- 104.Ding TP, Thode HG, Rees CE. 1983. Sulphur content and sulphur isotope composition of orange and black glasses in Apollo 17 drive tube 74002/1. Geochim. Cosmochim. Acta 47, 491–496. ( 10.1016/0016-7037(83)90271-5) [DOI] [Google Scholar]
- 105.Delano JW, McGuire J. 1992. Abundances of sodium, sulfur, and potassium in lunar volcanic glasses. LPI Technical Report, 92–09, Part 1, pp. 7–9. Lunar and Planetary Institute [Google Scholar]
- 106.Delano JW, Hanson BZ, Watson EB. 1994. Abundance and diffusivity of sulfur in lunar picritic magmas. In Proc. 25th Lunar and Planetary Science Conf., Houston, TX, 14–18 March, Abstract 1163, pp. 325–326. See http://www.lpi.usra.edu/meetings/lpsc1994/pdf/1163.pdf [Google Scholar]
- 107.Weitz CM, Rutherford MJ, Head JW, McKay DS. 1999. Ascent and eruption of a lunar high-titanium magma as inferred from the petrology of the 74001/2 drill core. Meteorit. Planet. Sci. 34, 527–540. ( 10.1111/j.1945-5100.1999.tb01361.x) [DOI] [Google Scholar]
- 108.Symonds RB, Rose WI, Reed MH, Lichte FE, Finnegan DL. 1987. Volatilization, transport and sublimation of metallic and non-metallic elements in high temperature gases at Merapi Volcano, Indonesia. Geochim. Cosmochim. Acta 51, 2083–2101. ( 10.1016/0016-7037(87)90258-4) [DOI] [Google Scholar]
- 109.Lofgren G, Donaldson CH, Williams RJ, Mullins O, Usselman TM. 1974. Experimentally reproduced textures and mineral chemistry of Apollo 15 quartz normative basalts. In Proc. 5th Lunar Science Conf., Houston, TX, 18–22 March, pp. 549–567 New York, NY: Pergamon. [Google Scholar]
- 110.Usselman TM, Lofgren GE, Donaldson CH, Williams RJ. 1975. Experimentally reproduced textures and mineral chemistries of high-titanium mare basalts. In Proc. 6th Lunar Science Conf., Houston, TX, 17–21 March, pp. 997–1020. New York, NY: Pergamon. [Google Scholar]
- 111.Day JMD, Taylor LA. 2007. On the structure of mare basalt lava flows from textural analysis of the LaPaz icefield and Northwest Africa 032 lunar meteorites. Meteorit. Planet. Sci. 42, 3–18. ( 10.1111/j.1945-5100.2007.tb00213.x) [DOI] [Google Scholar]
- 112.Giguere TA, Taylor GJ, Hawke BR, Lucey PG. 2000. The titanium contents of lunar mare basalts. Meteorit. Planet. Sci. 35, 192–200. ( 10.1111/j.1945-5100.2000.tb01985.x) [DOI] [Google Scholar]
- 113.Snyder GA, Taylor LA, Neal CR. 1992. A chemical model for generating the sources of mare basalts: combined equilibrium and fractional crystallization of the lunar magmasphere. Geochim. Cosmochim. Acta 56, 3809–3824. ( 10.1016/0016-7037(92)90172-F) [DOI] [Google Scholar]
- 114.Smith JA, Anderson AT, Newton RC, Olsen EJ, Wyllie PJ, Crewe AV, Isaacson MS, Johnson D. 1970. Petrologic history of the Moon inferred from petrography, mineralogy, and petrogenesis of Apollo 11 rocks. In Proc. Apollo 11 Lunar Science Conf., pp. 1149–1162. New York, NY: Pergamon. [Google Scholar]
- 115.Wood JA, Dickey JS, Marvin UB, Powell BN. 1970. Lunar anorthosites and a geophysical model of the Moon. In Proc. Apollo 11 Lunar Science Conf., pp. 965–988 New York, NY: Pergamon. [DOI] [PubMed] [Google Scholar]
- 116.Elkins-Tanton LT. 2012. Magma oceans in the inner Solar System. Annu. Rev. Earth Planet. Sci. 40, 113–139. ( 10.1146/annurev-earth-042711-105503) [DOI] [Google Scholar]
- 117.Liu Y, Spicuzza MJ, Craddock PD, Day JMD, Valley JW, Dauphas N, Taylor LA. 2010. Oxygen and iron isotope constraints on near-surface fractionation effects and the composition of lunar mare basalt source regions. Geochim. Cosmochim. Acta 74, 6249–6262. ( 10.1016/j.gca.2010.08.008) [DOI] [Google Scholar]
- 118.Sedaghatpour F, Teng FZ, Liu Y, Sears DWG, Taylor LA. 2013. Magnesium isotopic composition of the Moon. Geochim. Cosmochim. Acta 120, 1–16. ( 10.1016/j.gca.2013.06.026) [DOI] [Google Scholar]
- 119.Elkins-Tanton LT, Van Orman JA, Hager BH, Grove TL. 2002. Re-examination of the lunar magma ocean cumulate overturn hypothesis: melting of mixing is required. Earth Planet. Sci. Lett. 196, 239–249. ( 10.1016/S0012-821X(01)00613-6) [DOI] [Google Scholar]
- 120.O'Hara MJ. 2000. Flood basalts, basalt floods or topless bushvelds?: Lunar petrogenesis revisited. J. Petrol. 41, 1545–1651. ( 10.1093/petrology/41.11.1545) [DOI] [Google Scholar]