

ABSTRACT
Rates of infection with hospital-acquired Acinetobacter baumannii have exploded over the past decade due to our inability to limit persistence and effectively treat disease. A. baumannii quickly acquires antibiotic resistance, and its genome encodes mechanisms to tolerate biocides and desiccation, which enhance its persistence in hospital settings. With depleted antibiotic options, new methods to treat A. baumannii infections are desperately needed. A comprehensive understanding detailing A. baumannii cellular factors that contribute to its resiliency at genetic and mechanistic levels is vital to the development of new treatment options. Tools to rapidly dissect the A. baumannii genome will facilitate this goal by quickly advancing our understanding of A. baumannii gene-phenotype relationships. We describe here a recombination-mediated genetic engineering (recombineering) system for targeted genome editing of A. baumannii. We have demonstrated that this system can perform directed mutagenesis on wide-ranging genes and operons and is functional in various strains of A. baumannii, indicating its broad application. We utilized this system to investigate key gene-phenotype relationships in A. baumannii biology important to infection and persistence in hospitals, including oxidative stress protection, biocide resistance mechanisms, and biofilm formation. In addition, we have demonstrated that both the formation and movement of type IV pili play an important role in A. baumannii biofilm.
IMPORTANCE
Acinetobacter baumannii is the causative agent of hospital-acquired infections, including pneumonia and serious blood and wound infections. A. baumannii is an emerging pathogen and has become a threat to public health because it quickly develops antibiotic resistance, making treatment difficult or impossible. While the threat of A. baumannii is well recognized, our understanding of even its most basic biology lags behind. Analysis of A. baumannii cellular functions to identify potential targets for drug development has stalled due in part to laborious genetic techniques. Here we have pioneered a novel recombineering system that facilitates efficient genome editing in A. baumannii by single PCR products. This technology allows for rapid genome editing to quickly ascertain gene-phenotype relationships. To demonstrate the power of recombineering in dissecting A. baumannii biology, we use this system to establish key gene-phenotype relationships important to infection and persistence in hospitals, including oxidative stress protection, biocide resistance, and biofilm formation.
INTRODUCTION
Acinetobacter baumannii is an increasingly problematic hospital-associated opportunistic pathogen. It rapidly acquires antibiotic resistance, leading to the spread of multidrug-resistant strains impervious to nearly all antibiotic treatments (1–3). Exacerbating this problem, A. baumannii shows a robust ability to persist on a variety of clinical surfaces despite desiccation and biocide treatment, which facilitates its transfer to new patients (4, 5). While a substantial amount of clinical data has documented the global rise of A. baumannii as an antibiotic-resistant pathogen, dissection of the molecular details of A. baumannii biology that could be used to combat its spread are lagging. Aside from antibiotic resistance, very little in known about basic A. baumannii biology or the role of specific biological factors and cellular processes that directly facilitate its infections and pathogenesis (6). This discrepancy between the observed clinical impacts of A. baumannii and our mechanistic knowledge of its virulence is in part due to the lack of specific and rapid means to genetically manipulate A. baumannii to define and investigate precise gene-phenotype relationships.
Recombination-mediated genetic engineering (recombineering) is a quick and efficient way to perform rapid genome editing in vivo. Recombineering is mediated by phage proteins that belong to either the Red proteins of phage λ (Gam, Beta, and Exo) or the RecET proteins from the Rac prophage, which catalyze homologous recombination between short DNA segments. Both of these systems are functionally equivalent, with Exo/RecE exonucleases exposing 3′ single-stranded DNA that Beta/RecT bind and pair with the homologous genomic target region (7, 8). The Red system also encodes a Gam protein that inhibits host nucleases and is absent from RecET systems. The Escherichia coli λ Red system is the most well-studied recombineering system (9, 10). The development of recombineering revolutionized E. coli genetics, allowing rapid gene substitutions and deletions in a single step without the need for restriction enzymes and building editing constructs in vitro (9, 10). Recombineering rapidly accelerated our genetic analysis of E. coli, solidifying E. coli as a model bacterium and facilitating the prominence of E. coli for use in synthetic biology and biotechnology. The development of recombineering for A. baumannii would dramatically advance our ability to genetically interrogate this emerging pathogen.
Here we exploited a RecET recombinase system to recombineer genes in the A. baumannii genome. We demonstrate the utility of this simple, one-step system for rapidly creating marked or unmarked deletion of genes and operons. Importantly, this system is portable, and with the appropriate choice of selection cassettes, we show that it can be used in different A. baumannii strains. We establish the utility of this system by exploring gene-phenotype relationships in A. baumannii biology that are important to infection and persistence in hospitals using recombineering. These phenotypic mechanisms include oxidative stress protection, biocide resistance, and biofilm formation. Interestingly, our analysis demonstrates that both the biogenesis and functional role of type IV pili play an important part in A. baumannii biofilm formation.
RESULTS
Construction of a recombineering system for A. baumannii.
The purpose of this study was to develop a recombineering system in A. baumannii and show its utility in elucidating gene-phenotype relationships. We chose to begin with the ATCC clinical A. baumannii strain 17978 because of its wide use and well-annotated genome sequence (11). All strains and plasmids used in this study are listed in Table 1. Initially, we attempted to use the λ Red (gam, beta, and exo) recombineering system of E. coli (9). These genes were cloned into plasmid pMMB67EH, which we found allows for isopropyl β-d-1-thiogalactopyranoside (IPTG)-induced gene expression in A. baumannii (see Fig. S1A in the supplemental material). However, the E. coli λ Red system did not operate for recombineering in A. baumannii. This result is reasonable considering the organism-specific biases that have been observed for other phage recombinase systems (12, 13). Furthermore, we found that IPTG-induced expression of the E. coli λ Red system actually slowed the growth of A. baumannii, suggesting that it may be toxic when induced from pMMB67EH (see Fig. S1B).
TABLE 1 .
Strains and plasmids
| Strain/plasmid | Relevant genotype and property | Source and/or reference |
|---|---|---|
| Strains | ||
| A. baumannii strain 17978 | Wild type | ATCC (39) |
| A. baumannii strain 19606 | Wild type | ATCC |
| AT01 | A. baumannii 17978 carrying pAT01 | This study |
| AT02 | A. baumannii 17978 carrying pAT02 | This study |
| AT03 | A. baumannii 17978 carrying pAT03 | This study |
| AT04 | A. baumannii 19606 carrying pAT04 | This study |
| AT05 | A. baumannii 19606 Δlon::Kanr | This study |
| AT06 | A. baumannii 17978 Δ2456::Kanr | This study |
| AT07 | A. baumannii 17978 Δlon::Kanr | This study |
| AT08 | A. baumannii 17978 Δlon::FRT | This study |
| AT09 | A. baumannii 17978 ΔoxyR::Kanr | This study |
| AT10 | A. baumannii 17978 ΔoxyR::Kanr carrying pAT05 | This study |
| AT11 | A. baumannii 17978 carrying pABBR_MCS | This study |
| AT12 | A. baumannii 17978 ΔoxyR::Kanr carrying pABBR_MCS | This study |
| AT13 | A. baumannii 17978 ΔaceI::Kanr | This study |
| AT14 | A. baumannii 17978 ΔaceI::Kanr carrying pAT06 | This study |
| AT15 | A. baumannii 17978 ΔaceI::Kanr carrying pABBR_MCS | This study |
| AT16 | A. baumannii 17978 ΔadeB::Kanr | This study |
| AT17 | A. baumannii 17978 ΔadeB::FRT | This study |
| AT18 | A. baumannii 17978 ΔadeB::Kanr carrying pAT07 | This study |
| AT19 | A. baumannii 17978 ΔadeB ΔaceI::Kanr | This study |
| AT20 | A. baumannii 17978 ΔadeB::Kanr carrying pABBR_MCS | This study |
| AT21 | A. baumannii 17978 ΔpilH::Kanr | This study |
| AT22 | A. baumannii 17978 ΔpilH::Kanr carrying pMMB67EH | This study |
| AT23 | A. baumannii 17978 ΔpilH::Kanr carrying pAT08 | This study |
| AT24 | A. baumannii 17978 carrying pMMB67EH | This study |
| AT25 | A. baumannii 17978 ΔpilG::Kanr | This study |
| AT26 | A. baumannii 17978 ΔpilUT::Kanr | This study |
| Plasmids | ||
| pMMB67EH | Ampr | ATCC (40) |
| TOPO TA 2.1 | Kanr | Invitrogen |
| pKD4 | Kanr | 9 |
| pWH1266 | Acinetobacter plasmid | Lab stock |
| pBR322 | General cloning plasmid | Lab stock |
| pABBR_MCS | Ampr | This study |
| pAT01 | pMMB67EH with E. coli λ-Red system | This study |
| pAT02 | pMMB67EH with RecAb system | This study |
| pAT03 | pMMB67EH with FLP recombinase | This study |
| pAT04 | pMMB67EH with RecAb system, Tetr | This study |
| pAT05 | pABBR_MCS carrying oxyR with native promoter (p_oxyR) | This study |
| pAT06 | pABBR_MCS carrying aceI with native promoter | This study |
| pAT07 | pABBR_MCS carrying adeB with native promoter | This study |
| pAT08 | pMMB67EH carrying pilH (p_pilH) | This study |
Using the Basic Local Alignment Search Tool (BLAST) algorithm, we identified several RecT A. baumannii homologs, including a strong homolog to the E. coli λ Red Beta protein in the shotgun-sequenced A. baumannii strain IS-123 (gene ACINIS123_2461; E value 9e−75). The adjacent gene in IS-123, ACINIS123_2462, was part of the yqaJ/recE superfamily of exonucleases and had sequence similarity to the Exo protein from the E. coli λ Red system (E value 2e−16), suggesting that this gene pair may represent a recombination system in A. baumannii. We did not identify a Gam homolog in any A. baumannii strain. The RecET gene pair from IS-123 was cloned into the IPTG-inducible pMMB67EH vector for expression in A. baumannii. We call this the RecAb system. IPTG-induced expression of the RecAb system did not affect growth of A. baumannii strain 17978 (see Fig. S1B in the supplemental material). We used A. baumannii 17978 expressing the RecAb system for the majority of these experiments.
Application of RecAb for gene replacement.
Four regions of DNA that contain genes of various sizes and encoding different biological functions were selected from the A. baumannii 17978 genome for targeted deletion: A1S_2456 (LysR-type regulator), A1S_1750 (adeB), A1S_1030-A1S_1031 (lon), and A1S_2063 (aceI). We identified an error in the published A. baumannii 17978 genome that created an artificial frameshift in the gene encoding the Lon protease. This resulted in lon being annotated by two genes, A1S_1030 and A1S_1031. We chose this gene set because they represent genes of various lengths, location in the genome, and functions. A complete list of genes deleted in this study, along with their sizes and locations, is provided in Table 2. Through a commercial supplier (Integrated DNA Technologies), we synthesized oligonucleotides containing 125 bases flanking the coding sequence (CDS) of each targeted gene, including the ATG start codon or the stop codon (Fig. 1). We added an additional 18 to 25 nucleotides (nt) on the 3′ end of each primer to facilitate amplification of a PCR product with an antibiotic selection cassette flanked by the 125 bp upstream and downstream of the target coding sequence. A kanamycin resistance marker was amplified from TOPO TA 2.1 (Invitrogen) to create our recombineering PCR products.
TABLE 2 .
Deleted genes in this study
| Gene name | Gene location | Length (bp) | Locus tag | Accession no. |
|---|---|---|---|---|
| lon | 1191086–1188755 | 2,331 | A1S_1030 | YP_001084064.1 |
| A1S_1031 | YP_001084065.1 | |||
| lona | Contig sidl23403laccninz.GG704573 | 2,576 | HMPREF0010_01568 | GG704573.1 |
| HMPREF0010_01569 | ||||
| LysR family transcriptional regulator | 2844859–2845533 | 675 | A1S_2456 | YP_001085476.1 |
| aceI | 2406038–2406508 | 471 | A1S_2063 | YP_001085092.1 |
| adeB | 2039428–2036447 | 2,982 | A1S_1750 | YP_001084779.1 |
| oxyR | 1150365–1151153 | 789 | A1S_0992 | YP_001084026.1 |
| pilUT | 1036617–1038800 | 2,184 | A1S_0896 | YP_001083930.1 |
| A1S_0897 | YP_001083931.1 | |||
| pilH | 3259867–3260118 | 252 | A1S_2814 | YP_001085822.1 |
| pilG | 3260253–3260606 | 354 | A1S_2815 | YP_001085823.1 |
A. baumannii strain 19606.
FIG 1 .

Schematic diagram of the one-step recombineering method. Primers containing 125-bp homology flanking the gene of interest are used to amplify the PCR product. The insert is then recombined into the genome using our RecAb system. Screening primers outside the region of homology are used to confirm insertion of the kanamycin cassette.
Following transformation of the PCR product in A. baumannii cells expressing RecAb, we successfully isolated colonies where each targeted gene was replaced with the kanamycin cassette (Fig. 2A). We did not identify any positive colonies if the RecAb system was not expressed. Eighty to one hundred percent of screened colonies had successfully recombined at the desired locus with some variation of efficiency among the target genes. The few false-positive colonies that grew on kanamycin but still carried the parental copy of the target gene were universally smaller than their true-positive counterparts. PCR screening showed that none of the false-positive colonies contained a kanamycin cassette. Furthermore, none of the false-positive colonies grew when streaked on fresh kanamycin agar plates.
FIG 2 .

PCR verification of constructed mutants. (A) Following recombineering, PCR was used to verify the presence of the wild-type allele or the replacement of that allele with a kanamycin resistance cassette to create the mutant strain. The wild-type control is marked “WT.” Shown are gene replacements with a kanamycin resistance cassette for targets lon, A1S_2456, aceI, and adeB. (B) Engineering of markerless mutants using the FLP-FRT recombination system. Lane 1, adeB wild type; lane 2, ΔadeB::FRT-Kanr strain; lane 3, ΔadeB strain after excision of the kanamycin resistance cassette; lane 4, lon wild type; lane 5, Δlon::FRT-Kanr strain; lane 6, Δlon strain after excision of the kanamycin resistance cassette. (C) Δlon::Kanr mutant of A. baumannii strain 19606. The 19606 strain wild-type control is marked “WT.”
Using primers that flank the target coding sequences, we sequenced the recombination site of two positive colonies for each gene target. The majority had the correct sequence; however, we did identify two colonies that had errors in the region of chemically synthesized homology, indicating that sequencing of the recombineering junction should be performed to validate fidelity of the synthesized DNA. These results indicate that the RecAb system is applicable genome-wide, efficient, and accurate.
The plasmid carrying RecAb is unstable in the absence of ampicillin selection. Colonies identified as positive for gene replacement with the kanamycin cassette were streaked on kanamycin plates overnight, and the resulting single colonies were patched to kanamycin and ampicillin plates. Fifty to seventy percent of the colonies tested had lost the RecAb plasmid, indicating that this plasmid can be easily cured after recombineering (data not shown).
To optimize the recombineering parameters in A. baumannii, we tested the length of homology required for efficient recombineering using the RecAb system. While PCR products with less that 75-bp regions of homology did not produce any recombinants under the conditions tested, PCR products with 100 bp of homology did produce recombinants. However, fragments with 125 bp of homology displayed a 1- to 2-log increase in recombination efficiency. Next, we tested recombineering efficiency using 100 ng, 500 ng, 1 µg, 5 µg and 10 µg of PCR product, but we did not observe any recombinants using less than 5 µg of PCR product. Using 10 µg of PCR product did not increase the efficiency of recombinant production compared with that with 5 µg. When using electrocompetent cells at a density of 1010 CFU/reaction and 5 µg of PCR product with 125 bp of flanking homology, we obtained approximately 100 colonies from each transformation, with a range of 20 to 200 colonies, depending on the gene being targeted for replacement.
Engineering of markerless mutants using an FLP/FRT recombinase system.
The antibiotic resistance marker can be removed by incorporating FLP recognition target (FRT) sites that flank the antibiotic cassette and expressing the FLP recombinase (14). To do this, the FRT site-flanked kanamycin cassette from plasmid pKD4 is amplified and used for targeted gene replacement as described above (9). After selection and isolation of an A. baumannii mutant that was cured of RecAb, plasmid pMMB67EH carrying the FLP recombinase is transformed into the strain carrying the FRT-flanked gene replacement cassette. Colonies carrying the FLP recombinase are streaked on solid agar containing 1 mM IPTG, which induces expression of the FLP recombinase, to result in excision of the kanamycin resistance cassette and simultaneous loss of the unstable pMMB67EH plasmid. To demonstrate this technique, adeB and the lon genes were initially replaced with the FRT-flanked kanamycin cassette (Fig. 2B, lane 1 versus lane 2 and lane 4 versus lane 5, respectively). The kanamycin resistance cassette was removed by transformation of the pMMB67EH plasmid and activation of the FLP recombinase enzyme (Fig. 2B, lanes 3 and 6).
Application of RecAb in A. baumannii strain 19606.
To determine if this system could be used in other A. baumannii strains, we attempted to delete lon in another widely used clinical A. baumannii strain, ATCC 19606. We observed that strain 19606 is resistant to the β-lactams ampicillin and carbenicillin. To overcome this, we replaced the bla gene on RecAb with tet for tetracycline resistance. Using this derivative, we easily isolated colonies with replacement of lon by the kanamycin cassette in strain 19606 (Fig. 2C). Thus, with the appropriate selection of resistance cassettes, this system appears to have far-reaching applications among different A. baumannii strains.
Deletion of an A. baumannii oxyR homolog results in increased sensitivity to hydrogen peroxide.
To establish the relevance of this recombineering system in characterizing gene-phenotype relationships, we first focused on the well-studied response of bacteria to oxidative stress. Response to oxidative stress is important in order for bacteria to adapt and survive in the presence of biocides and antibiotics (15, 16). In E. coli, hydrogen peroxide triggers activation of the transcription factor OxyR, which induces expression of oxidative stress protection genes (17, 18). Deletion of oxyR in E. coli results in increased sensitivity to hydrogen peroxide. The protein A1S_0992 from A. baumannii strain 17978 shows 29% identity (E value, 2e−34) to E. coli OxyR by BLAST bioinformatics analysis, suggesting that A1S_0992 may play an OxyR-like role in A. baumannii. Consistent with studies of OxyR in E. coli, deletion of A1S_0992 using our recombineering system produced an A. baumannii strain that displays increased sensitivity to hydrogen peroxide (Fig. 3, top). Ectopic expression of A1S_0992 (oxyR) from a plasmid complements the mutant phenotype, demonstrating that A1S_0992 is very likely an OxyR homolog that plays an important role in oxidative stress protection in A. baumannii (Fig. 3, bottom).
FIG 3 .

Deletion of oxyR homolog A1S_0992 from A. baumannii results in increased sensitivity to hydrogen peroxide (H2O2). The parental A. baumannii strain and oxyR::Kanr mutant were struck on Luria-Bertani (LB) agar with or without 0.25 mM H2O2 (top). The oxyR::Kanr mutant H2O2 sensitivity phenotype is recovered by ectopic expression of the oxyR allele from a plasmid (p_oxyR) but not by the empty plasmid (pABBR_MCS) (bottom).
Application of RecAb to efflux pumps and biocide resistance.
Recently, an efflux pump (encoded by aceI) responsible for resistance to the biocide chlorhexidine was discovered in A. baumannii 17978 (19). However, the authors were unable to delete aceI from the A. baumannii chromosome using traditional genetic tools. In contrast, we were easily able to replace aceI with a kanamycin cassette using the RecAb system (Fig. 2A). This demonstrates an advantage in using the RecAb system over traditional mutagenesis strategies in A. baumannii. In their article, the authors demonstrated that removal of the aceI homolog in the closely related bacterium Acinetobacter baylyi resulted in a 2-fold decrease in resistance to chlorhexidine. In agreement with their data, we found a similar 2-fold decrease in resistance when aceI was deleted from A. baumannii 17978 (Table 3). Complementation of this mutant with expression of aceI from a plasmid restored chlorhexidine resistance to the parental level (Table 3).
TABLE 3 .
Chlorhexidine MICs
| Strain description | Chlorhexidine MIC (μg/ml) |
|---|---|
| Parental | 4 |
| ΔaceI::Kanr | 2 |
| ΔadeB::Kanr | 1 |
| Parental + empty vector | 4 |
| ΔaceI::Kanr + empty vector | 2 |
| ΔadeB::Kanr + empty vector | 1 |
| ΔaceI::Kanr complemented | 4 |
| ΔadeB::Kanr complemented | 2 |
| ΔadeBa | 1 |
| ΔadeBa ΔaceI::Kanr | 0.5 |
A. baumannii 17978 carrying deletion of adeB after FRT-mediated excision of the kanamycin resistance cassette.
In addition to aceI encoding a gene important for resistance to the biocide chlorhexidine, the AdeABC efflux pump is important for A. baumannii survival in the presence of chlorhexidine and has been shown to confer on A. baumannii resistance to a variety of other bactericidal compounds (20, 21). For example, our mutant strain carrying a deletion of adeB (A1S_1750) shows a 4-fold decrease in resistance to chlorhexidine. This efflux system has been previously shown to act synergistically with the AdeIJK efflux pump system in the presence of antibiotics (22). To investigate a possible relationship between AceI and AdeABC, we created a double aceI adeB mutant and tested its sensitivity to chlorhexidine. We observed an additive, not synergistic, effect between AceI and the AdeABC system for chlorhexidine resistance (Table 3). This suggests that these two systems act independently to mediate chlorhexidine resistance in A. baumannii.
Deletion of a type IV pilus retraction gene reveals importance for biofilm formation.
Type IV pili were recently shown to be involved in natural transformation and twitching motility in Acinetobacter nosocomialis strain M2 (23, 24). The authors demonstrated that the genes involved in both assembly and the dynamics of this system played a role. Type IV pili have been shown to be important for biofilm formation in Pseudomonas aeruginosa; however, the role of this surface appendage in biofilm formation has not been studied in A. baumannii (25).
To explore the potential role of type IV pili in A. baumannii biofilm formation, we began by deleting the putative A. baumannii 17978 operon encoding homologs of the type IV pilus proteins PilU (A1S_0896) and PilT (A1S_0897) (see Fig. S2 in the supplemental material). PilU and PilT have been shown to be important for pilus function in A. baumannii and P. aeruginosa and have been implicated in pilus depolymerization and retraction (25). Using a crystal violet staining assay, we found that the pilUT mutant showed a marked decrease in biofilm formation compared to the parental strain (Fig. 4). Transmission electron microscopy showed type IV pili present on the surface of the parental A. baumannii 17978 strain but absent from our pilUT mutant (Fig. 5A and B). This supports the conclusion that fully functional type IV pili are important for biofilm production in this strain. Interestingly, a pilT mutant in A. nosocomialis strain M2 was shown to be defective for type IV-dependent natural transformation and twitching motility, indicating impaired pilus function, but it still produced visible pili on the cell surface (23). This suggests either strain variation in pilus formation between A. baumannii 17978 and M2 or that deleting both pilU and pilT is required to lose pilus formation on the cell surface.
FIG 4 .
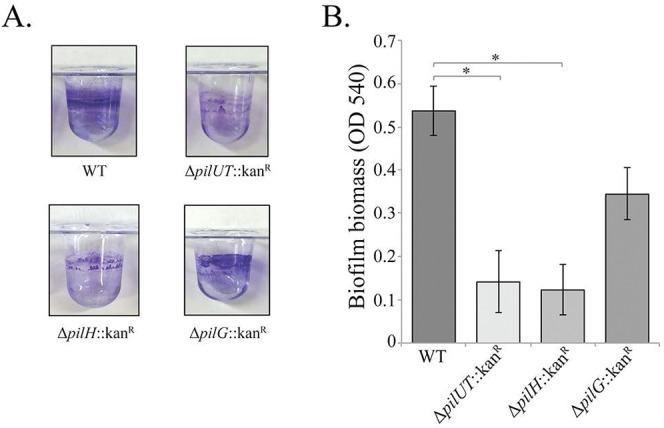
Biofilm formation assay of A. baumannii wild type, ΔpilUT::Kanr strain, ΔpilH::Kanr strain, and ΔpilG::Kanr strain, (A) Crystal violet staining of each strain in a PVC microtiter plate. (B) Biofilm formation measured by crystal violet staining for optical density at 540 nm (OD540). Asterisks denote significant differences in biofilm formation (t test; *, P < 0.0001; n = 8).
FIG 5 .
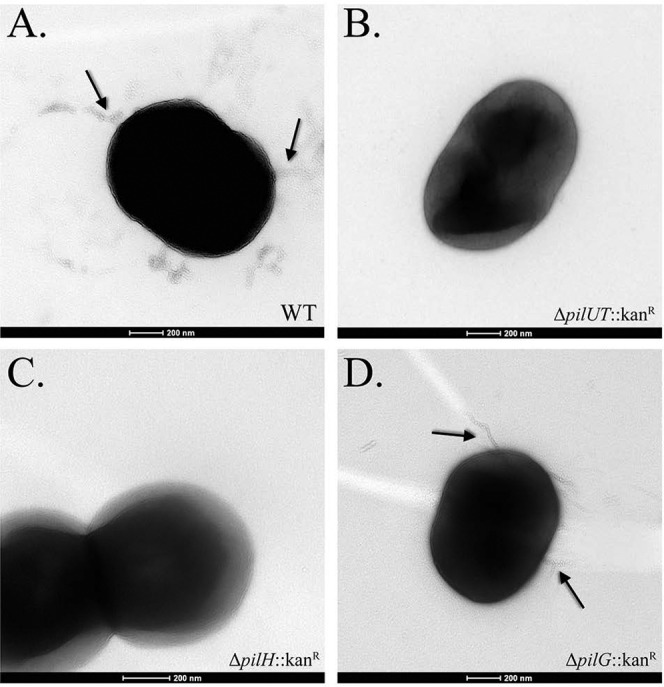
Transmission electron microscopy images of type IV pilus mutants of A. baumannii strain 17978. Black arrows specify pilus-like appendages. (A) Wild-type A. baumannii 17978; (B) ΔpilUT::Kanr strain; (C) ΔpilH::Kanr strain; (D) ΔpilG::Kanr strain.
Once assembled, pili undergo extension and retraction to mediate twitching motility. In P. aeruginosa, extension and retraction signaling is mediated by CheY-like response regulators encoded by pilG and pilH, respectively (26–28). To investigate the role of pilus movement and signaling in A. baumannii biofilm production, we deleted the pilH (A1S_2814) and pilG (A1S_2815) homologs in A. baumannii. Deletion of the extension response regulator, PilG, had no effect on biofilm formation (Fig. 4). Consistent with this observation, pili were readily observed on the surface of the pilG mutant and were indistinguishable from those of the parental strain (Fig. 5D). Interestingly, deletion of the retraction response regulator encoded by pilH resulted in a severe decrease in biofilm formation that resembled findings for the pilUT mutant. Further supporting the role for type IV pili in biofilm formation, we could not identify any pili on the surface of the pilH mutant (Fig. 5C).
The growth rates of pilUT, pilH, and pilG mutants were similar to that of the wild type (WT) (see Fig. S3A in the supplemental material), showing that the observed alterations in biofilm formation were not due to growth defects. Complementation of the pilH mutant by ectopic expression of pilH from a plasmid restored biofilm formation to the same level as that of the parental strain carrying the empty vector (see Fig. S3B). Real-time quantitative PCR analysis of our complemented strain showed that ectopic expression of pilH closely matched endogenous expression (see Fig. S3C).
The gene downstream of pilH is another putative type IV pilus gene, A1S_2813, known as the pilI homolog. While pilH and pilI do not appear to be cotranscribed, it was also possible that replacement of pilH with a kanamycin cassette created a polar effect on pilI that could influence our results (see Fig. S2 in the supplemental material). To test this, we measured the transcript levels of pilI in our parental strain and pilH mutant and found them to be identical, indicating that our pilH mutation does not affect expression of pilI (see Fig. S3D).
DISCUSSION
While high-throughput “omic”-level approaches have dramatically increased our knowledge of A. baumannii genome content, we have lacked tools for rapid genetic manipulation to identify and confirm specific gene-phenotype relationships. In this study, we have identified a RecET recombinase system in the A. baumannii strain IS-123 that we have engineered as a tool for recombineering in A. baumannii. To illustrate proof of principle, we successfully deleted and replaced genes of various sizes that encoded different biological functions using PCR products containing an optimized homology of 125 bp. The ability of the RecAb system to function in two A. baumannii strains and the presence of homologous RecAb systems in other sequenced A. baumannii genomes suggest that it could be widely used in A. baumannii. Using the FRT-FLP system, we further demonstrated the feasibility of constructing markerless mutants, which allows stepwise deletion of multiple genes using RecAb. Recombineering has catapulted the rate of genetic analysis of E. coli. It allowed for the creation of the widely used single-gene deletion Keio Library, solidified E. coli as the most tractable Gram-negative bacterium for studying genetics, and made E. coli the bacterium of choice for synthetic biology and biotechnology (29).
Aranda et al. recently described another method for creating stable gene replacements with PCR products (30). While similar in overall goal, our method diverges in several ways. To construct stable gene replacements, the method described by Aranda et al. uses PCR products with 500 bp of homology, which requires eight different primers and three separate PCR stages for synthesis. This method generates the desired mutants but is reported to be prone to significant illegitimate recombination between the PCR product and unknown locations in the genome. Incorporation of the PCR products at off-target locations could cause complications in evaluating phenotypes in subsequent studies. The RecAb system for stable gene replacement we describe here uses PCR products with 125 bp of homology, which requires two primers and one PCR stage for synthesis. The RecAb system has an apparent zero off-target rate. Furthermore, incorporation of the FRT sites allows for easy construction of unmarked mutants and subsequent construction of additional mutations in the same strain without the need for other selection markers.
Our understanding of A. baumannii biology lags far behind the clinical data outlining the emergence of this problematic hospital-associated pathogen. Genomic analysis indicates that even basic aspects of A. baumannii biology differ from that of model organisms like E. coli (31). Understanding how A. baumannii causes disease, persists in the clinical environment, and acquires antibiotic resistance is critical to preventing infections and managing disease. Adaptation of recombineering as a genetic technique to quickly introduce genetic mutations into A. baumannii will allow a rapid expedition in the gene-phenotype analysis of this extraordinary multidrug-resistant pathogen.
Our analysis of oxidative stress protection and biocide resistance demonstrates the utility of our system for gene-phenotype analysis, especially for previously difficult genetic targets, such as aceI. Our analysis supports the predicted role for the OxyR homolog in A. baumannii oxidative stress protection and the function of AceI in chlorhexidine resistance in A. baumannii. Additionally, we utilized the FRT-FLP system to explore potential genetic interactions between AceI and the AdeABC efflux system by creating this double mutant. Our data suggest an additive effect, indicating that the efflux transporters play an independent, not synergistic, role in resistance to chlorhexidine.
Type IV pili are found on a plethora of Gram-positive and Gram-negative bacteria and are important for motility and attachment during biofilm development (25, 32). The ability to form a biofilm is recognized as a virulence factor in A. baumannii and may support its persistence in clinic (3, 33, 34). Here, we demonstrate that type IV pili are required for biofilm formation in A. baumannii strain 17978 and identify specify genetic determinates of pilus formation. Deletion of pilU (A1S_0896) and pilT (A1S_0897) or pilH (A1S_2814) results in dramatically decreased biofilm formation and an associated loss of visible pili on the cell surface. pilU, pilT, and pilH all encode proteins involved with pilus retraction, while pilG, which does not affect biofilm formation, encodes the signal for pilus extension (28). This suggests that pilus retraction may play a greater role then extension in forming pili and biofilm in A. baumannii 17878.
The pilus genes studied here were classified by comparison with their well-studied P. aeruginosa homologs (28). Comparing our results to those published for P. aeruginosa, we identify a similar correlation between pilus abundance and biofilm density but an opposite trend with respect to specific mutations. Agreeing with our results, PilT and PilU have been shown to impact biofilm formation in P. aeruginosa (23, 35–37). In contrast, however, P. aeruginosa pilT and pilU mutants present with a dense biofilm and hyperpiliated phenotype (23, 35–37). Additionally, we show that the response regulator required for pilus retraction, PilH, but not that for extension, PilG, is required for pilus and biofilm formation in A. baumannii 17978. Meanwhile, recent studies of P. aeruginosa type IV pilus mutants have confirmed that pilG mutants had no surface pilin, whereas pilH mutants were hyperpiliated (38). The correlation between pilus abundance and biofilm density agrees between P. aeruginosa and A. baumannii, but the opposite impacts of pilU, pilT, pilG, and pilH mutations suggest an important difference in pilus regulation and biofilm formation between these two bacteria.
Our dwindling supply of effective antibiotics requires development of new treatment options to combat the rapid emergence of multidrug-resistant A. baumannii. Information gained from the explosion in high-throughput sequencing offers many potential cellular functions that could be targeted. However, specific gene-phenotype relationships must be established to validate our choices and streamline our efforts considering the very long timeline to actual production of a therapeutic. The recombineering system here provides a previously unavailable rapid and specific means to verify gene-phenotype relationships, and successful application in oxidative stress management, biocide tolerance, and biofilm formation demonstrates its applicability to a broad range of A. baumannii biology.
MATERIALS AND METHODS
Bacterial strains, plasmids, growth conditions, and antibiotics.
The bacterial strains and plasmids used in this study are listed in Table 1. The A. baumannii Rec genes (RecAb) were synthesized by Genewiz. The pABBR_MCS plasmid was generated as follows. The origin of replication from plasmid pWH1266 was PCR amplified and cloned into the PciI site of pBR322. We synthesized a multiple cloning site ending in a T7 transcriptional terminator and cloned this into the ZraI site of the above plasmid to create pABBR_MCS. Strains were grown in Luria-Bertani (LB) broth/agar at 37°C. The antibiotics carbenicillin (75 µg/ml) and kanamycin (25 µg/ml or 7.5 µg/ml) were added for selection as needed.
Gene inactivation using RecAb.
A. baumannii carrying RecAb on pMMB67EH was inoculated into rich medium (LB) with carbenicillin to maintain the plasmid. IPTG was added to a final concentration of 2 mM, and the bacteria were grown for 3 h to mid-log phase at 37°C. After 3 washes in ice-cold 10% glycerol and concentrating 1,000-fold, 100 µl of bacteria (~1010 bacteria) were mixed with 5 µg of recombineering PCR product and electroporated in a 2-mm cuvette at 1.8 kV. After outgrowth in 4 ml rich medium containing 2 mM IPTG, the bacteria were pelleted, plated on 7.5 µg/ml kanamycin, and incubated overnight at 37°C. This method routinely yielded between 20 and 200 colonies.
PCR verification.
Primers located outside the regions of homology were used to PCR amplify the genomic location of the genes of interest. These primers are listed in Table S1 in the supplemental material with the suffix “scr.” Briefly, a freshly isolated colony was suspended in 100 µl of water with a plastic tip from which 2-µl portions were used in separate 20-µl PCRs following a 2-min preincubation at 95°C. Deletion of the gene and insertion of a kanamycin cassette subsequently yield a PCR product of a different size than when the gene is present.
Deletion of kanamycin cassette.
Plasmid pKD4 was used as a template for generating a kanamycin cassette flanked by FRT sites. Gene inactivation was carried out as described above. Following confirmation and curing of the RecAb plasmid, the mutant strains were transformed with pMMB67EH expressing the FLP recombinase and induced with 2 mM IPTG. Single colonies carrying this plasmid were isolated and cured. A loss of kanamycin resistance was observed in these colonies, which agrees with the PCR verification that the kanamycin cassette was lost.
Hydrogen peroxide sensitivity.
The various strains were streaked on LB agar supplemented with 0.25 mM hydrogen peroxide. When necessary, carbenicillin was added at 75 µg/ml to agar plates to maintain plasmids; to maintain a control, plates containing only LB were also used. A fresh colony of the WT, a ΔoxyR::Kanr strain, and its complemented strain were streaked onto each plate. Plates were incubated at 37°C overnight.
MIC assay.
MIC assays for chlorhexidine were performed in triplicate at least twice as previously described (39).
Biofilm assay.
Biofilm formation by A. baumannii 17978 and mutant strains was tested by inoculating a 5-ml culture of Luria broth (LB) with a single colony and grown overnight to stationary phase. The overnight culture was then diluted with LB at 1:100, and 100 µl of the diluted culture was added to a polyvinylchloride (PVC) microtiter plate in replicates of eight. Strains were allowed to grow for 24 h at 30°C. Unbound cells were removed. Bound cells were washed twice with 200 µl sterile water and subsequently stained with 0.1% crystal violet for 15 min. Cells were then washed with 200 µl phosphate-buffered saline (PBS) three times and allowed to air dry for 30 min. Stain was then solubilized with the addition of 200 µl 95% ethanol. Next, 125 µl of solubilized stain was transferred to a new 96-well, polystyrene flat-bottom plate for optical density measurement at 540 nm using a SpectraMax Plus384 absorbance microplate reader with the software program SOFTmax Pro v. 6.2.2.
Genetic complementation of mutants.
Complementation vectors for the ΔoxyR::Kanr, ΔadeB::Kanr, and ΔaceI::Kanr strains were constructed using primer sets listed in Table S1 in the supplemental material. The full-length genes with their native promoters were amplified, cloned into vector pABBR_MCS, and subsequently electroporated into their corresponding mutant strain. Complementation of ΔpilH was carried out by amplifying the full-length gene with a strong Shine-Dalgarno sequence with primer sets listed in Table S1. The gene was then cloned into pMMB67EH and electroporated into the ΔpilH::Kanr strain
RNA extraction and qRT-PCR analysis.
RNAs from freshly streaked plates of wild-type and mutant strains were prepared using a Direct-zol RNA MiniPrep kit with TRI-Reagent (Zymo Research, Orange, CA). DNase treatment was carried out using a Turbo DNA-free kit (Ambion, Inc.) with RNA diluted to 10 ng/µl. Primers listed in Table S1 in the supplemental material were designed using the Primer3 software program (40) and synthesized by Integrated DNA Technologies, Inc. Final primer and RNA concentrations used were 200 nM and 30 ng, respectively. Reaction mixtures were prepared using the Kapa SYBR Fast quantitative PCR (qPCR) kit and monitored by using LightCycler 96 (Roche, Indianapolis, IN). Gene expression was normalized to A. baumannii 16S rRNA. Relative abundance was determined using the ΔΔCT method. For each gene, reverse transcription-quantitative PCR (qRT-PCR) was performed for three biological replicates.
Transmission electron microscopy.
Wild-type and pilH::Kanr, pilG::Kanr, and pilUT::Kanr mutant strains were streaked on LB agar plates and incubated overnight at 37°C. Several bacterial colonies were transferred to 30 µl of 8% glutaraldehyde and swirled gently. Four microliters of the bacterial suspension was pipetted onto Formvar film 150 square mesh copper grids (Electron Microscopy Sciences). After the cells were allowed to adhere to the grids for 5 min, the liquid was wicked away with filter paper. The grids were then washed with molecular-grade water, and excess water was removed with filter paper. The grids were stained with 2% uranyl acetate for 1 min, and liquid was removed with filter paper. The grids were dried for 1 h before storing for later imaging. Images were obtained on an FEI Tecnai transmission electron microscope at the Microscopy and Imaging Facility at the University of Texas at Austin.
SUPPLEMENTAL MATERIAL
Expression and growth curve analysis of recombineering systems in A. baumannii. (A) Expression of GFP from the IPTG-inducible vector pMMB67EH in A. baumannii using 0 mM to 5 mM IPTG. (B) Growth curve analysis of A. baumannii harboring λ Red and RecET systems with and without induced expression with IPTG. Download
Genetic organization of predicted type IV pili and flanking genes. Genes used in this study are shown as red arrows. Download
(A) Growth curve of wild-type (WT) and mutant strains from the biofilm assay. (B) Complementation of ΔpilH::Kanr. Asterisks denote significant differences in biofilm formation (t test, *, P < 0.0001; n = 8). (C) qRT-PCR analysis of the pilH gene in WT, ΔpilH::Kanr, and complemented ΔpilH strains. (D) qRT-PCR analysis of the pilI gene in WT and ΔpilH::Kanr strains. Download
Oligonucleotides used in this study
ACKNOWLEDGMENTS
We thank the Microscopy Facility at UT Austin.
This work was supported by start-up funds from UT Austin to B.W.D., NIH grants AI064184 and AI76322 to M.S.T., and Army Research Office grant W911NF-12-1-0390 to M.S.T.
Footnotes
Citation Tucker AT, Nowicki EM, Boll JM, Knauf GA, Burdis NC, Trent MS, Davies BW. 2014. Defining gene-phenotype relationships in Acinetobacter baumannii through one-step chromosomal gene inactivation. mBio 5(4):e01313-14. doi:10.1128/mBio.01313-14.
REFERENCES
- 1. Fournier PE, Vallenet D, Barbe V, Audic S, Ogata H, Poirel L, Richet H, Robert C, Mangenot S, Abergel C, Nordmann P, Weissenbach J, Raoult D, Claverie JM. 2006. Comparative genomics of multidrug resistance in Acinetobacter baumannii. PLoS Genet. 2:e7. 10.1371/journal.pgen.0020007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Perez F, Hujer AM, Hujer KM, Decker BK, Rather PN, Bonomo RA. 2007. Global challenge of multidrug-resistant Acinetobacter baumannii. Antimicrob. Agents Chemother. 51:3471–3484. 10.1128/AAC.01464-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roca I, Espinal P, Vila-Farres X, Vila J. 2012. The Acinetobacter baumannii oxymoron: commensal hospital dweller turned pan-drug-resistant menace. Front. Microbiol. 3:148. 10.3389/fmicb.2012.00148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rice LB. 2008. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J. Infect. Dis. 197:1079–1081. 10.1086/533452 [DOI] [PubMed] [Google Scholar]
- 5. Mortensen E, Trivedi KK, Rosenberg J, Cody SH, Long J, Jensen BJ, Vugia DJ. 2014. Multidrug-resistant Acinetobacter baumannii infection, colonization, and transmission related to a long-term care facility providing subacute care. Infect. Control Hosp. Epidemiol. 35:406–411. 10.1086/675612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gerischer U. 2008. Acinetobacter molecular biology. Caister Academic Press, Norfolk, United Kingdom [Google Scholar]
- 7. Cassuto E, Radding CM. 1971. Mechanism for the action of lambda exonuclease in genetic recombination. Nat. New Biol. 229:13–16. 10.1038/229013c0 [DOI] [PubMed] [Google Scholar]
- 8. Karakousis G, Ye N, Li Z, Chiu SK, Reddy G, Radding CM. 1998. The beta protein of phage lambda binds preferentially to an intermediate in DNA renaturation. J. Mol. Biol. 276:721–731. 10.1006/jmbi.1997.1573 [DOI] [PubMed] [Google Scholar]
- 9. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645. 10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. 2000. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 97:5978–5983. 10.1073/pnas.100127597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smith MG, Gianoulis TA, Pukatzki S, Mekalanos JJ, Ornston LN, Gerstein M, Snyder M. 2007. New insights into Acinetobacter baumannii pathogenesis revealed by high-density pyrosequencing and transposon mutagenesis. Genes Dev. 21:601–614. 10.1101/gad.1510307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Datta S, Costantino N, Zhou X, Court DL. 2008. Identification and analysis of recombineering functions from gram-negative and Gram-positive bacteria and their phages. Proc. Natl. Acad. Sci. U. S. A. 105:1626–1631. 10.1073/pnas.0709089105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Swingle B, Bao Z, Markel E, Chambers A, Cartinhour S. 2010. Recombineering using RecTE from Pseudomonas syringae. Appl. Environ. Microbiol. 76:4960–4968. 10.1128/AEM.00911-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. 10.1016/S0378-1119(98)00130-9 [DOI] [PubMed] [Google Scholar]
- 15. Nguyen D, Joshi-Datar A, Lepine F, Bauerle E, Olakanmi O, Beer K, McKay G, Siehnel R, Schafhauser J, Wang Y, Britigan BE, Singh PK. 2011. Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science 334:982–986. 10.1126/science.1211037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shatalin K, Shatalina E, Mironov A, Nudler E. 2011. H2S: a universal defense against antibiotics in bacteria. Science 334:986–990. 10.1126/science.1209855 [DOI] [PubMed] [Google Scholar]
- 17. Chiang SM, Schellhorn HE. 2012. Regulators of oxidative stress response genes in Escherichia coli and their functional conservation in bacteria. Arch. Biochem. Biophys. 525:161–169. 10.1016/j.abb.2012.02.007 [DOI] [PubMed] [Google Scholar]
- 18. Christman MF, Storz G, Ames BN. 1989. OxyR, a positive regulator of hydrogen peroxide-inducible genes in Escherichia coli and Salmonella typhimurium, is homologous to a family of bacterial regulatory proteins. Proc. Natl. Acad. Sci. U. S. A. 86:3484–3488. 10.1073/pnas.86.10.3484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hassan KA, Jackson SM, Penesyan A, Patching SG, Tetu SG, Eijkelkamp BA, Brown MH, Henderson PJ, Paulsen IT. 2013. Transcriptomic and biochemical analyses identify a family of chlorhexidine efflux proteins. Proc. Natl. Acad. Sci. U. S. A. 110:20254–20259. 10.1073/pnas.1317052110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Magnet S, Courvalin P, Lambert T. 2001. Resistance-nodulation-cell division-type efflux pump involved in aminoglycoside resistance in Acinetobacter baumannii strain BM4454. Antimicrob. Agents Chemother. 45:3375–3380. 10.1128/AAC.45.12.3375-3380.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wieczorek P, Sacha P, Hauschild T, Zorawski M, Krawczyk M, Tryniszewska E. 2008. Multidrug resistant Acinetobacter baumannii—the role of AdeABC (RND family) efflux pump in resistance to antibiotics. Folia Histochem. Cytobiol. 46:257–267. 10.2478/v10042-008-0056-x [DOI] [PubMed] [Google Scholar]
- 22. Damier-Piolle L, Magnet S, Brémont S, Lambert T, Courvalin P. 2008. AdeIJK, a resistance-nodulation-cell division pump effluxing multiple antibiotics in Acinetobacter baumannii. Antimicrob. Agents Chemother. 52:557–562. 10.1128/AAC.00732-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Harding CM, Tracy EN, Carruthers MD, Rather PN, Actis LA, Munson RS., Jr. 2013. Acinetobacter baumannii strain M2 produces type IV pili which play a role in natural transformation and twitching motility but not surface-associated motility. mBio 4(4):e00360-13. 10.1128/mBio.00360-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carruthers MD, Harding CM, Baker BD, Bonomo RA, Hujer KM, Rather PN, Munson RS., Jr. 2013. Draft genome sequence of the clinical isolate Acinetobacter nosocomialis strain. Genome Announc. 1(6):e00906-13. 10.1128/genomeA.00906-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burrows LL. 2012. Pseudomonas aeruginosa twitching motility: type IV pili in action. Annu. Rev. Microbiol. 66:493–520. 10.1146/annurev-micro-092611-150055 [DOI] [PubMed] [Google Scholar]
- 26. Darzins A. 1993. The pilG gene product, required for Pseudomonas aeruginosa pilus production and twitching motility, is homologous to the enteric, single-domain response regulator CheY. J. Bacteriol. 175:5934–5944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Darzins A. 1994. Characterization of a Pseudomonas aeruginosa gene cluster involved in pilus biosynthesis and twitching motility: sequence similarity to the chemotaxis proteins of enterics and the gliding bacterium Myxococcus xanthus. Mol. Microbiol. 11:137–153. 10.1111/j.1365-2958.1994.tb00296.x [DOI] [PubMed] [Google Scholar]
- 28. Bertrand JJ, West JT, Engel JN. 2010. Genetic analysis of the regulation of type IV pilus function by the Chp chemosensory system of Pseudomonas aeruginosa. J. Bacteriol. 192:994–1010. 10.1128/JB.01390-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008. 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aranda J, Poza M, Pardo BG, Rumbo S, Rumbo C, Parreira JR, Rodríguez-Velo P, Bou G. 2010. A rapid and simple method for constructing stable mutants of Acinetobacter baumannii. BMC Microbiol. 10:279. 10.1186/1471-2180-10-279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Robinson A, Brzoska AJ, Turner KM, Withers R, Harry EJ, Lewis PJ, Dixon NE. 2010. Essential biological processes of an emerging pathogen: DNA replication, transcription, and cell division in Acinetobacter spp. Microbiol. Mol. Biol. Rev. 74:273–297. 10.1128/MMBR.00048-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Conrad JC, Gibiansky ML, Jin F, Gordon VD, Motto DA, Mathewson MA, Stopka WG, Zelasko DC, Shrout JD, Wong GC. 2011. Flagella and pili-mediated near-surface single-cell motility mechanisms in P. aeruginosa. Biophys. J. 100:1608–1616. 10.1016/j.bpj.2011.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gaddy JA, Actis LA. 2009. Regulation of Acinetobacter baumannii biofilm formation. Future Microbiol. 4:273–278. 10.2217/fmb.09.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Espinal P, Martí S, Vila J. 2012. Effect of biofilm formation on the survival of Acinetobacter baumannii on dry surfaces. J. Hosp. Infect. 80:56–60. 10.1016/j.jhin.2011.08.013 [DOI] [PubMed] [Google Scholar]
- 35. Comolli JC, Hauser AR, Waite L, Whitchurch CB, Mattick JS, Engel JN. 1999. Pseudomonas aeruginosa gene products PilT and PilU are required for cytotoxicity in vitro and virulence in a mouse model of acute pneumonia. Infect. Immun. 67:3625–3630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chiang P, Burrows LL. 2003. Biofilm formation by hyperpiliated mutants of Pseudomonas aeruginosa. J. Bacteriol. 185:2374–2378. 10.1128/JB.185.7.2374-2378.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Whitchurch CB, Mattick JS. 1994. Characterization of a gene, pilU, required for twitching motility but not phage sensitivity in Pseudomonas aeruginosa. Mol. Microbiol. 13:1079–1091. 10.1111/j.1365-2958.1994.tb00499.x [DOI] [PubMed] [Google Scholar]
- 38. Fulcher NB, Holliday PM, Klem E, Cann MJ, Wolfgang MC. 2010. The Pseudomonas aeruginosa Chp chemosensory system regulates intracellular cAMP levels by modulating adenylate cyclase activity. Mol. Microbiol. 76:889–904. 10.1111/j.1365-2958.2010.07135.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wiegand I, Hilpert K, Hancock RE. 2008. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 3:163–175. 10.1038/nprot.2007.521 [DOI] [PubMed] [Google Scholar]
- 40. Rozen S, Skaletsky H. 2000. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 132:365–386. 10.1385/1-59259-192-2:365 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expression and growth curve analysis of recombineering systems in A. baumannii. (A) Expression of GFP from the IPTG-inducible vector pMMB67EH in A. baumannii using 0 mM to 5 mM IPTG. (B) Growth curve analysis of A. baumannii harboring λ Red and RecET systems with and without induced expression with IPTG. Download
Genetic organization of predicted type IV pili and flanking genes. Genes used in this study are shown as red arrows. Download
(A) Growth curve of wild-type (WT) and mutant strains from the biofilm assay. (B) Complementation of ΔpilH::Kanr. Asterisks denote significant differences in biofilm formation (t test, *, P < 0.0001; n = 8). (C) qRT-PCR analysis of the pilH gene in WT, ΔpilH::Kanr, and complemented ΔpilH strains. (D) qRT-PCR analysis of the pilI gene in WT and ΔpilH::Kanr strains. Download
Oligonucleotides used in this study