

ABSTRACT
Macropinocytosis is exploited by many pathogens for entry into cells. Coronaviruses (CoVs) such as severe acute respiratory syndrome (SARS) CoV and Middle East respiratory syndrome CoV are important human pathogens; however, macropinocytosis during CoV infection has not been investigated. We demonstrate that the CoVs SARS CoV and murine hepatitis virus (MHV) induce macropinocytosis, which occurs late during infection, is continuous, and is not associated with virus entry. MHV-induced macropinocytosis results in vesicle internalization, as well as extended filopodia capable of fusing with distant cells. MHV-induced macropinocytosis requires fusogenic spike protein on the cell surface and is dependent on epidermal growth factor receptor activation. Inhibition of macropinocytosis reduces supernatant viral titers and syncytia but not intracellular virus titers. These results indicate that macropinocytosis likely facilitates CoV infection through enhanced cell-to-cell spreading. Our studies are the first to demonstrate virus use of macropinocytosis for a role other than entry and suggest a much broader potential exploitation of macropinocytosis in virus replication and host interactions.
IMPORTANCE
Coronaviruses (CoVs), including severe acute respiratory syndrome (SARS) CoV and Middle East respiratory syndrome CoV, are critical emerging human pathogens. Macropinocytosis is induced by many pathogens to enter host cells, but other functions for macropinocytosis in virus replication are unknown. In this work, we show that CoVs induce a macropinocytosis late in infection that is continuous, independent from cell entry, and associated with increased virus titers and cell fusion. Murine hepatitis virus macropinocytosis requires a fusogenic virus spike protein and signals through the epidermal growth factor receptor and the classical macropinocytosis pathway. These studies demonstrate CoV induction of macropinocytosis for a purpose other than entry and indicate that viruses likely exploit macropinocytosis at multiple steps in replication and pathogenesis.
INTRODUCTION
Coronaviruses (CoVs), in addition to causing mild upper respiratory infections (1), have demonstrated the capacity to cause severe and deadly zoonotic diseases, including severe acute respiratory syndrome (SARS) (2, 3) and Middle East respiratory syndrome (4). However, virus-host interactions that allow for CoV adaptation and survival are not well understood. CoVs are a family of enveloped RNA viruses with large positive-strand genomes of 26 to 32 kb (5). The CoV life cycle is initiated by binding of the viral spike glycoprotein to a cellular receptor, followed by entry by direct fusion at the plasma membrane or by endosomal uptake (6). CoV replicase proteins extensively modify intracellular membranes to form sites of viral RNA synthesis, followed by virus assembly and maturation in the endoplasmic reticulum-Golgi intermediate compartment (ERGIC), with release by nonlytic secretory mechanisms (5, 7, 8).
For several CoVs, including murine hepatitis virus (MHV) and SARS CoV, cell surface expression of spike protein mediates interactions with receptors on adjacent cells, resulting in cell fusion and syncytium formation. Syncytium formation is a well-described cytopathic effect for many viruses in cell culture or in animal model systems and has been suggested to increase viral cell-to-cell spreading (9). However, for CoVs, syncytium formation has not been tested for a role in replication or cell-to-cell spreading. In this study, we demonstrate that CoVs induce plasma membrane changes consistent with macropinocytosis. Macropinocytosis is a type of endocytosis that is morphologically defined by the presence of membranous extensions of outwardly polymerizing actin termed membrane ruffles. Membrane ruffles nonspecifically surround and internalize fluid cargo into large vesicles or macropinosomes (10–12). Membrane ruffling is involved in cell migration, cell-cell interactions, environmental sampling, recycling of surface proteins and membranes, and delivery of bulk material to endosomes and lysosomes (13–16). Macropinocytosis can be transient, such as during the clearance of apoptotic bodies, or constitutive, like that associated with immune cells monitoring the environment for pathogens (17) or in cells transformed by the SRC oncogene (18). Macropinocytosis may be initiated by activation of the epidermal growth factor receptor (EGFR) and involves signaling though GTPases and kinases, including Rac1, Cdc42, and Pak1. Further, macropinocytosis requires sodium-hydrogen exchangers (NHE) and thus is specifically inhibited by 5-(N-ethyl-N-isopropyl)-amiloride (EIPA) (19, 20). Macropinocytosis has been identified as an entry mechanism for several pathogens, including Salmonella enterica, Neisseria gonorrhoeae, coxsackie B virus, influenza virus, Ebola virus, vaccinia virus, Nipah virus, and HIV (19, 21). In each of these studies, membrane ruffling or blebbing is induced by addition of the pathogen to a cell culture, and membrane modifications cease after pathogen internalization. To date, no role for macropinocytosis other than entry has been described as a host-pathogen interaction of viruses or bacteria.
In this study, we show that cells infected with MHV or SARS CoV induce macropinocytosis with the following well-defined characteristics: membrane ruffling and extensive filopodia; large vesicle internalization; increased bulk fluid uptake; actin polymerization; dependence on signaling through Cdc42, Rac1, and Pak1; and sensitivity to EIPA. We demonstrate that MHV-induced macropinocytosis is continuous once initiated ≥4 h postinfection (hpi), significantly after viral entry is completed. Replicating virus is required for induction of macropinocytosis, and inhibition of macropinocytosis impairs viral titers and cell-cell fusion. MHV-induced macropinocytosis requires fusogenic spike glycoprotein and is dependent on EGFR activation. The results support a role for CoV-induced macropinocytosis in cell fusion and virus cell-to-cell spreading, representing novel exploitation of macropinocytosis machinery for virus use.
RESULTS
Infection with MHV or SARS CoV induces continuous membrane ruffling.
During live-cell imaging of MHV-infected cells, we observed that MHV-infected cells displayed plasma membrane ruffling and extensive filopodia. We therefore examined the etiology and role of plasma membrane ruffling during CoV infection. Murine delayed brain tumor (DBT) astrocytoma cells were infected with MHV strain A59 (MHV A59) expressing green fluorescent protein (GFP) as a fusion with the replicase protein nsp3 (22), and cells were imaged continuously from 4 to 12 hpi, until the monolayer was entirely involved in syncytia or was lost (Fig. 1A; see Movie S1A to C in the supplemental material). Cell infection was confirmed both by the presence of GFP-nsp3 at replication complexes and by syncytium formation, a characteristic cytopathic effect of MHV infection. Membrane ruffling on multiple edges of the cell was noted to begin after 5 hpi, similar to the time of appearance of GFP-nsp3 fusion proteins at replication complexes, and was continuous throughout infection. The plasma membrane changes were extensive and included lamellipodia, filopodia, peripheral dorsal ruffles, and circular dorsal ruffles (Fig. 1A; see Movie S1A to C). The membrane ruffling associated with infection frequently resulted in the internalization of large vesicles. We measured the diameters of 386 vesicles in five live-imaging movies of MHV-infected cell monolayers. Vesicle diameters ranged from 0.49 to 4.14 µm with a mean diameter of 1.45 µm (Fig. 1B). This large vesicle diameter was similar to that reported for macropinosomes in multiple systems and much larger and more variable in size than that of endosomes generated by other forms of endocytosis (23). In addition to ruffling and vesicle formation, MHV-infected cells reproducibly manifested long filopodia that extended from infected cells and contacted distant cells, resulting in fusion at the point of contact and subsequent recruitment of cells into syncytia (see Movie S2A to C in the supplemental material).
FIG 1 .

Infection with MHV or SARS CoV induces continuous membrane ruffling. (A) DBT cells were infected with MHV-Δ2-GFP3 at an MOI of 1 PFU/cell and imaged from 4 to 12 hpi. The small panels to the right are enlargements showing details of the white-boxed area. Green fluorescence indicates nsp3. Black arrowheads denote vesicles. White arrowheads follow the evolution of a vesicle over time. See Movie S1A to C in the supplemental material. (B) Vesicle diameter was measured in five movies of infected cells. MOI = 1 PFU/cell, n = 386 vesicles. (C, D) DBT cells were infected with MHV A59 at an MOI of 1 PFU/cell for 8 h. DBT-hACE2 cells were infected with SARS CoV at an MOI of 0.1 PFU/cell for 24 h. Cells were fixed with 10% formalin and stained for F-actin (green). The arrow indicates a ruffle (C). Every third infected (or mock-infected) cell was imaged and scored for ruffling by three blinded reviewers. Data are represented as the means ± the standard errors of the means of two replicates performed in duplicate. Significance was assessed by one-way ANOVA with Dunnett’s post hoc test. n = ≥30 fields per replicate. *, P<0.05.
To determine the composition of these membrane ruffles, we tested for the presence of actin by staining MHV-infected, fixed cells with fluorescent phalloidin (Fig. 1C and D). Because macropinocytosis has not previously been reported in CoV-infected cells, we tested whether it occurred during infection with other CoVs, specifically, SARS CoV. To control for variations in cell type, DBT cells expressing the human ACE2 receptor for SARS CoV (DBT-hACE2) (24) were infected with SARS CoV for 24 h and compared with mock-infected DBT-hACE2 cells, mock-infected DBT cells, and MHV-infected DBT cells. Cells were fixed, stained with fluorescent phalloidin, imaged, and then scored for ruffling (Fig. 1C). Both MHV- and SARS CoV-infected cells demonstrated a significantly greater number of cells exhibiting membrane ruffles than mock-infected cells (Fig. 1D).
Infection with MHV or SARS CoV induces bulk fluid uptake consistent with macropinocytosis.
A hallmark of macropinocytosis is bulk fluid uptake from the surrounding environment. To test whether CoV-infected cells were inducing bulk fluid uptake, we used Nile Red neutral polystyrene nanoparticles with a diameter of 800 nm as markers, since their size is excluded from all endocytic pathways except macropinocytosis. MHV-infected DBT cells (Fig. 2A and B) and SARS CoV-infected DBT-hACE2 cells (Fig. 2B) were incubated with nanoparticles during the last 3 h of infection. Cell monolayers infected with either MHV or SARS CoV demonstrated a significantly higher percentage of cells with internalized nanoparticles than mock-infected cells (Fig. 2B). Macropinocytosis has been described as a means of pathogen entry in several different systems. However, since our data suggested that MHV-induced ruffling was occurring much later during infection, we next sought to determine the timing of bulk fluid uptake during MHV infection. Cells were mock infected or infected with MHV and then incubated with nanoparticles for 2-h intervals beginning at infection and fixed immediately afterward (Fig. 2C). Significantly increased nanoparticle uptake was first detected between 4 and 6 hpi and was also prominent at 6 to 8 hpi, indicating that increased fluid-phase uptake initiates at 4 h or later postentry and continues through the remainder of infection. This result was consistent with our visual and quantitative measurements of ruffling and vesicle formation. Because nanoparticle uptake occurred late during infection, we next tested whether replicating MHV is required to induce bulk fluid uptake. Cells were mock infected, infected with MHV, or incubated with an equal concentration of UV-inactivated or heat-inactivated MHV and incubated with nanoparticles for 2-h intervals, beginning at infection (Fig. 2C). UV-inactivated MHV did not result in increased nanoparticle uptake, suggesting that surface receptor interactions were insufficient to induce macropinocytosis. Heat-inactivated MHV also did not cause nanoparticle internalization, suggesting that the membrane-ruffling phenotype observed was not a cellular response to foreign particles. In addition, in order to confirm that macropinocytosis is not utilized for entry of CoVs, we labeled virions with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) and added them to DBT cells grown on a glass bottom dish at a high multiplicity of infection (MOI) of 25 PFU/cell (Fig. 2D). Virions were adsorbed for 30 min at 4°C to synchronize the infection before they were transferred to the 37°C chamber incubator surrounding the microscope. Cells were then live imaged for 1 h. While we did visualize viruses entering cells at the plasma membrane, we did not observe any evidence of cellular membrane ruffling at the site of entry or elsewhere on the cell. Thus, virus infection is required for induction of bulk fluid uptake, and MHV-induced macropinocytosis is not associated with virus entry.
FIG 2 .

Infection with MHV or SARS CoV induces bulk fluid uptake consistent with macropinocytosis. (A, B) DBT cells were infected with MHV A59 at an MOI of 1 PFU/cell for 8 h. DBT-hACE2 cells were infected with SARS CoV at an MOI of 0.1 PFU/cell for 24 h. Nanoparticles were added 3 h prior to fixation, and cells were washed, fixed, stained, and imaged. Arrows denote nanoparticles (red). DAPI is blue, and nsp8 is green. (B) Data are represented as the mean ± the standard error of the mean of two replicates performed in duplicate. n = ≥30 fields per replicate. (C) Cells were mock infected or infected with MHV, UV-inactivated MHV (UV), or heat-inactivated MHV (HI) at an MOI = 1 PFU/cell (or the equivalent volume of noninfectious virus) for 8 h. Nanoparticles were added in 2-h increments, as designated, and cells were washed, fixed, stained, and imaged. Data are represented as the mean ± the standard error of the mean of two replicates performed in duplicate. (D) DBT cells on a glass bottom dish were infected with DiI-labeled MHV (white arrows) at an MOI of 25 PFU/cell, incubated at 4°C for 30 min, and imaged in a 37°C chamber incubator for 1 h. Time is in minutes:seconds. Virus fusion with the cell was observed, but membrane ruffling was absent. An image representative of triplicate experiments is shown. Significance was assessed by one-way ANOVA with Dunnett’s post hoc test. **, P < 0.005; ***, P < 0.0001.
MHV-induced macropinocytosis is dependent on the classical macropinocytosis pathway.
We next determined whether MHV-induced macropinocytosis requires known mediators of cellular macropinocytosis. We selected Rac1, Cdc42, and Pak1 from the classical macropinocytosis pathway for small interfering RNA (siRNA) inhibition. Inhibition of RhoA was chosen as a negative control, since it is not associated with macropinocytosis. For each siRNA molecule, a target knockdown rate of ≥80% was confirmed by immunoblotting (Fig. 3A and B). Transfection efficiency was tested with siRNA-AllStars-GFP and found to be >96% (data not shown). Inhibition of Pak1, Cdc42, and Rac1 resulted in significantly decreased nanoparticle internalization following MHV infection, while nanoparticle uptake was unchanged in cells transfected with a scrambled siRNA or with a siRNA targeting RhoA (Fig. 3C). These results demonstrate that MHV-induced macropinocytosis signals through a known cellular macropinocytosis pathway.
FIG 3 .
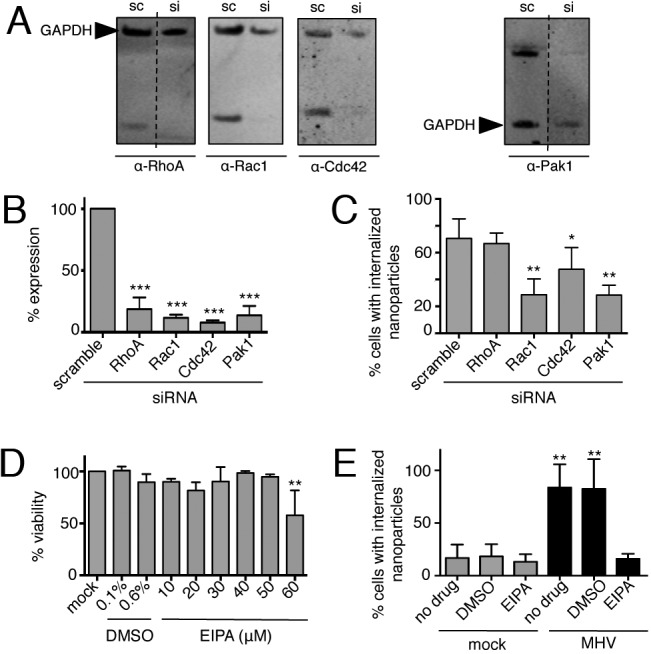
MHV-induced macropinocytosis is dependent on the classical macropinocytosis pathway. (A, B) Cells were reverse transfected with siRNA for 72 h, and protein knockdown was confirmed by immunoblotting (A) and standardized to GAPDH (B). Scrambled siRNA (sc)- and siRNA (si)-treated samples are from the same gel for each protein. RhoA and Pak1 are from discontinuous lanes separated by dashed lines. Data are represented as the means ± the standard errors of the means in triplicate assays. (C) Cells were reverse transfected for 68 h and infected with MHV for 8 h. Nanoparticles were added during the final 3 h, and cells were washed, fixed, stained, and imaged. Data are represented as the means ± the standard errors of the means of two replicates performed in duplicate, n = ≥30 fields per replicate. (D) The 12-h toxicity of EIPA was assessed with CellTiter-Glo. (E) Cells were mock infected or infected with MHV at an MOI of 1 PFU/cell for 8 h with no drug, DMSO, or 40 µM EIPA. Nanoparticles were added during the final 3 h of infection. Cells were washed, fixed, stained, and imaged, and the percentage of cells with internalized nanoparticles was calculated. Data are represented as the means ± the standard errors of the means of two replicates performed in duplicate. Significance was assessed by one-way ANOVA with Dunnett’s post hoc test. *, P < 0.05; **, P < 0.01; ***, P < 0.0001.
EIPA is an inhibitor of NHE and is the most specific known chemical inhibitor of macropinocytosis. Other endocytic pathways do not use NHE and are not impacted by treatment (25). We compared nanoparticle internalization in MHV-infected and uninfected cells treated with nontoxic doses of EIPA (Fig. 3D) or with dimethyl sulfoxide (DMSO), the vehicle control. EIPA-treated, infected cells internalized significantly fewer nanoparticles than DMSO-treated, infected cells (Fig. 3E). Thus, all of the data collected to this point are consistent with the hypothesis that MHV induces macropinocytosis during infection: membrane ruffling, bulk fluid uptake, dependence on classical pathway members, and sensitivity to EIPA.
Inhibition of macropinocytosis impairs MHV replication.
We next tested the requirement for macropinocytosis during MHV replication (Fig. 4). We utilized siRNA knockdown of Pak1, treatment with Clostridium difficile toxin A (CdtA), or treatment with EIPA and determined the effect on viral replication. CdtA decreases the ADP-ribosylation of Rho family proteins, and thus, we used this as an independent mechanism to confirm the relationship between macropinocytosis and viral replication (26). Inhibition of Pak1 with siRNA prior to infection caused a ≥80% decrease in MHV titers at 12 hpi, while the use of a scrambled siRNA or an siRNA against RhoA did not have a significant effect on titers (Fig. 4A). Treatment with CdtA also caused a significant decrease in MHV titers at 10 hpi (Fig. 4B and C). EIPA was added to MHV-infected cell monolayers at different concentrations and times postinfection (Fig. 4D and E). EIPA treatment resulted in a ≥75% decrease in virus titers when added at any time after 2 hpi. To test the effect of EIPA on different stages of the virus life cycle, we infected cells with MHV expressing firefly luciferase (FFL) as a fusion with the replicase protein nsp2 (22) and measured FFL activity in the presence or absence of EIPA added at 8 hpi (Fig. 4F). In replicate experiments, supernatant was collected at 10 hpi and cell-associated virus was collected by freezing and thawing infected cells three times. The titer of each sample was assessed by plaque assay (Fig. 4G). Intracellular FFL-nsp2 expression from the viral genome was not altered by EIPA treatment. In addition, the viral titer was significantly decreased in the supernatant of cells treated with EIPA, while the intracellular virus titer was not affected by the addition of EIPA. Together, these results indicate that EIPA does not affect intracellular virus replication, assembly, or maturation but alters the overall peak titer of the infectious virus in the supernatant, consistent with an effect on late stages of virus release or cell-to-cell spreading.
FIG 4 .

Inhibition of macropinocytosis impairs MHV replication. (A) Cells were reverse transfected for 68 h and infected with MHV for 12 h. The titers of supernatant samples were determined via plaque assay. Data are represented as the means ± the standard errors of the means of two replicates performed in duplicate. (B) The 12-h toxicity of CdtA was assessed with CellTiter-Glo. (C) Cells were treated with HEPES buffer or 10 µM CdtA for 2 h prior to infection with MHV A59 at an MOI of 1 PFU/cell. The viral titer was measured at 10 hpi. Data are represented as the mean ± the standard errors of the mean of an experiment done in triplicate. (D, E) Cells were infected with MHV at an MOI of 1 PFU/cell. At the times indicated postinfection, 0.4% DMSO or 10, 20, or 40 µM EIPA was added. The viral titer was measured at 12 hpi. EIPA was added at 6 hpi at 10, 20, or 40 µM (D) or at 2, 4, 6, or 8 hpi at 40 µM (E). Data are represented as the mean ± the standard error of the mean of two replicates performed in duplicate. (F, G) Cells were infected with MHV-FFL2 at an MOI of 1 PFU/cell. At 8 hpi, 0.4% DMSO or 40 µM EIPA was added. Supernatant was collected at 10 hpi, and the viral titer was determined. Cells were collected in luciferase lysis buffer and assessed for luminescence (F) or in DMEM and subjected to three rounds of freezing and thawing before the titer was determined (G). Data are represented as the mean ± the standard error of the mean of two replicates performed in duplicate. Significance was assessed by one-way ANOVA with Dunnett’s post hoc test. *, P < 0.05; **, P < 0.005; ***, P < 0.0005; n.s., not significant.
The presence of fusogenic spike protein at the plasma membrane is required to induce macropinocytosis.
Having determined that MHV-induced macropinocytosis requires active virus replication and occurs at a late stage of the virus life cycle, we investigated the role of the spike protein in CoV-induced macropinocytosis by using MHV A59 and recombinant MHV 2S. In MHV A59, the spike protein is cleaved by furin in the trans-Golgi network during virion maturation (27), resulting in fusogenic spike protein on nascent virions and on the plasma membrane of infected cells. Interaction of the MHV A59 spike protein with the cellular receptor results in either virus-cell fusion and cell entry or cell-cell fusion and syncytium formation (27). In contrast, the spike protein of another strain, MHV-2, is not cleaved by furin during the exit of nascent virions but rather is cleaved by cathepsins following endocytosis during entry (28). Thus, released infectious MHV-2 does not have a fusogenic spike protein, and cells infected with MHV-2 do not express a fusogenic spike protein on the cell surface or generate syncytia in culture.
To test for the role of spike protein and spike protein fusogenic activity in MHV-induced macropinocytosis, we mock infected or infected cells with MHV A59 or with recombinant MHV A59 encoding the MHV-2 spike protein (MHV 2S) (29) and tested nanoparticle internalization. MHV 2S-infected cells internalized significantly fewer nanoparticles than MHV A59-infected cells (Fig. 5A). To test the specific requirement for spike protein cleavage, we infected cells with an MHV A59 C12 mutant containing an amino acid mutation (H716D) that abolishes the furin cleavage site (30). During infection with MHV C12, there was no significant difference in nanoparticle internalization from that in mock-infected cells (Fig. 5A). We next used a furin inhibitor, peptidyl chloromethyl ketone (dec-RVKR-cmk) (27) to block furin cleavage of the MHV A59 spike protein. Treatment with dec-RVKR-cmk decreased both nanoparticle uptake and syncytium formation in a concentration-dependent manner (Fig. 5B to D). To test whether spike protein cleavage alone is sufficient to induce macropinocytosis, we treated cells infected with MHV 2S with L-1-tosylamide-2-phenylethyl chloromethyl ketone (TPCK)-trypsin in order to generate fusogenic spike protein on the cell surface and assessed nanoparticle uptake. Cells infected with MHV 2S and treated with trypsin recovered the capacity for nanoparticle internalization (Fig. 5E). Together, these results demonstrate that expression of fusion-competent spike protein at the plasma membrane is necessary for MHV macropinocytosis induction.
FIG 5 .

The presence of fusogenic spike protein at the plasma membrane is required to induce macropinocytosis. (A) Cells were mock infected or infected with MHV A59, 2S, or C12 at an MOI of 1 PFU/cell. Nanoparticles were added at 5 hpi for 3 h, and the cells were washed, fixed, stained, and imaged. (B to D) DBT cells were mock infected or infected with MHV A59 at an MOI of 1 PFU/cell in DMEM or in DMEM with DMSO or dec-RVKR-cmk (dRc) for 8 h. The cells were exposed to dec-RVKR-cmk for 12 h, and toxicity was assessed with CellTiter-Glo (B). Nanoparticles were added 3 h prior to fixation, and cells were washed, fixed, stained, and imaged. Percentages of syncytial cells (C) and cells with internalized nanoparticles (D) were measured. (E) Cells were mock infected or infected with MHV A59 or 2S at an MOI of 1 PFU/cell. At 5 hpi, cells were treated with TPCK trypsin for 5 min and washed and then nanoparticles were added for 3 h. Cells were washed, fixed, stained, and imaged. Data are represented as the mean ± the standard error of the mean of two replicates performed in duplicate. n = ≥30 cells per replicate. Significance was assessed by one-way ANOVA with Dunnett’s post hoc test; ***, P < 0.0001; **, P < 0.01; *, P < 0.05.
MHV-induced macropinocytosis is associated with, but independent of, syncytium formation.
The necessity for cleaved spike protein in macropinocytosis induction could be explained either by a requirement for spike protein-mediated cell-cell fusion or by a direct role for spike protein in macropinocytosis induction. To distinguish between these possibilities, we used two approaches: induction of syncytia by different methods and blockade of syncytium formation with antireceptor antibodies. To test the cell fusion hypothesis, cells were chemically treated with polyethylene glycol 1500 (PEG-1500) or transfected with the vesicular stomatitis virus G protein (VSV-G) (Fig. 6A) to induce cell-cell fusion and then incubated with nanoparticles. Both PEG-1500 treatment and VSV-G expression resulted in small syncytia with fewer than 10 nuclei. The nanoparticle uptake levels of syncytia with similar numbers of nuclei from MHV-infected cells, PEG-1500-treated cells, and VSV-G-transfected cells were compared. PEG-1500 treatment resulted in nanoparticle uptake greater than that of mock-infected cells but significantly less than that seen during MHV infection. Expression of VSV-G did not result in greater nanoparticle uptake than in mock-infected cells. We next blocked the interactions of MHV A59 spike protein with its cellular receptor, carcinoembryonic antigen (CEACAM), by adding anti-CEACAM blocking antibodies at 2 hpi and measured the effect on syncytium size and nanoparticle uptake (Fig. 6B). The anti-CEACAM antibodies resulted in a significant reduction in syncytial cell number and size but did not significantly decrease nanoparticle uptake by infected cells (Fig. 6C). Together, these results demonstrate that cleaved spike protein at the cell surface, and not cell fusion alone, is required to initiate and sustain macropinocytosis.
FIG 6 .

MHV-induced macropinocytosis is associated with, but independent of, syncytium formation. (A) DBT cells were mock infected or infected with MHV A59 at an MOI of 1 PFU/cell for 8 h, treated with PEG for 1 min, washed, and incubated for 3 h or transfected with VSV-G for 24 h. Nanoparticles were added 3 h prior to fixation, and cells were washed, fixed, stained, and imaged. Syncytia with ≤10 nuclei were analyzed. Data are represented as the mean ± the standard error of the mean of two replicates, each performed in duplicate. n = ≥30 cells per replicate. (B, C) Cells were mock infected or infected with MHV at an MOI of 1 PFU/cell. Anti-CEACAM blocking antibodies were added at 2 hpi, nanoparticles were added at 5 hpi, and cells were washed, fixed, stained, and imaged at 8 hpi. The number of nuclei per syncytium (B) and the percentage of cells with internalized nanoparticles (C) were measured. Significance was assessed by one-way ANOVA with Dunnett’s post hoc test. ***, P < 0.0001; **, P < 0.01; *, P < 0.05; n.s., not significant.
CoV-induced macropinocytosis is dependent on EGFR activation.
Signaling through the EGFR is essential for induction of macropinocytosis in several systems (31–33). To test whether EGFR activation is required for CoV-induced macropinocytosis, we utilized gefitinib, which specifically inhibits EGFR autophosphorylation and prevents EGFR activation (34). DBT cells were mock infected or infected with MHV A59, gefitinib was added after viral entry at 1.5 hpi, and cells were analyzed for nanoparticle uptake and syncytium size (Fig. 7A to C). Addition of gefitinib to MHV-infected cells significantly decreased the percentage of syncytia with internalized nanoparticles (Fig. 7B). We also observed that gefitinib significantly decreased the number of nuclei in a syncytium, with only half as many nuclei as in a DMSO-treated syncytium (Fig. 7C). We then evaluated the effect of EIPA on syncytium size and discovered that treatment with EIPA decreased both the percentage of infected cells involved in syncytia (Fig. 7D) and the size of the syncytia (Fig. 7E). These data suggest that CoVs utilize EGFR activation to induce macropinocytosis and to initiate cell-to-cell spreading.
FIG 7 .

CoV-induced macropinocytosis is dependent on EGFR activation. (A) Gefitinib was added to cells for 12 h, and toxicity was assessed with CellTiter-Glo. (B, C) DBT cells were mock infected or infected with MHV A59 at an MOI of 1 PFU/cell in DMEM or in DMEM supplemented with DMSO or gefitinib at 1.5 hpi. Cells were fixed at 8 hpi. Nanoparticles were added 3 h prior to fixation, and cells were washed, fixed, stained, and imaged. The percentage of cells with internalized nanoparticles (B) and the number of nuclei per syncytium (C) were determined. (D, E) DBT cells were mock infected or infected with MHV A59 at an MOI of 1 PFU/cell in DMEM or in DMEM supplemented with DMSO or EIPA at 1.5 hpi. Cells were fixed at 8 hpi, stained, and imaged. The percentage of infected cells involved in syncytia (D) and the number of nuclei per syncytium (E) were determined. Data are means ± standard deviations of triplicates. n = ≥30 fields per replicate. Statistical significance was assessed by one-way ANOVA with Dunnett’s post hoc test. ***, P < 0.0001; **, P < 0.01; *, P < 0.05.
DISCUSSION
In this study, we demonstrate that MHV infection induces macropinocytosis, as defined by characteristic membrane ruffling; internalization of large pleomorphic vesicles; increased fluid-phase uptake; dependence on Rac1, Cdc42, and Pak1; and sensitivity to EIPA. MHV-induced macropinocytosis requires cell surface expression of the cleaved, fusogenic spike protein, and spike protein-mediated induction of macropinocytosis is dependent on direct or indirect interactions of the spike protein with the EGFR. We have summarized these data in the model shown in Fig. 8.
FIG 8 .

Model of macropinocytosis during CoV infection. MHV 2S enters the cell via endocytosis after the spike protein interacts with the CEACAM receptor. Spike protein is cleaved to its fusogenic form by cathepsins after entry via endocytosis, and the virus fuses with the endosomal membrane to release the genome to the cytoplasm. Replication occurs, and virions are assembled in the ERGIC and then packaged and released via exocytosis. Spike protein that reaches the surface of the cell is uncleaved (black ball and stick) and cannot mediate syncytium formation with neighboring cells. MHV A59 enters the cell via fusion at the cell membrane after the spike protein interacts with the CEACAM receptor. The genome immediately enters the cytoplasm, and replication occurs. Packaging of nascent virions occurs in the ERGIC, and free spike proteins and spike protein incorporated into virions are cleaved by furin in the trans-Golgi compartment. Virions are packaged and released via exocytosis. Spike protein that reaches the cell surface is cleaved and fusogenically active (red ball and stick) and can mediate fusion events with neighboring cells. This cleaved, activated spike protein can also mediate interactions, potentially through the EGFR (purple ball and stick), that induce the macropinocytosis pathway within the cell, which relies on Rac1, Cdc42, and Pak1. Actin modifications at the cell surface then cause membrane ruffling and macropinosome internalization, in addition to filopodia that can facilitate spike protein-receptor interactions with neighboring cells. The location of spike protein cleavage is denoted by the red-striped regions.
CoVs have been shown previously to modify cytoplasmic membranes to form viral replication complexes (7). Here, we report that CoVs also have the capacity to modify the plasma membrane through activation of the macropinocytosis pathway. Prior to this study, the only role demonstrated for virus-induced macropinocytosis was in virus entry. MHV-associated macropinocytosis, in contrast, is initiated relatively late during infection and is continuous once activated. MHV 2S does not stimulate cell-cell fusion or macropinocytosis in infected cells but still causes disease in vivo, suggesting that macropinocytosis may not be required for the fitness or pathogenesis of all strains of MHV or for all CoVs. The disease phenotypes of MHV A59 and 2S differ, as infection with MHV A59, but not MHV 2S, causes spinal cord demyelination (35), suggesting that macropinocytosis-driven cell-to-cell spreading could be a tissue-specific strategy. The conservation of macropinocytosis during MHV and SARS CoV infections, however, suggests that macropinocytosis induction could be an important determinant of pathogenesis for viruses capable of maintaining cleaved spike protein on the surface of infected cells.
Our results demonstrate two consistent and dramatic phenotypes associated with MHV-induced macropinocytosis: ruffling with vesicle internalization and filopodia associated with cell fusion and recruitment into syncytia. The fact that inhibition of MHV-induced macropinocytosis impairs the overall virus titer while not affecting entry or intracellular infectious virus suggests the possibility of multiple functions that favor overall virus fitness, specifically, recruitment of membranes and nutrients into infected cells and syncytia, enhancement of virus release, or virus cell-to-cell spreading. MHV A59 acquires its viral envelope by budding through the ERGIC and is released from the cell without lysis via the exocytic pathway. No previous role for a traditionally endocytic pathway such as macropinocytosis has been reported to be used during viral release (36, 37). The observed increase in the formation of long filopodia from infected cells that contact and recruit uninfected cells into syncytia suggests a role for macropinocytosis as a mechanism by which CoVs potentiate cell communication over significant distances. Filopodia could act as mediators of CoV cell-cell fusion by concentrating spike protein and virus at the filopodium tip. Actin modifications have been previously implicated in the cell-to-cell spreading of other viruses. Vaccinia virus utilizes actin tails to infect distant cells at a rate faster than that at which it infects adjacent cells (38, 39). HIV and human T-cell lymphotropic virus use filopodia to create virological synapses to transfer infectious material from one cell to another (40, 41). Macropinocytosis can occur in polarized cells (42) and could represent a novel mechanism of virus cell-to-cell spreading over tight junctions within airway epithelium. Viral spreading in this manner has several advantages over viral spreading via exocytosis, specifically, immune evasion, concentration of reagents, speed, and the capacity to circumvent physical barriers. Increasing the size and duration of plasma membrane extensions could increase opportunities for interactions with adjacent or distant cells.
Our results also show that expression of cleaved, fusogenic spike protein on the cell surface is necessary to induce macropinocytosis, whether by furin-mediated cleavage in the cell or by exogenous cleavage by trypsin on the cell surface. Induction of macropinocytosis also requires EGFR activation and signaling through the known macropinocytosis cellular pathway. On the basis of our results, we propose a model in which fusogenic spike protein on the plasma membrane results in EGFR activation, leading to a signaling cascade that manifests as increased membrane ruffling and filopodium formation to facilitate cell-cell fusion and virus spread.
Spike protein-EGFR interactions could occur at several cellular locations and be either direct or indirect. Spike protein localized at the plasma membrane could directly interact with the neighboring EGFR. The EGFR is also a highly trafficked receptor that translocates to the mitochondria after activation (43) and then promptly back to the plasma membrane, providing opportunities for interactions between the spike protein and the EGFR within the cell. Proteins from other viruses have been previously shown to activate tyrosine kinase receptors in a ligand-independent fashion, such as E5 of bovine papillomaviruses (44), or through the production of a cellular ligand, as in human papillomavirus 16 (44).
Our results raise many important questions for future studies. What is the relationship of GTPase and EGFR activation to specific stages of the viral life cycle? Does the virus receptor play a role in facilitating EGFR activation and induction of macropinocytosis? How does the capacity to induce macropinocytosis favor the fitness of MHV A59? The results of our studies define novel roles for the CoV spike protein and for macropinocytosis during CoV infection and raise the possibility that other RNA viruses may usurp macropinocytosis machinery for purposes other than virus entry.
MATERIALS AND METHODS
Viruses, cells, plasmids, and antibodies.
Recombinant MHV A59 (wild type, GenBank accession no. AY910861) (45, 46) was used as the wild-type strain in MHV experiments. Work with SARS CoV was completed with the reverse genetics infectious clone based on the Urbani strain (47). Viral studies with SARS CoV were performed in a select-agent-certified biosafety level 3 laboratory by using protocols reviewed and approved by the Institutional Biosafety Committee of Vanderbilt University and the Centers for Disease Control for the safe study and maintenance of SARS CoV. MHV-Δ2-GFP3 and MHV-FFL2 have been previously reported (22). MHV 2S and C12 were kind gifts from Susan Weiss (University of Pennsylvania) and have been previously described (29, 48). DBT cells (49) were grown in Dulbecco’s modified Eagle medium (DMEM) with 10% fetal bovine serum, 1% 1 M HEPES, 100 U/ml penicillin, 100 µg/ml streptomycin, and 0.23 µl/ml amphotericin B. DBT cells expressing the SARS CoV receptor (DBT-hACE2) (24) were cultured as previously described. The VSV-G plasmid used was a kind gift from Michael Whitt (University of Tennessee Health Science Center [UTHSC]) (50). MHV nsp8-specific rabbit antiserum (VU123) has been previously described (51). Alexa Fluor 488 phalloidin (Molecular Probes) was used to stain for F-actin. Rabbit anti-Cdc42 C-terminal polyclonal antibody ab155940, rabbit anti-RhoA polyclonal antibody ab86297, mouse anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (6C5) monoclonal antibody ab8245, mouse anti-Rac1 (0.T.127) monoclonal antibody ab33186, and rabbit anti-Pak1 (phosopho-S144) (EP656Y) monoclonal antibody ab40795 were purchased from Abcam. Rabbit polyclonal antiserum specific for CEACAM was a kind gift from Tom Gallagher (Loyola University, Chicago, IL) (52). CdtA was a kind gift from Borden Lacy (Vanderbilt University) (53).
Live imaging of MHV-infected cells.
DBT cells were seeded onto 35-mm MatTek glass bottom culture dishes for 48 h. Cells were then infected with MHV-Δ2-GFP3 at an MOI of 1 PFU/cell. In a separate experiment, cells were transfected with a GFP-expressing plasmid before being infected with MHV A59 at an MOI of 1 PFU/cell. In the viral entry experiment, cells were infected with DiI-labeled MHV A59 at an MOI of 25 PFU/cell and the infection was synchronized at 4°C for 30 min before imaging. Plates were transferred to a live-cell incubator surrounding the objective stage of a Nikon Eclipse TE-2000S widefield fluorescence microscope. Cells were imaged with a 40× oil immersion lens through differential interference contrast and fluorescein isothiocyanate short-pass filters, and images were captured at 30-s intervals. The resulting images were assembled with Nikon Elements, ImageJ, Adobe Photoshop CS2, and QuickTime Pro.
Fluorescent labeling of virions.
Viral stocks of MHV A59 were grown on DBT cells, purified with a cushion of 5.5 ml of 20% sucrose (20% sucrose, 0.1 M MgSO4, 50 mM HEPES, 150 mM NaCl [pH 7.4], 0.2-µm filtered) layered over 1.5 ml of 60% sucrose (20% sucrose, 0.1 M MgSO4, 50 mM HEPES, 150 mM NaCl [pH 7.4], 0.2-µm filtered), and spun at 27,000 rpm at 4°C for 90 min. Virions were then incubated with 5 µM DiI (Life Technologies) for 1 h at room temperature, purified with a cushion of 1.5 ml 20% sucrose layered over 1 ml 60% sucrose, and spun at 28,000 rpm at 4°C for 90 min to remove the free dye.
Determination of bulk fluid uptake.
DBT cells grown to 80% confluence on 12-mm glass coverslips were infected with MHV A59 at an MOI of 1 PFU/cell. At designated times postinfection, 100 µg/ml 800-nm fluorescent polystyrene nanoparticles (Corpuscular Inc.) were added to the cells, which were then incubated for 3 h at 37°C. DBT-hACE2 cells were infected with SARS CoV for 24 h at an MOI of 0.1 PFU/cell, and 100 µg/ml nanoparticles were added for the final 3 h prior to fixation. Cells were washed with medium three times for 5 min. Drugs were added where indicated. Cells were fixed in 100% methanol or 10% formalin and then put at −20°C or 4°C, respectively, overnight. Cells were rehydrated in phosphate-buffered saline (PBS) for 10 min and blocked in PBS containing 5% bovine serum albumin (BSA). The following steps were performed with immunofluorescence assay (IFA) wash solution (PBS containing 1% BSA and 0.05% Nonidet P-40) at room temperature. Blocking solution was aspirated, and cells were incubated in primary antibodies where indicated (MHV, anti-nsp8 antibody at 1:250) for 45 min. Cells were then washed in IFA wash solution three times for 5 min per wash. Cells were then incubated in secondary antibodies (goat anti-rabbit– and anti-mouse–Alexa Fluor 488 [1:1,000] and 546 [1:2,000] [Molecular Probes]) for 30 min. Cells were washed again three times for 5 min per wash, stained with 0.005 mg/ml 4′,6-diamidino-2-phenylindole (DAPI), washed with PBS three times for 5 min per wash, and then rinsed in water. Coverslips were mounted with Aqua-Poly/Mount (Polysciences, Inc.) and visualized by confocal immunofluorescence microscopy on a Zeiss LSM 510 laser scanning confocal microscope or a by widefield microscopy with a Nikon Eclipse TE-2000S fluorescence microscope at 488 and 543 nm with a 40× oil immersion lens. Images were processed and assembled with Nikon Elements, ImageJ, and Adobe Photoshop CS2 (12.0 by 64), and infected and mock-infected cells were processed in parallel. Scores for ruffling events were calculated by encrypting the images and allowing three independent blinded reviewers to evaluate if a cell was positive or negative for ruffling by utilizing fixed criteria and a binary system. Cells were determined to have internalized nanoparticles on the basis of a binary scoring system.
Viral replication assays.
For viral replication analysis, DBT cells were infected in triplicate with MHV A59 at the MOI indicated for each experiment. Following 30 min of adsorption with rocking at room temperature, the medium was aspirated and the cells were washed three times with PBS and then incubated with prewarmed medium at 37°C. Aliquots of medium were collected at the indicated times postinfection, and virus titers were determined in duplicate by plaque assay on DBT cells as described previously (54). EIPA (Sigma) was applied during infection as indicated.
Toxicity assays.
The toxicity of CdtA (Lacy lab, Vanderbilt), EIPA (Sigma), dec-RVKR-cmk (Calbiochem), and gefitinib (Selleck Chemicals) for DBT cells was tested for 12 h by using the Cell Titer Glo (Promega) protocol in accordance with the manufacturer’s instructions.
siRNA assays.
SMARTpools for murine Cdc42, Rac1, Pak1, RhoA, and scrambled siRNA were obtained from Thermo Scientific. DBT cells were reverse transfected in accordance with the protocol for Lipofectamine RNAiMAX (Life Technologies). For siRNA knockdown and virus infection, cells were reverse transfected for 68 h and infected with MHV at an MOI of 1 for 8 h and nanoparticle and fixation protocols were completed as described above. To test protein knockdown, cells were reverse transfected, incubated for 72 h, harvested with non-SDS lysis buffer, subjected to immunoblot analysis, and imaged with the Odyssey Imaging System (LiCor). Band intensity was normalized to that of GAPDH. siRNA transfection efficiency was tested by transfection of AllStars Negative siRNA Alexa Fluor 488 (Qiagen). For replication assays with siRNA knockdown, cells were reverse transfected for 68 h before infection with MHV at an MOI of 1 for 12 h.
UV inactivation and heat inactivation assays.
MHV was UV inactivated by placing 1 ml of stock in an open 30-mm dish in a UV cross-linker for 45 min. MHV was heat inactivated by placing 1 ml of stock at 70°C for 45 min. Inactivated-virus titers were determined by plaque assay, and the virus was considered to be inactivated when plaque formation was absent. DBT cells were mock infected or infected with MHV A59, UV-inactivated virus, or heat-inactivated virus at an MOI of 1 or an equivalent volume of noninfectious virus for 8 h.
Cell-associated virus assay.
DBT cells were infected with MHV-FFL2 at an MOI of 1 PFU/cell. At 8 hpi, cells were washed twice with PBS and then DMSO or EIPA was added. At 10 hpi, supernatant was collected and cells were harvested in DMEM and subject to three rounds of freezing and thawing. The titer of each fraction was determined by plaque assay as previously described. Cells from a parallel experiment were collected in Luciferase Assay Buffer (Promega) and assessed for luminescence via the manufacturer’s protocol.
Furin inhibition assay.
DBT cells grown to 80% confluence on 12-mm glass coverslips were mock infected or infected with MHV A59 at an MOI of 1 PFU/cell. Cells were left untreated, treated with 0.25% DMSO, or treated with various concentrations of dec-RVKR-cmk (Sigma) at the time of infection. Nanoparticle assays were completed, and cells were fixed and stained as described above and then imaged and assessed for nanoparticle uptake and syncytium formation.
Fusion assays.
DBT cells grown to 80% confluence on 12-mm glass coverslips were treated for 1 min with PEG-1500 (Sigma) and rinsed three times with PBS before incubation with nanoparticles for 3 h. Nanoparticle assays were completed, and cells were fixed and stained, imaged, and assessed for nanoparticle uptake and syncytium formation. Alternatively, cells were grown to ~60% confluence and transfected with VSV-G in accordance with the protocol for Lipofectamine 2000 (Life Technologies) for 24 h before nanoparticle incubation for 3 h. DBT cells grown to ~80% confluence on 12-mm glass coverslips were infected with MHV A59 or 2S at an MOI of 1 PFU/cell. At 5 hpi, cells were treated with 2.5 µg/ml TPCK trypsin (Sigma) for 5 min and washed once with PBS and then nanoparticles were added and the cells were incubated for 3 h, washed, fixed, stained, and imaged. DBT cells grown to 80% confluence on glass coverslips were mock infected or infected with MHV at an MOI of 1 PFU/cell. Anti-CEACAM blocking antibodies were added at 2 hpi, nanoparticles were added at 5 hpi for 3 h of incubation, and the cells were washed, fixed, stained, and imaged.
Statistical analysis.
Statistical tests were used as noted in the figure legends and performed with GraphPad Prism software (GraphPad, La Jolla, CA). The statistical significance of differences from uninfected or vehicle-treated samples is denoted as stated in the figure legends and was determined by one-way analysis of variance (ANOVA) with Dunnett’s multiple-comparison test. P values of <0.05 were considered to be statistically significant.
SUPPLEMENTAL MATERIAL
Infection with MHV induces continuous membrane ruffling. DBT cells were infected with MHV-Δ2-GFP3 at an MOI of 1 PFU/cell and imaged from 4 to 12 hpi. This movie spans 8 to 8.6 hpi. Download
The left cells are enlarged. Download
The right cells are enlarged. Download
MHV-induced filopodia potentiate cell-cell interactions. DBT cells were transfected with a GFP-expressing plasmid and infected with MHV at an MOI of 1 PFU/cell. Cells were imaged from 7 to 12 hpi. This movie shows several interactions between the filopodia of the infected cell and neighboring uninfected cells. Download
Enlarged region of the movie. Download
Enlarged region of the movie. Download
ACKNOWLEDGMENTS
We thank members of the Denison laboratory for useful discussions and critical review of the manuscript. We thank Eric Prentice for technical assistance. We thank Susan Weiss (University of Pennsylvania) for the kind gift of MHV 2S and MHV C12 viruses, Michael Whitt (UTHSC) for the VSV-G plasmid, Tom Gallagher (Loyola) for the anti-CEACAM antibodies, and Borden Lacy (Vanderbilt) for CdtA.
This research was supported by Public Health Service awards T32 GM007347 (M.C.F.), R01 AI50083 (M.R.D.), and AI108197 (M.R.D.) and The Elizabeth B. Lamb Center for Pediatric Research. The Vanderbilt Cell Imaging Shared Resource (DK20593) provided additional support.
Footnotes
Citation Freeman MC, Peek CT, Becker MM, Smith EC, Denison MR. 2014. Coronaviruses induce entry-independent, continuous macropinocytosis. mBio 5(4):e01340-14. doi:10.1128/mBio.01340-14.
REFERENCES
- 1. Arden KE, Nissen MD, Sloots TP, Mackay IM. 2005. New human coronavirus, hcov-nl63, associated with severe lower respiratory tract disease in Australia. J. Med. Virol. 75:455–462. 10.1002/jmv.20288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, Tong S, Urbani C, Comer JA, Lim W, Rollin PE, Dowell SF, Ling AE, Humphrey CD, Shieh WJ, Guarner J, Paddock CD, Rota P, Fields B, DeRisi J, Yang JY, Cox N, Hughes JM, LeDuc JW, Bellini WJ, Anderson LJ, SARS Working Group 2003. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 348:1953–1966. 10.1056/NEJMoa030781 [DOI] [PubMed] [Google Scholar]
- 3. Kuiken T, Fouchier RA, Schutten M, Rimmelzwaan GF, van Amerongen G, van Riel D, Laman JD, de Jong T, van Doornum G, Lim W, Ling AE, Chan PK, Tam JS, Zambon MC, Gopal R, Drosten C, van der Werf S, Escriou N, Manuguerra JC, Stöhr K, Peiris JS, Osterhaus AD. 2003. Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet 362:263–270. 10.1016/S0140-6736(03)13967-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 367:1814–1820. 10.1056/NEJMoa1211721 [DOI] [PubMed] [Google Scholar]
- 5. Perlman S, Netland J. 2009. Coronaviruses post-SARS: update on replication and pathogenesis. Nat. Rev. Microbiol. 7:439–450. 10.1038/nrmicro2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Belouzard S, Millet JK, Licitra BN, Whittaker GR. 2012. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 4:1011–1033. 10.3390/v4061011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Angelini MM, Akhlaghpour M, Neuman BW, Buchmeier MJ. 2013. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio 4(4):e00524-13. 10.1128/mBio.00524-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Knoops K, Kikkert M, Worm SH, Zevenhoven-Dobbe JC, van der Meer Y, Koster AJ, Mommaas AM, Snijder EJ. 2008. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 6(9):e226. 10.1371/journal.pbio.0060226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yamada Y, Liu XB, Fang SG, Tay FP, Liu DX. 2009. Acquisition of cell-cell fusion activity by amino acid substitutions in spike protein determines the infectivity of a coronavirus in cultured cells. PLoS One 4(7):e6130. 10.1371/journal.pone.0006130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kerr MC, Teasdale RD. 2009. Defining macropinocytosis. Traffic 10:364–371. 10.1111/j.1600-0854.2009.00878.x [DOI] [PubMed] [Google Scholar]
- 11. Swanson JA, Watts C. 1995. Macropinocytosis. Trends Cell Biol. 5:424–428. 10.1016/S0962-8924(00)89101-1 [DOI] [PubMed] [Google Scholar]
- 12. Hansen CG, Nichols BJ. 2009. Molecular mechanisms of clathrin-independent endocytosis. J. Cell Sci. 122:1713–1721. 10.1242/jcs.033951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gu Z, Noss EH, Hsu VW, Brenner MB. 2011. Integrins traffic rapidly via circular dorsal ruffles and macropinocytosis during stimulated cell migration. J. Cell Biol. 193:61–70. 10.1083/jcb.201007003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Orth JD, McNiven MA. 2006. Get off my back! Rapid receptor internalization through circular dorsal ruffles. Cancer Res. 66:11094–11096. 10.1158/0008-5472.CAN-06-3397 [DOI] [PubMed] [Google Scholar]
- 15. Nobes C, Marsh M. 2000. Dendritic cells: new roles for cdc42 and rac in antigen uptake? Curr. Biol. 10:R739–R741. 10.1016/S0960-9822(00)00736-3 [DOI] [PubMed] [Google Scholar]
- 16. Hewlett LJ, Prescott AR, Watts C. 1994. The coated pit and macropinocytic pathways serve distinct endosome populations. J. Cell Biol. 124:689–703. 10.1083/jcb.124.5.689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Henson PM, Bratton DL, Fadok VA. 2001. Apoptotic cell removal. Curr. Biol. 11:R795–R805. 10.1016/S0960-9822(01)00474-2 [DOI] [PubMed] [Google Scholar]
- 18. Amyere M, Mettlen M, Van Der Smissen P, Platek A, Payrastre B, Veithen A, Courtoy PJ. 2002. Origin, originality, functions, subversions and molecular signalling of macropinocytosis. Int. J. Med. Microbiol. 291:487–494. 10.1078/1438-4221-00157 [DOI] [PubMed] [Google Scholar]
- 19. Mercer J, Helenius A. 2009. Virus entry by macropinocytosis. Nat. Cell Biol. 11:510–520. 10.1038/ncb0509-510 [DOI] [PubMed] [Google Scholar]
- 20. Mercer J, Helenius A. 2012. Gulping rather than sipping: macropinocytosis as a way of virus entry. Curr. Opin. Microbiol. 15:490–499. 10.1016/j.mib.2012.05.016 [DOI] [PubMed] [Google Scholar]
- 21. Pernet O, Pohl C, Ainouze M, Kweder H, Buckland R. 2009. Nipah virus entry can occur by macropinocytosis. Virology 395:298–311. 10.1016/j.virol.2009.09.016 [DOI] [PubMed] [Google Scholar]
- 22. Freeman MC, Graham RL, Lu X, Peek CT, Denison MR. 2014. Coronavirus replicase-reporter fusions provide quantitative analysis of replication and replication complex formation. J. Virol. 88:5319–5327. 10.1128/JVI.00021-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Swanson JA. 2008. Shaping cups into phagosomes and macropinosomes. Nat. Rev. Mol. Cell Biol. 9:639–649. 10.1038/nrm2447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sheahan T, Rockx B, Donaldson E, Corti D, Baric R. 2008. Pathways of cross-species transmission of synthetically reconstructed zoonotic severe acute respiratory syndrome coronavirus. J. Virol. 82:8721–8732. 10.1128/JVI.00818-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. West MA, Bretscher MS, Watts C. 1989. Distinct endocytotic pathways in epidermal growth factor-stimulated human carcinoma A431 cells. J. Cell Biol. 109:2731–2739. 10.1083/jcb.109.6.2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Just I, Selzer J, von Eichel-Streiber C, Aktories K. 1995. The low molecular mass GTP-binding protein rho is affected by toxin a from Clostridium difficile. J. Clin. Invest. 95:1026–1031. 10.1172/JCI117747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. de Haan CA, Stadler K, Godeke GJ, Bosch BJ, Rottier PJ. 2004. Cleavage inhibition of the murine coronavirus spike protein by a furin-like enzyme affects cell-cell but not virus-cell fusion. J. Virol. 78:6048–6054. 10.1128/JVI.78.11.6048-6054.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Qiu Z, Hingley ST, Simmons G, Yu C, Das Sarma J, Bates P, Weiss SR. 2006. Endosomal proteolysis by cathepsins is necessary for murine coronavirus mouse hepatitis virus type 2 spike-mediated entry. J. Virol. 80:5768–5776. 10.1128/JVI.00442-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Navas S, Seo SH, Chua MM, Das Sarma J, Lavi E, Hingley ST, Weiss SR. 2001. Murine coronavirus spike protein determines the ability of the virus to replicate in the liver and cause hepatitis. J. Virol. 75:2452–2457. 10.1128/JVI.75.5.2452-2457.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hingley ST, Gombold JL, Lavi E, Weiss SR. 1994. Mhv-a59 fusion mutants are attenuated and display altered hepatotropism. Virology 200:1–10. 10.1006/viro.1994.1156 [DOI] [PubMed] [Google Scholar]
- 31. Haigler HT, McKanna JA, Cohen S. 1979. Rapid stimulation of pinocytosis in human carcinoma cells A-431 by epidermal growth factor. J. Cell Biol. 83:82–90. 10.1083/jcb.83.1.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sánchez EG, Quintas A, Pérez-Núñez D, Nogal M, Barroso S, Carrascosa AL, Revilla Y. 2012. African swine fever virus uses macropinocytosis to enter host cells. PLoS Pathog. 8(6):e1002754. 10.1371/journal.ppat.1002754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mercer J, Knébel S, Schmidt FI, Crouse J, Burkard C, Helenius A. 2010. Vaccinia virus strains use distinct forms of macropinocytosis for host-cell entry. Proc. Natl. Acad. Sci. U. S. A. 107:9346–9351. 10.1073/pnas.1004618107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moasser MM, Basso A, Averbuch SD, Rosen N. 2001. The tyrosine kinase inhibitor zd1839 (“Iressa”) inhibits her2-driven signaling and suppresses the growth of her2-overexpressing tumor cells. Cancer Res. 61:7184–7188 [PubMed] [Google Scholar]
- 35. Das Sarma J, Fu L, Tsai JC, Weiss SR, Lavi E. 2000. Demyelination determinants map to the spike glycoprotein gene of coronavirus mouse hepatitis virus. J. Virol. 74:9206–9213. 10.1128/JVI.74.19.9206-9213.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tooze J, Tooze SA, Fuller SD. 1987. Sorting of progeny coronavirus from condensed secretory proteins at the exit from the trans-Golgi network of att20 cells. J. Cell Biol. 105:1215–1226. 10.1083/jcb.105.3.1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tooze J, Tooze S, Warren G. 1984. Replication of coronavirus mhv-a59 in sac- cells: determination of the first site of budding of progeny virions. Eur. J. Cell Biol. 33:281–293 [PubMed] [Google Scholar]
- 38. Cudmore S, Cossart P, Griffiths G, Way M. 1995. Actin-based motility of vaccinia virus. Nature 378:636–638. 10.1038/378636a0 [DOI] [PubMed] [Google Scholar]
- 39. Doceul V, Hollinshead M, van der Linden L, Smith GL. 2010. Repulsion of superinfecting virions: a mechanism for rapid virus spread. Science 327:873–876. 10.1126/science.1183173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McDonald D, Wu L, Bohks SM, KewalRamani VN, Unutmaz D, Hope TJ. 2003. Recruitment of HIV and its receptors to dendritic cell-T cell junctions. Science 300:1295–1297. 10.1126/science.1084238 [DOI] [PubMed] [Google Scholar]
- 41. Igakura T, Stinchcombe JC, Goon PK, Taylor GP, Weber JN, Griffiths GM, Tanaka Y, Osame M, Bangham CR. 2003. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science 299:1713–1716. 10.1126/science.1080115 [DOI] [PubMed] [Google Scholar]
- 42. Mettlen M, Platek A, Van Der Smissen P, Carpentier S, Amyere M, Lanzetti L, de Diesbach P, Tyteca D, Courtoy PJ. 2006. Src triggers circular ruffling and macropinocytosis at the apical surface of polarized MDCK cells. Traffic 7:589–603. 10.1111/j.1600-0854.2006.00412.x [DOI] [PubMed] [Google Scholar]
- 43. Demory ML, Boerner JL, Davidson R, Faust W, Miyake T, Lee I, Hüttemann M, Douglas R, Haddad G, Parsons SJ. 2009. Epidermal growth factor receptor translocation to the mitochondria: regulation and effect. J. Biol. Chem. 284:36592–36604. 10.1074/jbc.M109.000760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. DiMaio D, Petti LM. 2013. The E5 proteins. Virology 445:99–114. 10.1016/j.virol.2013.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sperry SM, Kazi L, Graham RL, Baric RS, Weiss SR, Denison MR. 2005. Single-amino-acid substitutions in open reading frame (ORF) 1b-nsp14 and ORF 2a proteins of the coronavirus mouse hepatitis virus are attenuating in mice. J. Virol. 79:3391–3400. 10.1128/JVI.79.6.3391-3400.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Denison MR, Yount B, Brockway SM, Graham RL, Sims AC, Lu X, Baric RS. 2004. Cleavage between replicase proteins p28 and p65 of mouse hepatitis virus is not required for virus replication. J. Virol. 78:5957–5965. 10.1128/JVI.78.11.5957-5965.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yount B, Curtis KM, Fritz EA, Hensley LE, Jahrling PB, Prentice E, Denison MR, Geisbert TW, Baric RS. 2003. Reverse genetics with a full-length infectious cDNA of severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. U. S. A. 100:12995–13000. 10.1073/pnas.1735582100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gombold JL, Hingley ST, Weiss SR. 1993. Fusion-defective mutants of mouse hepatitis virus A59 contain a mutation in the spike protein cleavage signal. J. Virol. 67:4504–4512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hirano N, Fujiwara K, Matumoto M. 1976. Mouse hepatitis virus (MHV-2). Plaque assay and propagation in mouse cell line DBT cells. Jpn. J. Microbiol. 20:219–225. 10.1111/j.1348-0421.1976.tb00978.x [DOI] [PubMed] [Google Scholar]
- 50. Robison CS, Whitt MA. 2000. The membrane-proximal stem region of vesicular stomatitis virus G protein confers efficient virus assembly. J. Virol. 74:2239–2246. 10.1128/JVI.74.5.2239-2246.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bost AG, Carnahan RH, Lu XT, Denison MR. 2000. Four proteins processed from the replicase gene polyprotein of mouse hepatitis virus colocalize in the cell periphery and adjacent to sites of virion assembly. J. Virol. 74:3379–3387. 10.1128/JVI.74.7.3379-3387.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Thorp EB, Gallagher TM. 2004. Requirements for CEACAMs and cholesterol during murine coronavirus cell entry. J. Virol. 78:2682–2692. 10.1128/JVI.78.6.2682-2692.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chumbler NM, Farrow MA, Lapierre LA, Franklin JL, Haslam DB, Goldenring JR, Lacy DB. 2012. Clostridium difficile toxin B causes epithelial cell necrosis through an autoprocessing-independent mechanism. PLoS Pathog. 8(12):e1003072. 10.1371/journal.ppat.1003072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lavi E, Gilden DH, Wroblewska Z, Rorke LB, Weiss SR. 1984. Experimental demyelination produced by the A59 strain of mouse hepatitis virus. Neurology 34:597–603. 10.1212/WNL.34.5.597 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Infection with MHV induces continuous membrane ruffling. DBT cells were infected with MHV-Δ2-GFP3 at an MOI of 1 PFU/cell and imaged from 4 to 12 hpi. This movie spans 8 to 8.6 hpi. Download
The left cells are enlarged. Download
The right cells are enlarged. Download
MHV-induced filopodia potentiate cell-cell interactions. DBT cells were transfected with a GFP-expressing plasmid and infected with MHV at an MOI of 1 PFU/cell. Cells were imaged from 7 to 12 hpi. This movie shows several interactions between the filopodia of the infected cell and neighboring uninfected cells. Download
Enlarged region of the movie. Download
Enlarged region of the movie. Download