

Abstract
Affinity purification coupled to mass spectrometry (AP-MS) is an effective means of identifying protein-protein interactions to better understand biological functions. However, issues associated with sample preparation still limit the success of AP-MS for specific classes of proteins, including those associated with chromatin that exhibit overall poor solubility in the protocols normally used for AP-MS analysis. Here, we wanted to provide a generally applicable method to simultaneously identify interactors for the chromatin-bound and the soluble fractions of a given bait protein. Using four FLAG-tagged canonical histone proteins (H2A, H2B, H3.1 and H4) we demonstrate that the chromatin solubility issue can be robustly alleviated by fragmenting DNA prior to AP-MS using a combination of sonication and nuclease treatment. We show that – in comparison to a commonly used AP-MS method – our optimized protocol greatly improves the recovery of chromatin-associated interactors for core histones. Critically, this is achieved while preserving the interaction partners associated with the soluble portion of the histones. Detailed protocols amenable to the study of both histone and non-histone baits are presented here.
Keywords: systems biology, chromatin, protein-protein interactions, Affinity purification coupled to mass spectrometry
Text body
A fundamental requirement of living organisms is proper organisation and utilisation of their genetic material. Chromatin, the protein and DNA fractions composing chromosomes, is essential to these processes. The basic structural unit of chromatin is the nucleosome, which consists of two copies of each core histone (histones H2A, H2B, H3 and H4) around which 147 base pairs of DNA are wrapped. Nucleosomes are key not only in preserving the integrity of a cell's genetic material but also in ensuring its proper use, as they serve as docking sites for hundreds of proteins [1]. However, proteins physically embedded in chromatin (such as histones), or tightly interacting with it, are poorly solubilized in conventional affinity purification (AP) approaches, which most often employ passive lysis in mild detergents followed by centrifugation of the insoluble material, thereby hampering the analysis of their protein-protein interactions. Solutions for specifically analyzing the protein-protein interactions associated with chromatin have been developed by a number of groups and reviewed elsewhere [2, 3]. In general, most of these studies begin by the preparation of a nuclear extract, centrifugation of the chromatin, and re-solubilisation of chromatin-associated proteins by the use of high-salt extraction, sometimes aided by nuclease digestion or sonication [4-6]. This type of approach is powerful in that it focuses specifically on the interactions that occur on chromatin (by opposition to interactions that occur in the nucleoplasm or in other subcellular fractions), but they require several steps. We further reasoned that unless only chromatin-associated interactions were of interest, it should be possible to obtain in a single experiment a complete picture of the interactome of a protein that partitions to both chromatin-bound and unbound fractions.
Over the past several years, we and others have been utilizing stable ectopic expression of a tagged bait of interest [7] coupled to gentle single-step AP and one-dimensional LC-MS/MS analysis to map protein-protein interactions (here referred to as AP-MS; Supplementary Figure 1; [8]). Our recently optimized method was selected as the starting point for improvements aiming to better characterize chromatin-associated protein complexes without the need for increasing the number of steps needed for the process. Briefly, in this method, ∼50-100 million cells are harvested, re-suspended in a mild lysis buffer (50 mM Hepes-NaOH pH 8.0, 100 mM KCl, 2 mM EDTA, 0.1% NP40, 10% glycerol) and submitted to a freeze/thaw cycle to improve lysis. The lysate is clarified by centrifugation before the tagged protein is affinity purified from the supernatant and interactors identified by MS (see supplementary material for complete method details). To test the efficiency of this protocol for chromatin-associated proteins, four HEK293 cell lines stably expressing N-terminally 3XFLAG tagged core histones (H2A, H2B, H3.1 and H4), and another HEK293 cell line expressing 3XFLAG alone (negative control), were harvested in biological triplicates. The tagged protein was purified, digested on-beads with trypsin and analyzed using a 5600 TripleTOF mass spectrometer. The resulting files were handled using the ProHits LIMS system [9] and submitted to database searches with Mascot and Comet [10] through the iProphet pipeline [11]. Statistically significant interaction partners for each histone were identified by SAINT (Significance Analysis of INTeractome; [12]). As expected, the results generated from this approach revealed that the standard AP-MS approach enables efficient recovery of soluble interactors (i.e. not chromatin associated). This included protein complexes involved in the nuclear import of histone H3.1/H4 dimers (supplementary Table 1) [13], and nucleosome assembly proteins (PPM1G, NASP, etc.) that associated with histones H2A and H2B. However, many known chromatin-bound interaction partners for histones were not recovered.
Because the AP-MS protocol outlined above only utilises the cellular supernatant after lysis while the pellet is discarded, we hypothesized that the lack of chromatin-associated protein complexes co-purifying with the tagged histones may be due to the low solubility of these complexes and their partitioning to the pellet. Chromatin solubilisation has been previously reported to be achieved with high salt protein extraction [4], nuclease digestion [5] and sonication [14]. The two last methods are particularly interesting to us, as they are in principle compatible with a standard AP-MS purification approach, and we therefore tested whether they improved our ability to recover protein complexes associated with chromatin. To this end, we used sonication to rapidly break chromatin into ∼1000 base pair fragments, followed by treatment with a promiscuous nuclease, benzonase, to further reduce chromatin fragments to a few hundred base pairs (approximately 1 to 2 nucleosomes of coiled DNA (Figure 1A)). A direct comparison of our “standard” approach, sampling soluble protein complexes only, against this chromatin optimized one (herein termed “chromatin”) sampling soluble and chromatin associated protein complexes, revealed a significant improvement in the solubility of histones after sonication and nuclease treatment as detected by Western blot (Figure 1B). Critically, this gain was conserved following affinity purification, and the chromatin optimized approach led to a major increase in the recovery of bait histone proteins (Figure 1B). In addition to looking at extraction and immunopurification of the histone baits across our two approaches, we further tested the impact of chromatin solubilisation on solubility and co-precipitation of known histone interactors. As with the histones themselves, the solubility of the Bromodomain and Extra Terminal domain (BET) protein BRD4L[15] was greatly enhanced following chromatin shearing, while only modest gains were observed for another BET family member, BRD2 (Figure 1C). Importantly however, in both cases, we were only able to detect the association of BET proteins with histones when chromatin shearing was incorporated into our sample preparation. This shows the value of our optimized protocol for the identification of interactions within both soluble and chromatin-associated complexes that were previously undetectable using our “standard” AP purification protocol targeting soluble complexes only.
Figure 1.
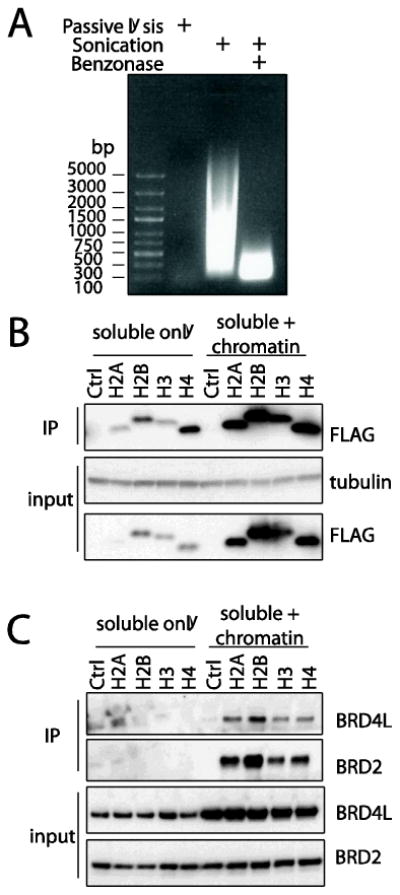
DNA shearing facilitates the affinity purification of chromatin-associated proteins. (A) Phenol/chloroform extracted DNA from passively lysed cells, lysed cells which were further sonicated or both sonicated and treated with benzonase, was resolved on a 1% agarose gel and stained with ethidium bromide. Note that DNA from passively lysed cells did not enter the agarose gel due to its large size. (B) Chromatin shearing increases the solubility of 3XFLAG tagged histone baits. Cell extracts and affinity purified samples from HEK293 cells stably expressing 3XFLAG histones or an empty control vector were resolved on a 4-15% Criterion SDS-PAGE gels and immunoblotted with antibodies as indicated. (C) Chromatin shearing increases the solubility of chromatin-associated proteins and their recovery as histone interactors.
To determine whether the increased histone solubility of our optimized solubilisation protocol for chromatin-associated complexes would translate into increased histone-interactor identification by MS, an additional set of core histone purifications was performed by modifying the “standard” AP-MS approach to include chromatin shearing by sonication and nuclease treatment (Supplementary Figure 1, see supplementary materials for method details). After performing analysis of this new dataset with SAINT, 180 proteins were found to be associated with at least one core histone, as compared to 63 using our “standard” AP-MS procedure (AvgP ≥0.8; supplementary Table 1). Importantly, direct comparison of both datasets revealed that including chromatin shearing as part of our sample preparation did not results in significant loss of interaction partners (Figure 2) and, critically, allowed for large gains to be made in the detection of chromatin-associated proteins such as the E3 ubiquitin ligase UHRF1 and the DNA helicase CHD4. This trend was further reinforced when enriched GO terms from the interactors produced by both methods were compared (Supplementary Figure 2). Consistent with this, when the H3.1 interactome mapped here was compared to previous report of H3.1 AP-MS datasets [5, 13, 16] a large overlap was observed (Supplementary Figure 3).
Figure 2.
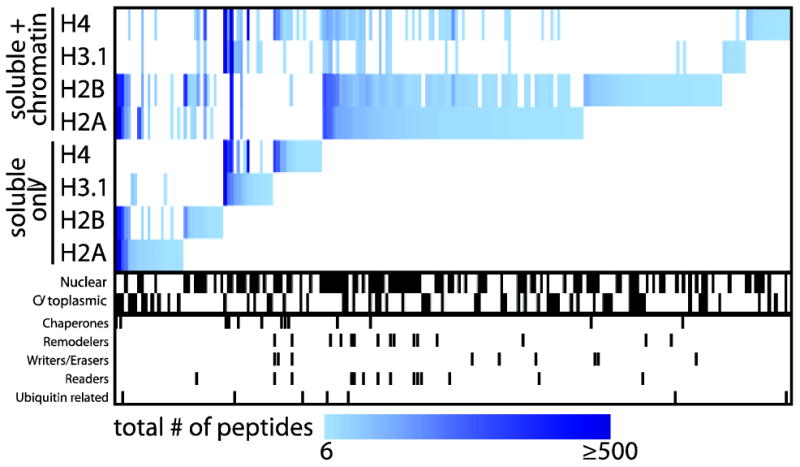
The use of DNA shearing in AP sample preparation enhances the identification by mass spectrometry of chromatin-bound interactors for core histone proteins. A heat map of the interaction partners identified with each histone bait using the “standard” or chromatin optimized AP approach (Avg SAINT ≥ 0.8) was generated using MeV 4.81. The pixel color corresponds to the total number of peptides (spectral counts) detected for a given prey across all replicates. Interactor localization and functions, as defined by GO annotation, is highlighted below the heat map.
As with our initial experiments using the “standard” AP-MS approach, significant differences in the interactomes of the four purified bait histones were retained, reinforcing the usefulness of our modified AP-MS workflow for resolving distinct chromatin-associated complexes. For instance, purification of either histone H2A and H2B (but not H3.1 or H4) enabled MS detection of most members of the NuRD complex [17]. Conversely, purification of H3.1 and H4 resulted in the identification of shared interaction partners, such as ASF1A/B and IPO4 that are known to occur with H3.1/H4 dimers but not with H2A/H2B. A number of bait-specific interaction partners were also detected. For example, the E3 ubiquitin ligase UBR7 was found with high peptide counts only in H3.1 purifications, which is consistent with a previous report [18]. Purification of 3XFLAG tagged H4 resulted in the identification of interaction partners shared with either H3.1 (e.g. CHAF1A/B) or H3.3 (e.g. DAXX) [16] showcasing our ability to access subcomplexes associated with histone proteins in a reproducible manner (Supplementary Figure 4).
While in the examples mentioned above, specificity for one or a few baits was detected, many proteins (NUCKS1, HMGN1, MSH6, etc.) were also deemed by SAINT analysis to be interaction partners for all four histones. This is certainly expected based on our experimental design, which would enrich for proteins strongly associated with chromatin – or at least with components of the nucleosomes which we have profiled here (as per [14]). We foresee that this list of interaction partners associated with each of the histone proteins will become very useful when trying to score specific interactions for other (non-histone) proteins using this AP-MS pipeline. Enrichment of interactors over this general “histone-associated proteins” would enable determination of specificity for chromatin-associated proteins. Toward this end, we have deposited all MS files generated here in the MassIVE raw file repository housed at the Center for Computational Mass Spectrometry at UCSD (http://massive.ucsd.edu) to facilitate reanalysis. In addition, all negative controls were deposited in the Contaminant Repository of Affinity Purification ([19]; www.crapome.com) to support community-based analysis of AP-MS data. Lastly, we have included in our searchable online database (http://prohits-web.lunenfeld.ca) both the complete and SAINT filtered protein datasets generated here. Of note, while we initially selected histones for our protocol optimization, the application of the protocol was also successful for the analysis of non-histone proteins (data not shown).
In summary, the workflow presented here is flexible in its nature and can be used to study a wide array of chromatin-associated proteins. This approach can also be adapted for the analysis of wild type and mutant forms of these proteins, for example to profile the effects on the interactome of histone mutations as were recently uncovered in glioblastoma multiforme patients to affect histone H3.3 (K27M and G34R/G34V) [20]. Lastly, the reproducibility of the AP protocol outlined here is amenable to quantitative mass spectrometry and thus can allow for study of chromatin-associated protein complex dynamics by AP-MS.
Supplementary Material
Sup Table 1 - Complete interaction partner list with an average SAINT score ≥ 0.8.
Sup Fig 1 - Overview of experimental procedure.
Sup Fig 2 - GO annotation of histone interaction partners identified using the standard and chromatin optimized AP-MS approach.
Sup Fig 3 - Comparison of histone H3 interactome identified by AP-MS in four distinct studies.
Sup Fig 4 - High reproducibility of chromatin optimized AP-MS approach accross three biological replicates.
Highlights.
Chromatin-bound proteins suffer from low solubility, precluding identification of their interaction partners in standard AP-MS.
We report a streamlined approach coupling standard AP-MS to DNA shearing to solubilize chromatin-bound proteins.
Our method provides a holistic view of chromatin bound bait proteins interactomes.
While optimized here on histone proteins, our approach is generally applicable to other proteins that associate with chromatin.
Acknowledgments
We want to thank Guoci Teo and Hyungwon Choi (National University of Singapore) for early access to SAINTexpress algorithm and to Jeremy Carver and Nuno Bandeira with submission to the MassIVE data repository. We also wish to thank Wade Dunham and James Knight for manuscript editing and the Gingras and Pawson labs for helpful discussion. The website for the supplementary material was designed by Guomin Liu and Jianping Zhang. This work was supported by funding from the Canadian Institutes of Health Research (CIHR) to A-C.G. and T.P. (MOP 123322) and from the National Institutes of Health to A-C.G. (5R01GM94231). A.-C.G. holds the Canada Research Chair in Functional Proteomics and the Lea Reichmann Chair in Cancer Proteomics. J.-P.L was supported by a post-doctoral fellowship from CIHR.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–95. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lambert JP, Pawson T, Gingras AC. Mapping physical interactions within chromatin by proteomic approaches. Proteomics. 2012;12:1609–22. doi: 10.1002/pmic.201100547. [DOI] [PubMed] [Google Scholar]
- 3.Sidoli S, Cheng L, Jensen ON. Proteomics in chromatin biology and epigenetics: Elucidation of post-translational modifications of histone proteins by mass spectrometry. J Proteomics. 2012;75:3419–33. doi: 10.1016/j.jprot.2011.12.029. [DOI] [PubMed] [Google Scholar]
- 4.Mak AB, Ni Z, Hewel JA, Chen GI, Zhong G, Karamboulas K, et al. A lentiviral functional proteomics approach identifies chromatin remodeling complexes important for the induction of pluripotency. Mol Cell Proteomics. 2010;9:811–23. doi: 10.1074/mcp.M000002-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foltz DR, Jansen LE, Black BE, Bailey AO, Yates JR, 3rd, Cleveland DW. The human CENP-A centromeric nucleosome-associated complex. Nat Cell Biol. 2006;8:458–69. doi: 10.1038/ncb1397. [DOI] [PubMed] [Google Scholar]
- 6.Lambert JP, Fillingham J, Siahbazi M, Greenblatt J, Baetz K, Figeys D. Defining the budding yeast chromatin-associated interactome. Mol Syst Biol. 2010;6:448. doi: 10.1038/msb.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glatter T, Wepf A, Aebersold R, Gstaiger M. An integrated workflow for charting the human interaction proteome: insights into the PP2A system. Mol Syst Biol. 2009;5:237. doi: 10.1038/msb.2008.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dunham WH, Mullin M, Gingras AC. Affinity-purification coupled to mass spectrometry: basic principles and strategies. Proteomics. 2012;12:1576–90. doi: 10.1002/pmic.201100523. [DOI] [PubMed] [Google Scholar]
- 9.Liu G, Zhang J, Larsen B, Stark C, Breitkreutz A, Lin ZY, et al. ProHits: integrated software for mass spectrometry-based interaction proteomics. Nat Biotechnol. 2010;28:1015–7. doi: 10.1038/nbt1010-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eng JK, Jahan TA, Hoopmann MR. Comet: an open-source MS/MS sequence database search tool. Proteomics. 2013;13:22–4. doi: 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- 11.Shteynberg D, Deutsch EW, Lam H, Eng JK, Sun Z, Tasman N, et al. iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol Cell Proteomics. 2011;10:M111 007690. doi: 10.1074/mcp.M111.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi H, Larsen B, Lin ZY, Breitkreutz A, Mellacheruvu D, Fermin D, et al. SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat Methods. 2011;8:70–3. doi: 10.1038/nmeth.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campos EI, Fillingham J, Li G, Zheng H, Voigt P, Kuo WH, et al. The program for processing newly synthesized histones H3.1 and H4. Nat Struct Mol Biol. 2010;17:1343–51. doi: 10.1038/nsmb.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lambert JP, Mitchell L, Rudner A, Baetz K, Figeys D. A novel proteomics approach for the discovery of chromatin-associated protein networks. Mol Cell Proteomics. 2009;8:870–82. doi: 10.1074/mcp.M800447-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–31. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drane P, Ouararhni K, Depaux A, Shuaib M, Hamiche A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010;24:1253–65. doi: 10.1101/gad.566910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, LeRoy G, Seelig HP, Lane WS, Reinberg D. The dermatomyositis-specific autoantigen Mi2 is a component of a complex containing histone deacetylase and nucleosome remodeling activities. Cell. 1998;95:279–89. doi: 10.1016/s0092-8674(00)81758-4. [DOI] [PubMed] [Google Scholar]
- 18.Foltz DR, Jansen LE, Bailey AO, Yates JR, 3rd, Bassett EA, Wood S, et al. Centromere-specific assembly of CENP-a nucleosomes is mediated by HJURP. Cell. 2009;137:472–84. doi: 10.1016/j.cell.2009.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis N, Li T, et al. The CRAPome: a Contaminant Repository for Afffinity Purification mass spectrometry data. Nat Methods. 2013 doi: 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–31. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sup Table 1 - Complete interaction partner list with an average SAINT score ≥ 0.8.
Sup Fig 1 - Overview of experimental procedure.
Sup Fig 2 - GO annotation of histone interaction partners identified using the standard and chromatin optimized AP-MS approach.
Sup Fig 3 - Comparison of histone H3 interactome identified by AP-MS in four distinct studies.
Sup Fig 4 - High reproducibility of chromatin optimized AP-MS approach accross three biological replicates.