

Abstract
A gram scale synthesis of the glucuronide metabolites of curcumin were completed in four steps. The newly synthesized curcumin glucuronide compounds 2 and 3 along with curcumin 1 were tested and their anti-proliferative effects against KBM-5, Jurkat cell, U266, and A549 cell lines were reported. Biological data revealed that as much as 1 μM curcumin 1 exhibited anticancer activity and almost 100% cell kill was noted at 10 μM on two out of four cell lines; while curcumin mono-glucuronide 2 as well as diglucuronide 3 displayed no suppression of cell proliferation.
1. Introduction
Curcumin 1 (1,7-bis[4-hydroxy-3-methoxyphenyl]-1,6-heptadiene-3,5-dione/diferuloyl methane), the main yellow pigment of the Curcuma longa L, has been reported to have antioxidant, antiproliferative and various biological properties.1-13 Curcumin has also been reported to decrease total cholesterol and LDL cholesterol level in serum and to increase the beneficial HDL cholesterol level.14 It has been claimed that curcumin inhibits HIV-I integrase protein, thus potentially preventing HIV-I from infecting CD-4 and CD-8 cell.15 Curcumin is a non-nutritive, non-toxic compound in turmeric, a spice that has been used for centuries in India and elsewhere as a herbal medicinal treatment for wounds, jaundice, and rheumatoid arthritis.16 It exerts cancer chemo-preventive efficacy in a wide variety of rodent models of carcinogenesis.17 In common with several other diet-derivative polyphenols, curcumin has very poor bioavailability,13 cellular uptake is slow and it gets metabolized fast, once inside the cell, requiring repeated oral doses in order to achieve significant concentration inside the cells for therapeutic activity. This poor bioavailability is attributed to one of the major metabolites, curcumin glucuronide, that is accumulated in the intestine18 when curcumin is administered orally.19 Interestingly enough, in the miliéu intérieur of studies done on curcumin, the critical central question which has remained unanswered or even addressed, is that of whether or not curcumin glucuronides exhibit any biological activity. In an effort to address this central question, curcumin glucuronides were synthesized and its potency against human cell lines was compared to the inhibitory activities with that of curcumin thereby providing irrefutable evidence demonstrating complete biological inactivity.
While the chemistry and biology of glucuronides is well known and plays a central role in detoxification pathway,20 the synthesis of curcumin glucuronides have been largely overlooked with only one briefly description in the literature via enzymatic synthesis with no chemical characterization.21 Thus, to date no chemical synthesis of curcumin glucuronides has not yet been reported nor the products chemically characterized. Herein we report an efficient synthesis of curcumin glucuronides starting from vanillin.
Results and Discussion
The syntheses of curcumin derivatives are shown in schemes 1-3. Conversion of vanillin to its corresponding β-D-glucuronide was performed with acetobromo-α-D-glucuronic acid methyl ester and silver oxide in quinoline to give the key aldehyde β-D-glucopyranosiduronic acid derivative 6 in 70% yield. Keto-enol compounds (7 and 9) were synthesized from the corresponding aldehydes with two equivalents of acetylacetone-boron complex in presence of n-butylamine (Scheme 1).22 A preliminary purification by flash column over silica gel followed by recrystallization from ethanol and water gave products that were visualized by fluorescence and UV illumination. Compound 8 was synthesized by coupling keto-enol compound 7 and the vanillin-boron complex in the presence of piperidine which was found to give better yields than when n-butylamine was used Subsequent hydrolysis of acetyl and ester groups at the glucuronide moiety 8 was achieved by treatment with aqueous 1 N NaOH in methanol (1:1) followed by acidification with 50% formic acid in ice water to give curcumin mono-glucuronide 2 in 70% yield.
Scheme 1.

Reagents and conditions: (a) Ag2O, quinoline, 0 °C to rt, 1.5 h, 70%; (b) 2,4-pentanedione, B2O3, (n-BuO)3B, EtOAc, 85 °C, 30 min, n-BuNH2, 105 °C, 30 min, 1 N HCl, 50%; (c) B2O3, (n-BuO)3B, EtOAc, 85 °C, 30 min, piperidine, 85 °C, 30 min, 0.5 N HCl, 50 °C, 30 min, 55%; (d) MeOH, 1 N NaOH, 50 °C, 1 h, 50% formic acid, 70 %.
Scheme 3.
Reagents and conditions: (a) 2,4-pentanedione, B2O3, (n-BuO)3B, EtOAc, 85 °C, 30 min, n-BuNH2, 105 °C, 30 min, 1 N HCl, 25%.
Similarly compound 9 was converted to the curcumin di-glucuronide 10 starting with the treatment of aldehyde 6 using piperidine as condensation reagent as shown in Scheme 2. Compound 8 was also synthesized from the keto-enol compound 9 using vanillin 4 as the aldehyde substrate for the condensation reaction. Hydrolysis of methyl ester and removal of acetyl groups was done using the same method described in scheme 1 affording curcumin di-glucuronide 3 as a yellow solid. In scheme 3, an equivalent molar mixture of 6 and vanillin 4 was reacted with the acetylacetone-boron complex in the same manner as described in Scheme 1 as well as in Scheme 2.23 After treatment of the reaction mixture with 1 N HCl and work up, the crude mixture was purified by silica gel flash chromatography to give the compounds 8 (25%) and 10 (<5%) along with the curcumin 1 (10%) as yellow products.
Scheme 2.

Reagents and condition: (a) 2, 4-pentanedione, B2O3, (n-BuO)3B, EtOAc, 85 °C, 30 min, n-BuNH2, 105 °C, 30 min, 1 N HCl, 50%; (b) B2O3, (n-BuO)3B, ethyl acetate, 85 °C, 30 min, piperidine, 85 °C, 30 min, 0.5 N HCl, 50 °C, 30 min, 55% (c) MeOH, 1 N NaOH, 50 °C, 1 h, 50% formic acid, 75 %.
Curcumin and curcumin glucuronides were evaluated for their ability to suppress the transcription factor, NK-κB in human leukemic KBM-5 cells as they expressed TNF receptors and were response to TNF activation. The KBM-5 cells were pre-incubated for 4 h with curcumin 1 and curcumin glucuronides (compound 2 and 3) and were treated 0.1 nM TNF for 30 min. The nuclear extracts were prepared and assayed for the NK-κB activation by electrophoretic mobility shift assay (EMSA). Curcumin inhibited TNF-induced NK-κB activation at 25 μM concentration; however curcumin glucuronides had no effects on NK-κB suppression (Figure. 2).
Figure 2.

Evaluation of curcumin 1 and its glucuronide derivatives 2 and 3 on TNF-induced NK-κB DNA binding in KBM-5 human myeloid cells. Cells were pretreated with curcumins for 4 h, then, stimulated with TNF (0.1 nmol/L) for 30 min, and analyzed for NK-κB DNA binding by EMSA.
Because NK-κB activation is known to regulate proliferation of tumor cells, we examined the effect of curcumin 1, and compounds 2 and 3 on proliferation of tumor cell lines. The effect of curcumin 1 and compounds 2 and 3 were compared at equimolar concentrations. The anti-proliferative effects (cytotoxicities) against different cell lines are shown in Figure 3.
Figure 3.
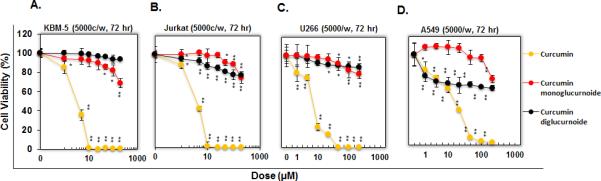
Evaluation of curcumin 1 and its glucuronide derivatives 2 and 3 on cell proliferation. Five thousand KBM-5 (A) Jurkat (B) U266 (C) and A549 (D) cells per well were seeded in triplicate onto 96-well plates; treated with the each compound at 1, 5, 10, 25, 50, 100 or 200 μM. After 72 the cell viability was measured by the MTT method and presented as % cell viability. The data were expressed as the means ± SD of triplicate samples. *P < 0.05 and **P < 0.01 vs. untreated cells.
In all four KBM-5, Jurkat, U266 and A549 cells, curcumin 1 showed anti-proliferative effects with IC50 values of 3.84, 4.29, 7.57 and 17.3 μM respectively. In contrast to curcumin 1, compounds 2 and 3 have no suppression against cell proliferation and IC50's were more than 200 μM concentrations (Figure 3).
2. Conclusion
Curcumin mono-glucuronide 2 and curcumin-di-glucuronide 3 were synthesized in four steps with overall yields of 61% and 62% respectively. It was also shown that both the acetylated compounds 8 and 10 along with curcumin 1 can be synthesized in one-pot condensation of 6 and 4 in presence of acetylacetone-B2O3 complex. The newly synthesized curcumin glucuronide compounds 2 and 3 along with curcumin 1 were tested and their anti-proliferative effects against KBM-5, Jurkat, U266 and A549 cell lines were reported. The biological data revealed that curcumin glucuronides showed very little anti-proliferative activity and no effect on NK-κB; while, curcumin displayed NK-κB inhibitory effects at 25 μM. Thus, confirming that curcumin mono-glucuronide 2 and curcumin-di-glucuronide 3 are non-toxic and non-antiinflammatory.
3. Experimental
4.1 Reagents, Instrumentation and Cell lines
All reagents and solvents were purchased from Aldrich Chemical Co. (Milwaukee, WI), and used without further purification. Melting points were measured in open capillary tubes on a Hoover capillary apparatus and are uncorrected. 1H-NMR and 13C-NMR spectra were recorded on an IBM-Bruker Avance 300 (300 MHz for 1H-NMR and 75.48 MHz for 13C-NMR), and IBM-Bruker Avance 500 (500 MHz for 1H-NMR and 125.76 MHz for 13C-NMR), spectrometers. Chemical shifts (δ) are determined relative to CDCl3 (referenced to 7.27 ppm (δ) for 1H-NMR and 77.0 ppm for 13C-NMR) or DMSO-d6 (referenced to 2.49 ppm (δ) for 1H-NMR and 39.5 ppm for 13C-NMR). Proton-proton coupling constants (J) are given in Hertz and spectral splitting patterns are designated as singlet (s), doublet (d), triplet (t), quadruplet (q), multiplet or overlapped (m), and broad (br). Low resolution mass spectra (ion spray, a variation of electrospray) were acquired on a Perkin-Elmer Sciex API 100 spectrometer or Applied Biosystems Q-trap 2000 LC-MS-MS.
Flash chromatography was performed using Merk silica gel 60 (mesh size 230-400 ASTM) or using an Isco (Lincon, NE) combiFlash Companion or SQ16x flash chromatography system with RediSep columns (normal phase silica gel (mesh size 230-400ASTM) and Fisher Optima TM grade solvents. Thin-layer chromatography (TLC) was performed on E.Merk (Darmstadt, Germany) silica gel F-254 aluminum-backed plates with visualization under UV (254nm) and by staining with potassium permanganate or ceric ammonium molybdate. Penicillin, streptomycin, RPMI 1640 medium, Dulbecco's modified eagles medium and Iscove's modified Dulbecco's medium, and fetal bovine serum were obtained from Invitrogen. Bacteria-derived human recombinant tumor necrosis factor (TNF), purified to homogeneity with a specific activity of 5 × 107 units/mg, and was kindly provided by Genentech (South San Francisco, CA). Solvents and reagents were purchased from Sigma-Aldrich, Acros organics and Fluka.
The KBM-5 (human chronic myeloid leukemia) cell lines were obtained from Dr. Nicholas J. Donato (University of Michigan Comprehensive Cancer Center), Jurkat (human T cell leukemia), U266 (multiple myeloma) and A549 (lung adenocarcinoma) cells were obtained from American Type Culture Collection (Manassas, VA). The Jurkat and U266 cells were cultured in RPMI 1640 medium, A549 cells were cultured in Dulbecco's modified eagles medium with 10% fetal bovine serum. KBM-5 cells were cultured in Iscove's modified Dulbecco's medium with 15% fetal bovine serum. All culture media were supplemented with 100 units/ml penicillin and 100 μg/mL streptomycin.
4.1.1 β-D-glucopyranosiduronic acid, 4-formy-2-methoxyphenyl, methyl ester, triacetate (6)
Vanillin 4 (0.5 g, 3.3 mmol) and acetobromo-α-D-glucuronic acid methyl ester 5 (2.3 g, 5.9 mmol) were dissolved in distilled quinoline (20 mL) and silver oxide (0.8 g, 4 mmol) was added in portions at 0 °C. The reaction mixture was stirred in the dark at 0 °C for 30 min. and at room temperature for 90 min. After the addition of acetic acid (40 mL), the mixture was poured into water (400 mL) and passed through celite pad and the filtrate was extracted with ethyl acetate (2 × 200 mL), washed with brine and then dried. Purification by flash chromatography using dry-packed silica gel and elution with hexane–EtOAC (70:30, first 25 min, and then 20:80 for 40 min) gave 6 (1.08 g, 70%) as a white solid: m.p. 100-102 °C. 1H NMR (500 MHz CDCl3) δ 9.90 (s, 1H), 7.44 (s, 1H), 7.43 (d, J = 7.8 Hz, 1H), 7.25 (d, J = 7.8 Hz, 1H), 5.35 (m, 3H), 5.19 (d, J = 6.6 Hz, 1H), 4.17 (d, J = 9 Hz, 1H), 3.89 (s, 3H), 3.73 (s, 3H), 2.08 (s, 3 H), 2.06 (s, 3H), 2.05 (s, 3H); 13C NMR (125 MHz CDCl3) 188.5, 167.7, 166.9, 166.7, 164.4, 148.6, 148.3, 130.6, 123.1, 116.4, 108.2, 97.2, 70.3, 69.1, 68.4, 66.6, 53.6, 50.4, 18.2, 18.2, 18.1; IR (neat) 1752.87, 1686.3, 1594.47; HRMS (C21H24O12) calcd. 468.12 found 486.4 (M+NH4).
4.1.2 5-Hydroxy-1-(4-hydroxy-3-methoxyphenyl)-1,4-hexadien-3-one (7)
2,4-Pentanedione (3.3 mL, 32.07 mmol) and B2O3 (2 g, 28.7 mmol) were dissolved in ethyl acetate (30 mL), and the solution was stirred at 80 °C for 30 min. To this mixture was added an ethyl acetate solution of (40 mL) of the vanillin 4 (2.2 g, 14.4 mmol) and (n-BuO)3 (1.6 mL, 5.96 mmol). After stirring for 30 min at 85 °C, n-butylamine (0.5 mL, 5.04 mmol) was added drop wise to the mixtures, which was allowed to stir at 105 °C for 1.5 h. Then the reaction mixture was treated with 1 N HCl (10 mL) at 50 °C and stirred at the same temperature for 1 h. Reaction mixture was cooled and then extracted with ethyl acetate, washed with water and dried over Na2SO4. Flash chromatography over silica gel (2:1, hexanes/EtOAc) followed by recrystallization from ethanol and water, gave the 7 as yellow crystals (1.69 g, 50%): mp 143.1- 145.1 °C; 1H NMR (CDCl3) δ 15.5 (bs, 1H), 7.52 (d, J = 0.6 Hz, 1H), 7.08 (d, J = 8.4 Hz, 1H), 7.01 (s, 1H), 6.92 (d, J = 8.4 Hz, 1H), 6.32 (d, J = 15.6 Hz, 1H), 5.90 (bs, 1H), 5.63 (s, 1H), 3.93 (s, 3H); 13C NMR (CDCl3) δ 196.9, 178.0, 147.8, 146.8, 140.1, 127.7, 122.7, 120.4, 114.8, 109.6, 100.7, 56.0, 26.8; IR (neat) 1752.87, 1686.3, 1594.47; HRMS (C13H14O4) calcd. 234.08 found 235.2 (M+H).
4.1.3 Curcumin β-D-glucopyranosiduronic acid 2,3,4-Tri-O-acetyl, methyl ester (8)
Compound 7 (1.35 g, 5.7 mmol) and B2O3 (600 mg, 8.6 mmol) were dissolved in ethyl acetate (15 mL) at 85 °C. To this mixture was added an ethyl acetate solution (30 mL) of 6 (1.6 g, 3.4 mmol) and (n-BuO)3B (2.28 mL, 8.49 mmol). After stirring for 1 h, the mixture was treated with piperidine (200 μL, mmol) at 85 °C for 30 min, and then with 0.5 N HCl (20 mL) at 50 °C for 30 min. Reaction mixture was cooled and then extracted with ethyl acetate, washed with water and dried over Na2SO4. Flash chromatography over silica gel (1:2 to 1:9 hexanes/EtOAc) gave 8 as yellow foam (2.17 g, 55%). IR (neat) 1755.68, 1626.44, 1586.48, 1509.51; 1H NMR (CDCl3) δ 7.59 (d, J = 15.6 Hz, 1H), 7.56 (d, J = 15.6 Hz, 1H), 7.10 (m, 5H), 6.92 (d, J = 7.8 Hz, 1H), 6.49 (d, J = 16.2 Hz, 1H), 6.48 (d, J = 16.2 Hz, 1H), 5.81 (s, 1H), 5.34 (m, 3H), 5.10 (d, J = 7.2 Hz, 1H), 3.94 (s, 3H), 3.86 (s, 3H), 3.74 (s, 3H), 2.08 (s, 3H), 2.05 (s, 3H), 2.04 (s, 3H); 13C NMR (CDCl3) δ 184.2, 182.2, 170.1, 169.4, 169.3, 166.9, 150.8, 148.0, 147.3, 147.3, 146.8, 141.0, 139.5, 131.9, 127.6, 123.6, 122.9, 121.7, 121.6, 120.2, 114.9, 111.7, 109.7, 101.4, 100.3, 72.7, 71.8, 71.1, 69.2, 56.1, 55.9, 52.9, 20.6, 20.6, 20.5; HRMS (C34H36O15) calcd. 684.2054 found 685.5 (M+H).
4.1.4 β-D-Mono-glucuronidecurcumin (2)
To a cooled (ice-water bath) solution of the glucuronate ester (500 mg, 0.74 mmol) in methanol (14 mL) was added aqueous 1 N NaOH solution (14 mL) drop wise. The resulting solution was then stirred for 3h at 0 °C and then pH of the solution was adjusted to 3-4 by adding 50% aqueous formic acid. The yellow solid was filtered, collected and purified by reversed phase chromatography using water and methyl alcohol as eluent (gradient 20 to 80, 50 min) to give 2 as yellow solid (0.28 g, 70%); IR (Neat): 3400, 1624.4, 1585.9. 1H NMR (DMSO-d6) δ 7.56 (d, J = 7.2 Hz, 1H), 7.56 (d, J = 6.6 Hz, 1H), 7.37 (s, 1H), 7.31 (s, 3H), 7.23 (d, J = 8.4 Hz, 1H), 7.14 (d, J = 9.6 Hz), 7.12 (d, J = 8.4 Hz, 1H), 6.84 (m, 2H), 6.75 (d, J = 15.6 Hz, 1H), 6.08 (s, 1H), 5.21 (bs, 1H), 5.10 (bs, 1H), 4.95 (d, J = 7.2 Hz, 1H), 3.84 (s, 3H), 3.83 (s, 3H), 3.4-3.12 (m, 4H); 13C NMR (DMSO-d6) δ 184.3, 182.9, 172.2, 166.2, 150.2, 149.7, 149.2, 148.5, 141.3, 140.3, 129.1, 126.6, 123.7, 122.9, 121.6, 116.2, 115.8, 111.8, 111.8, 101.5, 100.2, 77.4, 74.1, 73.5, 72.5, 56.3, 56.2; HRMS (C27H28O12) calcd. 544.1581 found 545.5 (M+H) and 567.4 (M+Na).
4.1.5 1-[4-(2,3,4-Tri-O-acetyl, methyl ester, β-D-glucuronides of-3-methoxyphenyl)-5-hydroxy-1,4-hexadien-3-one (9)
1H NMR (CDCl3) δ 7.58 (d, J = 15.6 Hz, 1H), 7.11 (m, 3H), 6.52 (d, J = 15.6 Hz, 1H), 5.33 (m, 3H), 5.1 (d, J = 7.2 Hz, 1H) 3.87 (s, 3H), 2.08 (s, 3H), 2.06 (s, 3H), 2.04 (s, 3H); 13C NMR (CDCl3) δ 197.7, 190.9, 176.9, 170.1, 169.4, 169.2, 166.9, 150.9, 147.4, 139.1, 131.8, 125.5, 122.2, 121.3, 120.2, 111.6, 111.8, 101.1, 100.3, 72.7, 71.8, 69.2, 60.4, 56.1, 52.9, 20.6, 20.5; HRMS (C26H30O13) calcd. 550.16 found 551.5(M+4H) and 568.5(M+NH4).
4.1.6 Bis-[2,3,4-Tri-O-acetyl, β-D-glucopyranosiduronic acid methyl ester]-curcumin (10)
IR (Neat): 2255, 1756, 1629; 1H NMR (CDCl3) δ 7.58 (d, J = 15.6 Hz, 2H), 7.10 (m, 6H), 7.31 (s, 3H), 6.53 (d, J = 15.6 Hz, 2H), 5.83 (bs, 1H), 5.33 (m, 6H), 5.10 (d, J = 7.2 Hz, 2H), 3.86 (s, 6H), 3.74 (s, 6H), 2.08 (s, 6H), 2.06 (s, 6H), 2.04 (s, 6H); 13C NMR (CDCl3) δ 183.1, 170.1, 169.3, 169.2, 166.9, 150.9, 147.5, 140.0, 131.8, 123.6, 121.7, 120.2, 111.8, 100.3, 100.1, 72.7, 71.8, 71.1, 69.2, 56.2, 52.9, 20.6, 20.5; HRMS (C47H52O24) calcd. 1000.9014 found 1001.9 (M+H) and 1018.1 (M+NH4).
4.1.7 β-D-Di-glucuronidecurcumin (3)
IR (Neat): 2816, 2777, 2682, 1614; 1H NMR (DMSO-d6) δ7.52 (d, J = 15.6 Hz, 2H), 7.30 (s, 2H), 7.22 (d, J = 8.4 Hz, 2H), 7.10 (d, J = 8.4 Hz, 2H), 6.79 (d, J = 16.2 Hz, 2H), 5 (d, J = 6.6 Hz, 2H), 3.80 (s, 6H), 3.64 (d, J = 9.0 Hz, 2H), 3.34 (m, 6H); 13C NMR (DMSO-d6) δ 183.9, 173.3, 150.1, 149.1, 141.5, 130.2, 123.8, 123.7, 117.1, 116.5, 112.3, 100.7, 76.8, 76.2, 73.8, 72.6, 56.9; HRMS (C33H36O18) calcd. 720.6281 found 721.6 (M+H) and 743.4 (M+Na).
4.2 Preparation of Nuclear Extracts and Electrophoretic Mobility Shift Assays for NF-κB
Nuclear extracts were prepared as described previously.24 Briefly, 1.0 × 106 cells were washed with cold PBS and suspended in 0.1 mL hypotonic lysis buffer containing protease inhibitors for 30 min. The cells were then lysed with 5 μL of 10% NPδ40. The homogenate was centrifuged, and supernatant containing the cytoplasmic extracts was removed and stored frozen at –80 °C. The nuclear pellet was resuspended in 20 μL ice-cold nuclear extraction buffer. After 1 h of intermittent mixing, the extract was centrifuged, and supernatants containing nuclear extracts were secured. To determine NK-κB activation, we did electrophoretic mobility shift assay as described previously, with the following exceptions. Briefly, nuclear extracts were incubated with 32P-end-labeled 45-mer double-stranded NK-κB oligonucleotide (15 μg protein with 16 fmol DNA) from the HIV long terminal repeat, 5'-TTGTTACAAGGGACTTTCCGCTGGGGACTTTCCAGGGAGGCGTGG-3' (boldface indicates NK-κB-binding sites), for 30 min at 37 °C, and the DNA-protein complex formed was separated from free oligonucleotide on 6.6% native polyacrylamide gels. The dried gels were visualized with a Storm820 and radioactive bands were quantified using ImageQuant software (GE Healthcare, Piscataway, NJ).
4.2.1 MTT Assay
The cell growth effects of curcumin and curcumin glucuronides were determined by the MTT uptake method as described. Briefly, 5 × 103 cells were seeded in triplicate in 96-well plates and then treated with various concentrations of curcumins for 72 h at 37 °C. Thereafter, MTT solution was added to each well. After a 2 h incubation at 37 °C, an extraction buffer (20% SDS and 50% dimethylformamide) was added; the cells were then incubated overnight at 37 °C, and the absorbance was measured at 570 nm by using a 96-well multiscanner (MRX Revelation, Dynex Technologies, Chantilly, VA).
4.2.2 Statistical analysis
All data values were presented as mean ± SD. Statistical comparisons were measured using Student's t-test. Differences were considered significant at a P < 0.05.
Supplementary Material
Figure 1.

Chemical structures of curcumin, curcumin monoglucuronide and curcumin diglucuronide.
Acknowledgements
This work was carried out with the startup funds to William G. Bornmann and collaboration with Bharat B. Aggarwal from the University of Texas M D Anderson Cancer Center. We dearly thank Asha Nadipuram, Zhenghong Peng, Yunming Ying and David Maxwell for their assistance and encouragement.
Footnotes
Supplementary Material
Supplementary data associated with this article can be found, at http://
References and notes
- 1.Sharma OP. Biochem. Biopharmacol. 1976;25:1811. doi: 10.1016/0006-2952(76)90421-4. [DOI] [PubMed] [Google Scholar]
- 2.Toda S, Miyase T, Arichi H, Tanizawa H, Takino Y. Chem. Pharm. Bull. Tokyo. 1985;33:1725. doi: 10.1248/cpb.33.1725. [DOI] [PubMed] [Google Scholar]
- 3.Lahiri M, Maru GB, Amonkar AJ, Bhide SV. Modulation of benzo(a)pyrene induced DNA damage by some chemopreventive agents. In: Bhide SV, Maru GB, editors. Chemoprevention of Cancer. Omega Scientific Publishers; New Delhi: 1992. p. 152. [Google Scholar]
- 4.Nagabhushan M, Amonker AJ, Bhide SV. Food. Chem. Toxicol. 1987;25:545. doi: 10.1016/0278-6915(87)90207-9. [DOI] [PubMed] [Google Scholar]
- 5.Nagabhushan M, Bhide SVJ. Nutr. Growth Cancer. 1987;4:82. [Google Scholar]
- 6.Soudamini KK, Kuttan RJ. Ethnopharmacol. 1989;27:227. doi: 10.1016/0378-8741(89)90094-9. [DOI] [PubMed] [Google Scholar]
- 7.Azuine MA, Bhide SV. Nutr. Cancer. 1992;17:77. doi: 10.1080/01635589209514174. [DOI] [PubMed] [Google Scholar]
- 8.Haung MT, Wang YZ, Georgiadis CA, Laskin JD, Conney AH. Carcinogenesis. 1992;13:2183. doi: 10.1093/carcin/13.11.2183. [DOI] [PubMed] [Google Scholar]
- 9.Singh AK, Sidhu GS, Deepa T, Maheshwari RK. Cancer Lett. 1996;107:109. doi: 10.1016/0304-3835(96)04357-1. [DOI] [PubMed] [Google Scholar]
- 10.Jee SH, Shen SC, Tseng CR, Chiu HC, Kuo MLJ. Invest. Dermatol. 1998;111:656. doi: 10.1046/j.1523-1747.1998.00352.x. [DOI] [PubMed] [Google Scholar]
- 11.Kawamori T, Lubert R, Steele VE, Kello GJ, Kaskey RB, Rao CV, Reddy BS. Cancer Res. 1999;59:597. [PubMed] [Google Scholar]
- 12.Mehta K, Pantazis P, McQueen T, Aggrawal BB. Anticancer Drugs. 1987;5:470. doi: 10.1097/00001813-199706000-00010. [DOI] [PubMed] [Google Scholar]
- 13.Arau'jo CAC, Leon LL. Mem. Inst. Oswaldo Cruz, Rio de Janeiro. 2001;96(5):723. doi: 10.1590/s0074-02762001000500026. [DOI] [PubMed] [Google Scholar]
- 14.Aggarwal BB, Kumar A, Aggarwal MS, Shishodia S. In: Phytochemicals in Cancer Chemoprevention. Bagchi D, Preuss HG, editors. Culinary & Hospitality Industry Publications Services; 2003. [Google Scholar]
- 15.Gilden D. John SJ, editor. Protease Inhibitors, Overview and Analysis. AIDS Treatment News. 1994 Mar;III6:243. GMHC Treatment Issue 1994. [Google Scholar]
- 16.Ammon HP, Wahl MA. Planta. Med. 1991;57:1. doi: 10.1055/s-2006-960004. [DOI] [PubMed] [Google Scholar]
- 17.Kelloff JS, Crowell JA, Hawk ET. J. Cell Biochem. 1994;26s:54. [Google Scholar]
- 18.Dempe JS, Pfeiffer E, Grimm AS, Metzler M. Mol. Nutr. Food. Res. 2008;52:1074. doi: 10.1002/mnfr.200800029. [DOI] [PubMed] [Google Scholar]; a Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB. Mol. Pharm. 2007;4:807. doi: 10.1021/mp700113r. [DOI] [PubMed] [Google Scholar]; b Irving GR, Howells LM, Sale S, Kralj-Hans I, Atkin WS, Clark SK, Britton RG, Jones DJ, Scott EN, Berry DP, Hemingway D, Miller AS, Brown K, Gescher AJ. Cancer. Prev. Res (Phila) 2013;6:119. doi: 10.1158/1940-6207.CAPR-12-0281. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Vareed SK, Kakarala M, Ruffin MT, Crowell JA, Normolle DP, Djuric Z, Brenner DE. Cancer. Epidemiol. Biomarkers. Prev. 2008;17:1411. doi: 10.1158/1055-9965.EPI-07-2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hergenhahn M, Bertram B, Wiessler M, Sorg BL. 2003 US Patent 20030153512. Mishra S, Narain U, Mishra R, Misra K. Bioorg. Med. Chem. 2005;13:1477. doi: 10.1016/j.bmc.2004.12.057. Mohri K, Watanable Y, Yoshida Y, Satoh M, Isobe K, Sugimoto N, Tsuda Y. Chem. Pharm. Bull. 2003;51:1268. doi: 10.1248/cpb.51.1268. Parvathy KS, Srinivas P. Ultrason. Sonochem. 2008;15:571. doi: 10.1016/j.ultsonch.2007.09.007. Recent review article on Glucuronides see: Stachulski AV, Meng X. Nat. Prod. Rep. 2013;30:806. doi: 10.1039/c3np70003h.
- 20.a Kumar GRV, Divakar S. Biotechnol. Lett. 2005;27:1411. doi: 10.1007/s10529-005-3691-8. [DOI] [PubMed] [Google Scholar]; b Kaminaga Y, Nagatsu A, Akiyama T, Sugimoto N, Yamazaki T, Maitani T, Mizukami H. FEBS Lett. 2003;555:311. doi: 10.1016/s0014-5793(03)01265-1. [DOI] [PubMed] [Google Scholar]
- 21.a Pabon HJJ. Recl. Trav. Chim. Pays-Bas. 1964;83:379–385. [Google Scholar]; b Masuda T, Matsumura H, Oyama Y, Takeda Y, Jitoe A, Kida A, Hidaka K. J. Nat. Prod. 1998;61:609. doi: 10.1021/np970555g. [DOI] [PubMed] [Google Scholar]
- 22.Weber WM, Hunsaker LA, Roybal CN, Bobrovnikova-Marjon EV, Abcouwer AF, Royer RE, Deck LM, Vander Jagt DL. Bioorg. Med. Chem. 2006;14:2450. doi: 10.1016/j.bmc.2005.11.035. [DOI] [PubMed] [Google Scholar]
- 23.Chaturvedi MM, Mukhopadhyay A, Aggarwal BB. Meth. Enzymol. 2000;319:585. doi: 10.1016/s0076-6879(00)19055-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.