

Abstract
From a γ-AApeptide-based one-bead-one-compound (OBOC) combinatorial library, we identified γ-AApeptides that can selectively inhibit STAT3/DNA interaction and suppress the expression levels of STAT3 target genes in intact cells. Our results demonstrate that in addition to the SH2 domain, the DNA binding domain of STAT3 is targetable for the development of new generation of anti-cancer therapeutics.
The Signal Transducer and Activator of Transcription 3 (STAT3) is a transcription factor that regulates many biological processes including cell proliferation, differentiation and survival.1-4 Under normal physiological condition, the activation of STAT3 is transient and tightly regulated, and is only triggered by the stimulation of extracellular cytokines and growth factors such as IL-6, EGF and PDGF, which leads to the phosphorylation of a specific tyrosine (Y-705) on STAT3.5, 6 This phosphorylation subsequently induces the dimerization of STAT3-STAT3 which is stabilized by two reciprocal phosphotyrosine-SH2 binding interactions. The phosphorylated STAT3 dimers translocate to the cell nucleus and bind to promoter regions in DNA, resulting in regulation of specific gene expression.7, 8 However, STAT3 is constitutively activated in a variety of cancers including both solid tumors (i.e. breast, prostate, lung, pancreatic) and hematological cancers (i.e. lymphoma, leukemia, melanoma).9-11 Such hyperactivation of STAT3 leads to uncontrolled cell proliferation by activating cell cycle regulators such as c-Myc and cyclin D1, and enhancement of cell survival by selectively inducing the expression of anti-apoptotic proteins including Bcl-xL and survivin. As such, STAT3 mediated signaling pathways are recognized as valid cancer targets.
Many approaches have been adopted to inhibit constitutive activation of STAT3. Among the domains of STAT3 that regulate its function are SH2 domain (dimerization domain) and the DNA-binding domain (Figure 1). Thus, STAT3 signaling can be suppressed by either inhibition of STAT3 dimerization or STAT3-DNA binding.1-4 Significant effort has been devoted to the development of STAT3/STAT3 dimerization inhibitors that disrupt the phosphotyrosine-SH2 binding.7, 8, 12-17 Because STAT3-DNA binding is downstream of phosphorylated STAT3 dimerization, most STAT3 dimerization inhibitors also exhibit inhibitory activity against STAT3-DNA binding.1-4 However, molecules that specifically recognize the STAT3 DNA binding domain, and therefore directly disrupt STAT3-DNA binding interactions, are rare. This is because the STAT3-DNA binding interface is large and unlike in other transcription factor/DNA interactions the STAT3 DNA binding domain is complex involving residues from multiple α-helices and β-sheets.18 As such, the rational design of inhibitors is difficult. However, disruption of STAT3-DNA binding may be an alternative approach in the regulation of gene transcription compared to the inhibition of SH2 domain dimerization. Buettner et al 19 used a virtual screening to identify NSC-368262 that inhibits STAT3-DNA binding by covalently alkylating Cys468, a residue on the DNA-binding surface of STAT3. The exploration of new and non-covalent molecular ligands that selectively inhibit STAT3-DNA binding are therefore very significant, as such an effort will not only lead to novel anti-cancer therapeutics, but also provide a new tool to further dissect the functional role of STAT3 in the regulation of cell proliferation and apoptosis.
Figure 1.

The domains of STAT3 protein.18
Based on chiral PNA backbone, we, have recently developed a new class of peptidomimetics termed “γ-AApeptides” (Figure 2),20 as they are oligomers of N-acylated-N-aminoethyl amino acids. This class of peptidomimetics can project the same number of side chains compared to a peptide of the same length.21, 22 Additionally, they are highly amendable for the generation of chemically diverse libraries because half of their side chains can be selected from an endless set of acylating agents.23, 24 Moreover, they are highly resistant to proteolytic degradation.20, 25 These features make γ-AApeptides a promising platform for the identification and development of potential molecular ligands and drug candidates. This has been evidenced by our recently developed one-bead-one-compound (OBOC) γ-AApeptide-based combinatorial library, from which we have successfully identified one γ-AApeptide capable of preventing the aggregation of Aβ peptides.24 Thus, we believed the similar approach of γ-AApeptide combinatorial library could be used to identify molecular ligands that specifically disrupt STAT3/DNA interaction.
Figure 2.
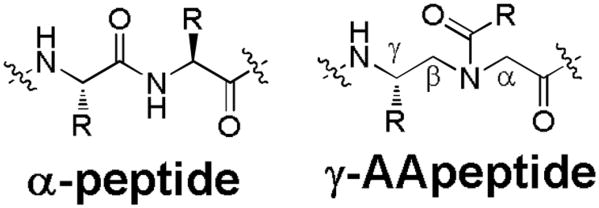
The structure of γ-AApeptide in comparison to α-peptide.
To test our hypothesis, the OBOC γ-AApeptide combinatoriallibrary was prepared as previously reported (Scheme 1).24 Briefly, TentaGel beads were attached with a Met residue, whichallows the cleavage of the γ-AApeptides from the beads upon thetreatment with CNBr.24, 26, 27 Four alloc-γ-AA building blocks andfive acylating agents (Figure 3b) were used to construct the OBOC γ-AApeptide library containing 192,000 different γ-AApeptides. The sequences in the library are composed of four γ-AApeptide building blocks, which are equivalent to 8-merpeptides in length. A library containing γ-AApeptides of samelengths has been previously reported to be successful in theidentification of the unnatural ligand disrupting Aβ aggregation.24
Scheme 1.

a, the preparation of γ-AApeptide OBOC library. b, The diversity of the library: Totally 4 × 5 × 4 × 5 × 4 × 5 × 4 × 6 = 192,000 compounds. Beads: TentaGel MB NH2, particle size: 140 - 170 μm, capacity: 0.5 nmol/bead.
Figure 3.

The structures of sequences identified from γ-AApeptide OBOC library against STAT3 screening.
To maximize chemodiversity, hydrophobic, cationic and anionic groups were used in the library. The on-bead library was treated with TFA to remove any protecting groups before use for screening.
A screening protocol was then carried out to identify ligands that potentially target STAT3-DNA binding. In brief, the library was first incubated with STAT3 (full length STAT3 protein) (see experimental for details), followed by the incubation with anti-STAT3 antibody (Figure S1). This antibody specifically recognizes the c-terminus of STAT3, and therefore potentially either disrupts the interaction of SH2 domain-binding beads with STAT3, or does not bind to STAT3 if such an interaction is too strong. In either case, anti-STAT3 antibody is not capable of sticking on beads recognizing SH2 domain of STAT3. Next, the library was incubated with Alexa Fluor 594 labeled secondary antibody, and the red-colored fluorescent beads were isolated by a micropipette under the microscope. Out of 192,000 beads, six positive beads were identified, suggesting the specificity of the library is high. The γ-AA peptides were cleaved off the beads by CNBr and sequenced by MS/MS (Figure S2).24 Four sequences were identified unambiguously (Figure 3).
In an effort to test the ability of the lead γ-AApeptides to inhibit STAT3-STAT3 dimerization, we conducted fluorescence polarization assays to determine whether these molecules disrupt the binding of STAT3 to fluorescein-labelled GpYLPQTV phosphotyrosine peptide which is known to bind the STAT3-SH2 domain.7 None of these molecules show any inhibitory activity (Figure S4), suggesting that these γ-AApeptides do not bind to STAT3-SH2 domain and therefore do not prevent STAT3 dimerization.
To assess if these γ-AApeptides bind to STAT3 and therefore inhibit STAT3-DNA binding, an in vitro STAT3 filter assay was carried out (Figure 4a). In this assay, nuclear extract from MDA-MB-468 human breast cancer cells were incubated with a biotin-conjugated STAT3 probe (please see experimental methods for more details) to allow the formation of STAT3/DNA probe complex, which could be detected with streptavidin-HRP by luminescence. The SH2-binding phosphotyrosine peptide, GpYLPQTV (IC50 of 150 nM for inhibition of dimerization of STAT3-STAT3 in vitro in FP assays), 28 inhibited STAT3-DNA binding by 40% and 60% at 10 μM and 100 μM in this STAT3 filter assay (Figure 4a), respectively. This is because dimerized STAT3 has higher DNA-binding affinity compared to monomeric STAT3, and therefore prevention of dimerization exhibits inhibitory effect on STAT3-DNA binding. Intriguingly, Figure 4a shows that all lead γ-AApeptides, although didn't bind to STAT3 SH2 domain, disrupted DNA-STAT3 binding effectively. Except for γ-AApeptide 4, the other sequences exhibited IC50s of 10-30 μM, with γ-AApeptide 1, inhibiting DNA-STAT3 binding at 30 μM. The fact that 1 was the most and 4 was the least potent suggest that a negative charge was not tolerated on the N-acetylated C-terminus of the AApeptides. However, the negative charge is tolerated on the N-aminoethyl at the C-terminus and on the N-acetylated N-terminus.
Figure 4.

DNA-STAT3 binding assays. a, STAT3 DNA binding filter plate assay with nuclear extract from MDA-MB-468 human breast cancer cells. Compounds (0-100 μM) were added to the mixture of nuclear extract and STAT3 probe and detected by chemiluminescence. b, STAT3 DNA binding filter plate assay on whole cells. Intact MDA-MB-468 cells were first treated with compounds then nuclear extracts were prepared, and incubated with STAT3 probe and detected by chemiluminescence. S3I-1757, a previously reported inhibitor of STAT3 dimerization, is included as a control. The concentration used for all the compounds is 100 μM. c, the structure of S3I-1757.
We next determined if the γ-AApeptides can retain STAT3-DNA binding inhibitory activity on whole cells. As phosphotyrosine peptide GpYLPQTV is not cell permeable, a previously reported small molecular inhibitor of STAT3 dimerization, S3I-1757, was included as a positive control.7 Figure 4b shows all γ-AApeptides are able to permeate cell membranes and disrupt STAT3-DNA binding. γ-AApeptide 1 was the most potent and its activity was comparable to S3I-1757. The results suggest γ-AApeptides might be promising candidates for the development of drug candidates targeting STAT3 signaling pathway.
To assess the ability of lead γ-AApeptides to modulate the expression of STAT3 regulated genes, we have carried out western immunoblotting to determine the effect of γ-AApeptides on the expression levels of survivin and cyclin D1 (Figure 5). Consistent with FP results, none of γ-AApeptides inhibit STAT3 phosphorylation, while S3I-1757, blocks STAT3 phosphorylation as expected. Figure 5 also shows that all of γ-AApeptides decreased to protein levels of survivin and cyclic D1, as potent as did S3I-1757.
Figure 5.

Treatment of MDA-MB-468cells with γ-AApeptides decreases the levels of survivin and cyclin D1 but not P-STAT3. MDA-MB-468 cells were treated overnight with 100 μM compounds and processed for western immunoblotting with the indicated antibodies as described in Methods. S3I-1757 was included as positive control. The results are representative of three independent experiments.
To rationalize the findings that these γ-AApeptides can disrupt STAT3/DNA binding, a computer molecular modeling was carried out by docking the most effective inhibitor 1 onto the STAT3 domain that binds DNA (Figure 6).
Figure 6.

Docking of γ-AApeptide 1 on the STAT3 DNA-binding domain (PDB 1BG1) using Glide program. a, STAT3 is shown as the surface representation and 1 is shown as the stick representation. b, Interactions between 1 and residues from STAT3. 1 is shown as the stick representation, and amino acid residues in STAT3 are shown in line representation.
As shown in Figure 6a, γ-AApeptide 1, containing multiple negatively charged carboxylate groups, is highly complementary to the STAT3 binding domain in which many cationic and polar amino acid residues are present. The three carboxylate groups interact with positively charged residues R423, R382 and K340 respectively through electrostatic attraction, which may account for the most critical force for the binding affinity of 1 towards STAT3 DNA-binding domain (Figure 6b). In addition, the phenyl ring near the N-terminus inserts deeply into the hydrophobic pocket formed by L430, I431 and V432. The hydrophobic interaction may further contribute to the binding specificity and affinity. Furthermore, the backbone of 1, including its C-terminus, make a few contacts with other polar and charged residues including E415, R417, N466 and Q409. Overall, the modeling suggests that as the STAT3 DNA binding domain is highly positively charged, the most negatively charged sequence in the identified γ-AApeptides, 1 binds to STAT3 DNA binding domain through a range of charge-charge interactions and hydrophobic interactions. Interestingly, the least potent γ-AA-peptide 4 has only one carboxylate that is not optimally positioned to interact with residue K340. The modeling also provides some insights into future rational design and optimization of molecules for the inhibition of STAT3/DNA binding.
In summary, we have developed a γ-AApeptide OBOC combinatorial library, and successfully identified lead compounds that disrupt STAT3-DNA interaction in nuclear extracts. The fact that these γ-AApeptides do not inhibit the binding of GpYLPQTV to STAT3 distinguishes them from STAT3-STAT3 dimerization inhibitors (REFs). Furthermore, despite their fairly large size, these γ-AApeptide were taken up by human cancer cells and inhibited STAT3-DNA binding and STAT3-regulated gene expression. This is not only the first report of γ-AApeptides inhibiting STAT3 function but also that γ-AApeptides are among the first few molecules that bind to STAT3 DNA-binding domain non-covalently and disrupt STAT3-DNA interaction. The results herein strongly suggests that STAT3 DNA-binding domain is a novel target for inhibiting STAT3 function, and inspires the future development of novel anti-cancer agents targeting STAT3 signaling. In addition, our strategy also demonstrates that the approach of γ-AApeptide OBOC library can be used to identify chemical probes or drug candidates against targets traditionally believed “undruggable”. Thus, with appropriate modification and further development of γ-AApeptide libraries, this strategy could be employed to develop more potent and specific ligands that bind to a variety of medicinally relevant targets. The further development of γ-AApeptide libraries, as well as the optimization of lead γ-AApeptides for their inhibitory activity against STAT3-DNA binding, is currently underway.
Supplementary Material
Acknowledgments
This work is supported by Elsa Pardee Foundation and Florida Bankhead-Coley program (JC) and NIH RO1 CA140681-05 (SS).
Footnotes
Electronic Supplementary Information (ESI) available: [development of γ-AApeptide library, ligand screening and identification, experimental details]. See DOI: 10.1039/b000000x/
Notes and references
- 1.Debnath B, Xu SL, Neamati N. J Med Chem. 2012;55:6645–6668. doi: 10.1021/jm300207s. [DOI] [PubMed] [Google Scholar]
- 2.Masciocchi D, Gelain A, Villa S, Meneghetti F, Barlocco D. Future Med Chem. 2011;3:567–597. doi: 10.4155/fmc.11.22. [DOI] [PubMed] [Google Scholar]
- 3.Lavecchia A, Di Giovanni C, Cerchia C. Expert Opin Ther Pat. 2014 doi: 10.1517/13543776.2014.877443. ASAP. [DOI] [PubMed] [Google Scholar]
- 4.Page BD, Ball DP, Gunning PT. Expert Opin Ther Pat. 2011;21:65–83. doi: 10.1517/13543776.2011.539205. [DOI] [PubMed] [Google Scholar]
- 5.Sun JZ, Blaskovich MA, Jove R, Livingston SK, Coppola D, Sebti SDM. Oncogene. 2005;24:3236–3245. doi: 10.1038/sj.onc.1208470. [DOI] [PubMed] [Google Scholar]
- 6.Turkson J, Kim JS, Zhang S, Yuan J, Huang M, Vultur AM, Raptis L, Sebti S, Hamilton AD, Jove J. Eur J Cancer. 2002;38:S98–S98. [Google Scholar]
- 7.Zhang XL, Sun Y, Pireddu R, Yang H, Urlam MK, Lawrence HR, Guida WC, Lawrence NJ, Sebti SM. Cancer Res. 2013;73:1922–1933. doi: 10.1158/0008-5472.CAN-12-3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang XL, Yue PB, Page BDG, Li TS, Zhao W, Namanja AT, Paladino D, Zhao JH, Chen Y, Gunning PT, Turkson J. Proc Natl Acad Sci U S A. 2012;109:9623–9628. doi: 10.1073/pnas.1121606109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Urlam MK, Pireddu R, Ge YY, Zhang XL, Sun Y, Lawrence HR, Guida WC, Sebti SM, Lawrence NJ. MedChemComm. 2013;4:932–941. doi: 10.1039/C3MD20323A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Siddiquee KAZ, Gunning PT, Glenn M, Katt WP, Zhang SM, Schrock C, Sebti SM, Jove R, Hamilton AD, Turkson J. ACS Chem Biol. 2009;4:309–309. doi: 10.1021/cb7001973. [DOI] [PubMed] [Google Scholar]
- 11.Cheng FD, Wang HW, Horna P, Wang Z, Shah B, Sahakian E, Woan KV, Villagra A, Pinilla-Ibarz J, Sebti S, Smith M, Tao JG, Sotomayor EM. Cancer Res. 2012;72:4440–4448. doi: 10.1158/0008-5472.CAN-11-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fletcher S, Page BDG, Zhang XL, Yue PB, Li ZH, Sharmeen S, Singh J, Zhao W, Schimmer AD, Trudel S, Turkson J, Gunning PT. ChemMedChem. 2011;6:1459–1470. doi: 10.1002/cmdc.201100194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siddiquee KAZ, Gunning PT, Glenn M, Katt WP, Zhang S, Schroeck C, Sebti SM, Jove R, Hamilton AD, Turkson J. ACS Chem Biol. 2007;2:787–798. doi: 10.1021/cb7001973. [DOI] [PubMed] [Google Scholar]
- 14.Gunning PT, Glenn MP, Siddiquee KAZ, Katt WP, Masson E, Sebti SM, Turkson J, Hamilton AD. ChemBioChem. 2008;9:2800–2803. doi: 10.1002/cbic.200800291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mandal PK, Liao WS, McMurray JS. Org Lett. 2009;11:3394–3397. doi: 10.1021/ol9012662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mandal PK, Gao FQ, Lu Z, Ren ZY, Ramesh R, Birtwistle JS, Kaluarachchi KK, Chen XM, Bast RC, Liao WS, McMurray JS. J Med Chem. 2011;54:3549–3563. doi: 10.1021/jm2000882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mandal PK, Ren ZY, Chen XM, Kaluarachchi K, Liao WSL, McMurray JS. Int J Pep Res Ther. 2013;19:3–12. doi: 10.1007/s10989-012-9313-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levy DE, Darnell JE., Jr Nat Reviews. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 19.Buettner R, Corzano R, Rashid R, Lin J, Senthil M, Hedvat M, Schroeder A, Mao A, Herrmann A, Yim J, Li H, Yuan YC, Yakushijin K, Yakushijin F, Vaidehi N, Moore R, Gugiu G, Lee TD, Yip R, Chen Y, Jove R, Horne D, Williams JC. ACS Chem Biol. 2011;6:432–443. doi: 10.1021/cb100253e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Niu YH, Hu YG, Li XL, Chen JD, Cai JF. New J Chem. 2011;35:542–545. [Google Scholar]
- 21.Niu Y, Padhee S, Wu H, Bai G, Harrington L, Burda WN, Shaw LN, Cao C, Cai J. Chem Commun. 2011;47:12197–12199. doi: 10.1039/c1cc14476f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, Smith C, Wu H, Padhee S, Manoj N, Cardiello J, Qiao Q, Cao C, Yin H, Cai J. ACS Chem Biol. 2014;9:211–217. doi: 10.1021/cb4006613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu H, Teng P, Cai J. Eur J Org Chem. 2014:1760–1765. [Google Scholar]
- 24.Wu H, Li Y, Ge B, Niu Y, Qiao Q, Tipton J, Cao C, Cai J. Chem Commun. 2014;50:5206–5208. doi: 10.1039/c3cc46685j. [DOI] [PubMed] [Google Scholar]
- 25.Yang Y, Niu Y, Hong H, Wu H, Zhang Y, Engle J, Barnhart T, Cai J, Cai W. Chem Commun. 2012;48:7850–7852. doi: 10.1039/c2cc33620k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aquino C, Sarkar M, Chalmers MJ, Mendes K, Kodadek T, Micalizio GC. Nat Chem. 2012;4:99–104. doi: 10.1038/nchem.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Udugamasooriya DG, Dineen SP, Brekken RA, Kodadek T. J Am Chem Soc. 2008;130:5744–5752. doi: 10.1021/ja711193x. [DOI] [PubMed] [Google Scholar]
- 28.Ren ZY, Cabell LA, Schaefer TS, McMurray JS. Bioorg Med Chem Lett. 2003;13:633–636. doi: 10.1016/s0960-894x(02)01050-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.