

Abstract
The use of copper and rhodium catalysts separately and in combination directs reactions between vinyldiazoacetates 3 and cinnamaldehydes 2 to from formal [4+3]-cycloaddition (epoxidation followed by Cope rearrangement), intramolecular cyclopropanation, and Mukaiyama-aldol reactions selectively and in high yield.
Keywords: Divergent Outcome, Rhodium and Copper, Formal [4+3]-Cycloaddition, Intramolecular Cyclopropanation, Mukaiyama-Aldol Reactions, Cope Rearrangement
That a reaction pathway can be redirected to a different product by changing a reactant or reaction conditions is well known and widely practiced. Such processes are referred to as “divergent”, and this term is broadly applied to methodology,1 synthesis,2 reactivity and selectivity,3 among others.4 However, processes in which the same reactant(s) form different products in synthetically meaningful yields by application of different catalysts are rare.5 We and others have reported exceptionally efficient catalyst-dependent processes that occur with the same diazo substrates to form structurally different compounds.5b,6–8 In the most well-documented cases, dirhodium(II) catalysts having different ligands differentiate between cyclopropanation and C-H insertion, ylide formation and aromatic substitution, or aromatic cycloaddition and C-H insertion with the same diazoacetates.6 The exclusive formation of one of two regioisomeric dihydropyrroles in modest yields from the reactions between vinyldiazoacetates and imines using dirhodium(II) acetate or copper(II) triflate (Scheme 1), due to either electrophilic metal carbene formation (intermediate 1a in the case of rhodium) or to initial iminium ion formation (intermediate 1b in the case of copper),7 exemplifies the critical role of catalyst in product selection. Other examples include the copper(I)/rhodium(II) product differences in macrocyclization with diazoacetates.8 Although both dirhodium and copper catalysts are well established catalysts for dinitrogen extrusion from diazo compounds, there can be striking differences between them in product outcomes from the same reactant(s).
Scheme 1.
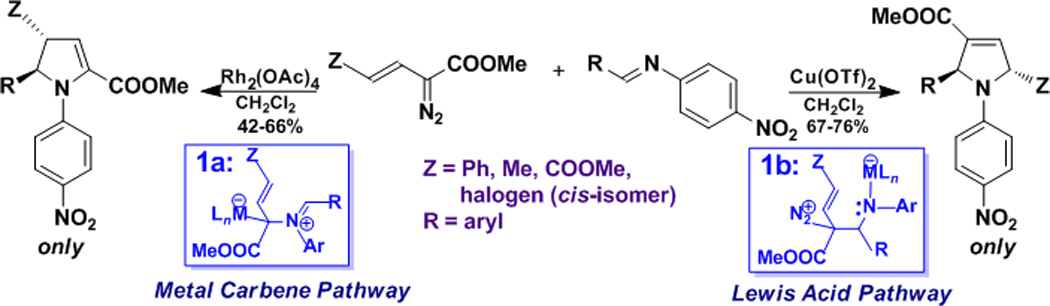
Divergent pathways to isomeric dihydropyrroles from the same reactants, but different catalysts.
We wish to report divergent copper and/or dirhodium catalyzed, synthetically-relevant processes that occur between cinnamaldehydes and siloxyvinyl-α-diazoacetates which clearly reveal fundamental differences between these two catalytic systems. Their applications include the syntheses of oxepin, furanone, and cyclopropane compounds by intramolecular cyclization (Scheme 2) with extraordinary efficiency and atom economy.
Scheme 2.
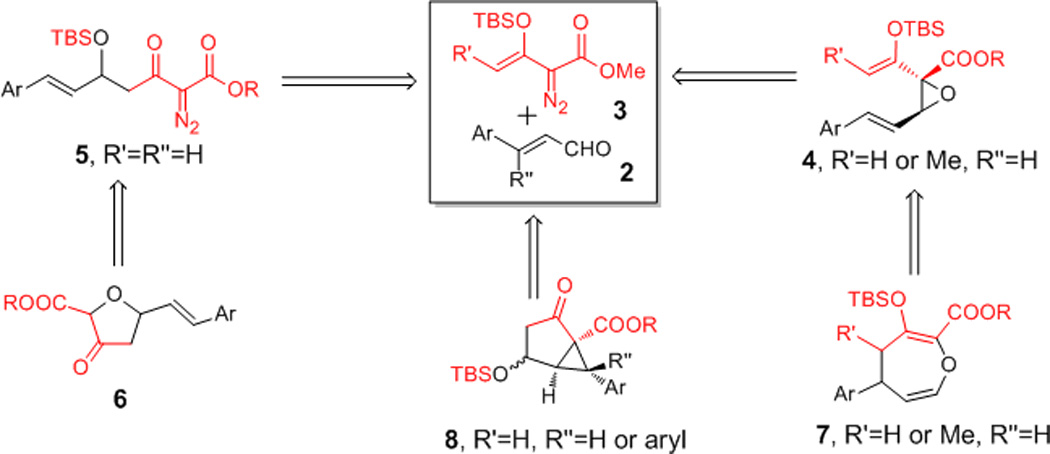
Divergent outcomes from copper- and rhodium-catalyzed reactions of 3 with cinnamaldehydes.
We have previously reported that methyl styryldiazoacetate undergoes ylide-induced epoxide formation in reactions with cinnamaldehydes catalyzed by dirhodium tetraacetate, but product yields were only modest, and mixtures were formed with the substituted aldehydes.9 Consequently, we were surprised when treatment of methyl siloxyvinyldiazoacetate (3-Me) with 4-nitrocinnamaldehyde at room temperature, catalyzed by dirhodium carboxylates, gave epoxide 4a with trans-divinyl substutuents in excellent yield (Scheme 3). Use of chiral catalysts that included Hashimoto’s Rh2(S-PTPA)410 and Davies’ Rh2(S-DOSP)411 did not give evidence of enantiocontrol in this transformation, and dirhodium carboxamidates exhibited very low reactivity towards dinitrogen extrusion of 3. Surprisingly, use of Cu(CH3CN)4PF6, which is even more reactive towards dinitrogen extrusion from diazo compounds than is rhodium acetate,12 gave the Mukaiyama-aldol addition product 5 in high yield when the reaction performed at 0°C without effecting diazo decomposition (Scheme 3).13 In this case, CuPF6 acts as Lewis acid for activation of the aldehyde towards electrophilic addition to 3 and does not cause decomposition of the diazoacetate in either the reactant or the product.
Scheme 3.
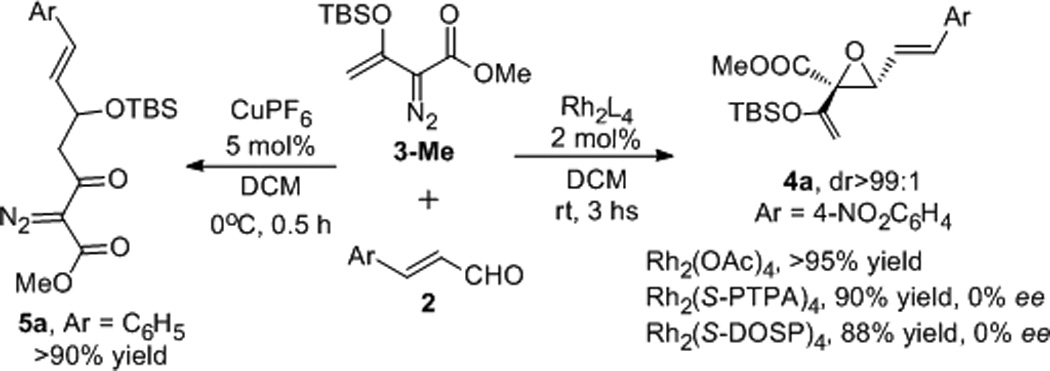
Divergent catalyst-dependent pathways from cinnamaldehydes and methyl siloxyvinyldiazoacetate 3-Me.
In contrast to their cis-divinylcyclopropane analogues that undergo [4+3]-cycloaddition at or below room temperature,14 and have been key steps in several total syntheses,15 trans-divinylepoxides are thermally stable at room temperature but slowly rearrange to 4,5-dihydrooxepins 7.16 This is obviously the reason why the conversion of 4 to dihydrooxepin does not occur under more moderate conditions. Hence a major challenge of this study has been to develop conditions suitable to form 4,5-dihydrooxepins in high yield. We initially applied thermal conditions to effect rearrangement of the trans-divinylepoxide 4a, and this approach did provide the 4-nitrophenyl derivative 7a, but the thermal transformation only occurred at or above 150°C and required long reaction times. To find the catalytic version of this rearrangement, we investigated the effects of an array of Lewis acids [CuPF6, CuOTf, Cu(OTf)2, Sc(OTf)3, Zn(OTf)2] to discover that with the application of a catalytic amount of Cu(hfacac)2 the reaction temperature could be decreased to 100°C while the reaction rate was dramatically increased. Further optimization by carrying out the two-step reaction in one-pot by performing the catalytic epoxidation in dichloromethane, replacing that solvent with toluene, adding Cu(hfacac)2, and heating at 125°C for 0.5 h gave 4,5-dihydrooxepin 7a in 95% isolated yield.17 The structure of 7a was confirmed by single-crystal X-ray diffraction analysis (Figure 1).
Figure 1.
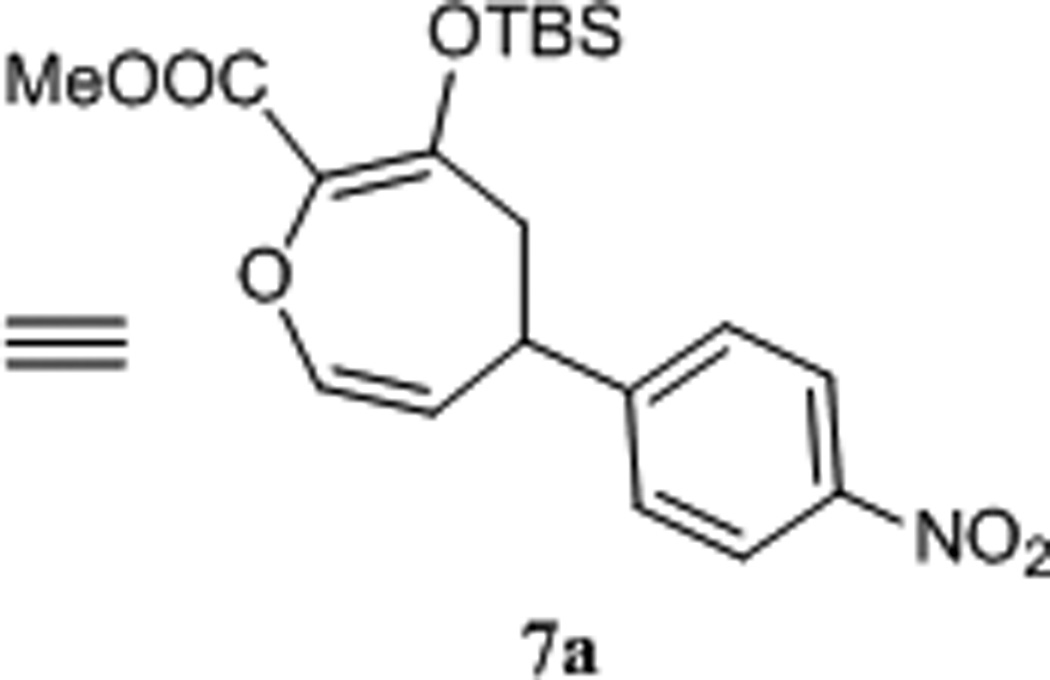
X-ray crystal structure of 7a.
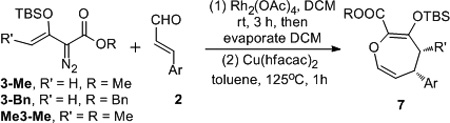
Using these optimum conditions we examined this rearrangement reaction for substrate generality in the two-step one-pot formal [4+3] cycloaddition of 3 with substituted cinnamaldehydes, and these results are summarized in Table 1. Product yields from reactions with cinnamaldehydes having electron-donating substituents were comparable to those from the nitrocinnamaldehydes (entries 4–9). Benzyl ester analogues (3-Bn) underwent the same two-step process with isolated yields that were identical to those obtained with the methyl esters (entries 10–12). That the reaction occurred with the stereospecificity of an electrocyclization reaction, whether from a direct Cope rearrangement of rearranged cis-divinyl epoxide18 or from the corresponding ylide, with propenyldiazoacetate Me3-Me as the reactant, cis-4,5-disubstituted-dihydrooxepine 7m was formed stereospecifically in very high yield.
Table 1.
Substrate Generality for the One-pot Tandem Epoxidation/Rearrangement Reaction.[a]
 | ||||
|---|---|---|---|---|
| Entry | 3 | 2, Ar | 7 | yield (%)[b] |
| 1 | 3-Me | 4-NO2C6H4 | 7a | 95 |
| 2 | 3-Me | 3-NO2C6H4 | 7b | 95 |
| 3 | 3-Me | 2-NO2C6H4 | 7c | 95 |
| 4 | 3-Me | C6H5 | 7d | 82 |
| 5 | 3-Me | 4-CF3C6H4 | 7e | 95 |
| 6 | 3-Me | 4-BrC6H4 | 7f | 90 |
| 7 | 3-Me | 4-ClC6H4 | 7g | 88 |
| 8 | 3-Me | 4-FC6H4 | 7h | 90 |
| 9 | 3-Me | 4-MeC6H4 | 7i | 81 |
| 10 | 3-Bn | 4-NO2C6H4 | 7j | 95 |
| 11 | 3-Bn | 2-NO2C6H4 | 7k | 95 |
| 12 | 3-Bn | C6H5 | 7l | 85 |
| 13 | Me3-Me | 4-NO2C6H4 | 7m | 95[c] |
The reaction was carried out with 3 (0.36 mmol), aldehyde (0.30 mmol), Rh2(OAc)4 (2 mol%), Cu(hfacac)2 (5 mol%), respective solvent (2 mL).
Isolated yield of 7 after chromatography.
The 1H NMR spectrum of the reaction mixture showed only one diasteroisomer, whose stereochemistry was confirmed by NOe analysis, see SI.
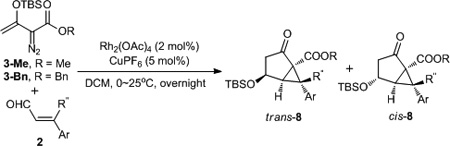
With the assumption that copper and rhodium catalysts act independently, so that the combination with vinyldiazoacetate 3-Me and cinnamaldehydes could achieve the formation of 7 under one set of reaction conditions, we treated 3-Me with cinnamaldehyde in the presence of catalytic amounts of rhodium acetate and copper(I) hexafluorophosphate at room temperature. However, instead of observing either epoxidation or dihydrooxepine products, use of this catalyst combination unexpectedly produced the two diastereoisomers19 of bicyclo[3.1.0]hexane 8a20 in high yield (Table 2). Substrates with electron-donating substituents showed higher reactivity (entry 2 versus entry 3), and this reactivity pattern was opposite to that observed for epoxidation. Reversal of diastereoselectivity from up to 9:1 to <1:20 was achieved by using a β-substituted cinnamaldehyde in this transformation (entry 5); interaction with the syn-phenyl substituent that occupies position 6 in 8 forces the OTBS group to favor the cis geometry. With dirhodium catalysts and Ar = Ph, diastereoselectivity ranged from 82:18 (with rhodium caprolactamate) to 27:73 (with rhodium trifluoroacetate).
Table 2.
Cooperative Rhodium and Copper Catalyzed Bicyclization of Vinyldiazoacetates 3 with α,β-Unsaturated Aldehydes.[a]
 | |||||
|---|---|---|---|---|---|
| entry | 3 | 2, Ar / R’’ | 8 | dr[b] (trans :cis) |
yield (%)[c] |
| 1 | 3-Me | C6H4 / H | 8a | 85:15 | 80 |
| 2 | 3-Me | 4-MeOC6H4 / H | 8b | 75:25 | 87 |
| 3 | 3-Me | 2-NO2C6H4 / H | 8c | 90:10 | 32 |
| 4 | 3-Bn | C6H4 / H | 8d | 70:30 | 86 |
| 5 | 3-Bn | C6H4 / C6H4 | 8e | <5:95 | 65 |
Reaction in 0.3 mol scale: 3 (0.36 mmol), aldehyde (0.30 mmol), Rh2(OAc)4 (2 mol%), CuPF6 (5 mol%), solvent (2 mL).
Determined by 1H NMR of the crude reaction mixture.
Isolated yield of 8 (trans and cis) after chromatography.
Treatment of 3-Me and cinnamaldehyde with both copper(I) and copper(II) compounds in catalytic amounts resulted in the formation of the Mukaiyama-aldol reaction product 5 to the complete exclusion of epoxide 4. The product of the Mukaiyama-aldol reaction, catalyzed by CuPF6, which gave optimum results, when treated with rhodium acetate gave cyclopropanation products 8a. Alternatively, by performing the reaction of 3-Me and cinnamaldehyde with CuPF6 as catalyst at 40°C (3 h), instead of at or below room temperature, the two step process can be accomplished in one pot (77% yield, dr = 90:10).17 Obviously, the role of CuPF6 as a Lewis acid in these reactions is pronounced, and the possibility exists that coordination of 5 with CuPF6 inhibits its use as a catalyst for dinitrogen extrusion. The advantage of the cooperative rhodium and copper catalyzed bicyclization is that the overall transformation can be conducted at room temperature. Furanone 6 formation (Scheme 2) from diazoacetoacetates has been previously reported,21 although not with the structural diversity that is available by this methodology.
In summary, the use of copper and rhodium catalysts separately and in combination directs the reaction between vinyldiazoacetates 3 and cinnamaldehydes to a broad diversity of products selectively and in high yield (Scheme 4). The basis for this catalyst-based selectivity lies in the differences in Lewis acidity between copper and rhodium catalysts and the bidentate coordinating ability of copper catalysts. That copper and dirhodium catalysts can work cooperatively for product formation is demonstrated.
Scheme 4.
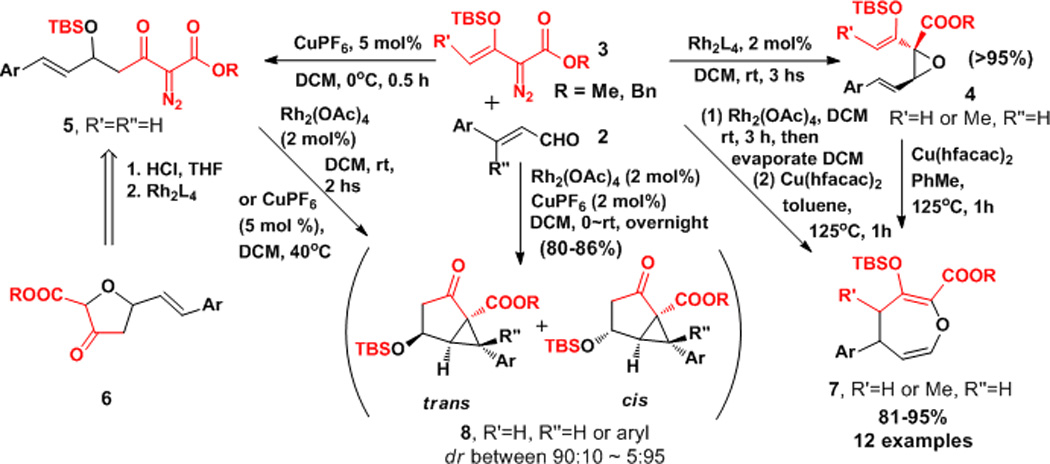
Divergent outcomes from copper- and rhodium-catalyzed reactions of 3 with cinnamaldehydes.
Experimental Section
To an oven-dried flask containing a magnetic stirring bar, cinnamaldehyde 2 (0.3 mmol), and Rh2(OAc)4 (2.0 mol%) in DCM (1.0 mL), was added diazo compound 3 (0.36 mmol) in DCM (1.0 mL) over 1 h via a syringe pump at room temperature. The reaction mixture was stirred for another 2 hours, then DCM was removed under reduced pressure, and toluene (2.0 mL) was added. The solution was transferred to the reaction tube containing a magnetic stirring bar; the tube was suited for use under high pressure. The reaction tube was sealed after Cu(hfacac)2 (5.0 mol%) was added, and the temperature of the reaction was warmed to 125°C in an oil bath with stirring. After complete consumption of the intermediate epoxide 4 (about 1 hour), monitored by 1H NMR (epoxide and the product oxepin overlap on thin layer chromatography), the reaction mixture was purified by column chromatography on silica gel (eluent: hexanes : EtOAc = 50:1 to 30:1) to give the pure products oxepin 7 in greater than 80% yield.
Acknowledgments
Support for this research to MPD from the National Institutes of Health (GM 46503) and National Science Foundation (CHE- 0748121) is gratefully acknowledged. WH thanks the National Science Foundation of China (20932003), and the MOST of China (2011CB808600).
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the Author.
Contributor Information
Xinfang Xu, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742; Institute of Drug Discovery and Development, East China Normal University, 3663 Zhongshan Bei Road, Shanghai 200062, China.
Wen-Hao Hu, Institute of Drug Discovery and Development, East China Normal University, 3663 Zhongshan Bei Road, Shanghai 200062, China, whu@chem.ecnu.edu.cn.
Peter Y. Zavalij, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742
Michael P. Doyle, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742.
References
- 1.a) Dai H-X, Stepan AF, Plummer MS, Zhang Y-H, Yu J-Q. Am. Chem. Soc. 2011;133:7222. doi: 10.1021/ja201708f. [DOI] [PubMed] [Google Scholar]; b) Jabre ND, Respondek T, Ulku SA, Korostelova N, Kodanko JJ. J. Org. Chem. 2010;75:650. doi: 10.1021/jo9021953. [DOI] [PubMed] [Google Scholar]; c) Pohlhaus PD, Bowman RK, Johnson JS. J. Am. Chem. Soc. 2004;126:2294. doi: 10.1021/ja0397963. [DOI] [PubMed] [Google Scholar]; d) Percec V, Barboiu B, Grigoras C, Bera TK. J. Am. Chem. Soc. 2003;125:6503. doi: 10.1021/ja034746j. [DOI] [PubMed] [Google Scholar]
- 2.a) Mizoguchi H, Oguri H, Tsug K, Oikawa H. Org. Lett. 2009;11:3016. doi: 10.1021/ol901020a. [DOI] [PubMed] [Google Scholar]; b) Medina SH, El-Sayed MEH. Chem. Rev. 2009;109:3141. doi: 10.1021/cr900174j. [DOI] [PubMed] [Google Scholar]; c) Wang K, Xiang D, Liu J, Pan W, Dong D. Org. Lett. 2008;10:1691. doi: 10.1021/ol800178x. [DOI] [PubMed] [Google Scholar]; d) Delest B, Nshimyumukiza P, Fasbender O, Tinant B, Marchand-Brynaert J, Darro F, Robiette R. J. Org. Chem. 2008;73:6816. doi: 10.1021/jo801256b. [DOI] [PubMed] [Google Scholar]
- 3.a) Whited MT, Grubbs RH. Acc. Chem. Res. 2009;42:1607. doi: 10.1021/ar900103e. [DOI] [PubMed] [Google Scholar]; b) Zhang G, Catalano VJ, Zhang L. J. Am. Chem. Soc. 2007;129:11358. doi: 10.1021/ja074536x. [DOI] [PubMed] [Google Scholar]; c) Lautens M, Han W. J. Am. Chem. Soc. 2002;124:6312. doi: 10.1021/ja011110o. [DOI] [PubMed] [Google Scholar]
- 4.a) Vaidya T, Manbeck GF, Chen S, Frontier AJ, Eisenberg R. J. Am. Chem. Soc. 2011;133:3300. doi: 10.1021/ja111317q. [DOI] [PubMed] [Google Scholar]; b) Hilt G. Angew. Chem. Int. Ed. 2009;48:6390. doi: 10.1002/anie.200901939. [DOI] [PubMed] [Google Scholar]; c) Wise EL, Rayment I. Acc. Chem. Res. 2004;37:149. doi: 10.1021/ar030250v. [DOI] [PubMed] [Google Scholar]; d) Dehli JR, Gotor V. Chem. Soc. Rev. 2002;31:365. doi: 10.1039/b205280f. [DOI] [PubMed] [Google Scholar]
- 5.a) Soriano E, Marco-Contelles J. Acc. Chem. Res. 2009;42:1026. doi: 10.1021/ar800200m. [DOI] [PubMed] [Google Scholar]; b) Panne P, Fox JM. J. Am. Chem. Soc. 2007;129:22. doi: 10.1021/ja0660195. [DOI] [PubMed] [Google Scholar]; c) Tanaka K, Fu GC. J. Am. Chem. Soc. 2003;125:8078. doi: 10.1021/ja035489l. [DOI] [PubMed] [Google Scholar]
- 6.a) Padwa A, Austin DJ, Hornbuckle SF, Semones MA, Doyle MP, Protopopova MN. J. Am. Chem. Soc. 1992;114:1874. [Google Scholar]; b) Padwa A, Austin DJ, Price AT, Semones MA, Doyle MP, Protopopova MN, Winchester WR, Tran A. J. Am. Chem. Soc. 1993;115:8669. [Google Scholar]; c) Padwa A, Austin DJ. Angew. Chem. Int. Ed. 1994;33:1797. [Google Scholar]; d) Bykowski D, Wu W-K, Doyle MP. J. Am. Chem. Soc. 2006;128:16038. doi: 10.1021/ja066452e. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Davies HML, Coleman MG, Ventura DL. Org. Lett. 2007;9:4971. doi: 10.1021/ol702218w. [DOI] [PubMed] [Google Scholar]
- 7.Doyle MP, Yan M, Hu W, Gronenberg LS. J. Am. Chem. Soc. 2003;125:4692. doi: 10.1021/ja029745q. [DOI] [PubMed] [Google Scholar]
- 8.a) Doyle MP, Protopopova MN, Poulter CD, Rogers DH. J. Am. Chem. Soc. 1995;117:7281. [Google Scholar]; b) Doyle MP, Peterson CS, Parker DL., Jr Angew. Chem. Int. Ed. 1996;35:1334. [Google Scholar]; c) Doyle MP, Protopopova MN, Peterson CS, Vitale JP, McKervey MA, Garcia CF. J. Am. Chem. Soc. 1996;118:7865. [Google Scholar]; d) Doyle MP, Peterson CS, Protopopova MN, Marnett AB, Parker DL, Jr, Ene DG, Lynch V. J. Am. Chem. Soc. 1997;119:8826. [Google Scholar]; e) Doyle MP, Hu W. J. Org. Chem. 2000;65:8839. doi: 10.1021/jo005589z. [DOI] [PubMed] [Google Scholar]
- 9.Doyle MP, Hu W, Timmons DJ. Org. Lett. 2001;3:3741. doi: 10.1021/ol016703i. [DOI] [PubMed] [Google Scholar]
- 10.a) Kitagaki S, Anada M, Kataoka O, Matsuno K, Umeda C, Watanabe N, Hashimoto S. J. Am. Chem. Soc. 1999;121:1417. [Google Scholar]; b) Goto T, Takeda K, Shimada N, Nambu H, Anada M, Shiro M, Ando K, Hashimoto S. Angew. Chem. Int. Ed. 2011;50:6803. doi: 10.1002/anie.201101905. [DOI] [PubMed] [Google Scholar]; c) Takeda K, Oohara T, Anada M, Nambu H, Hashimoto S. Angew. Chem. Int. Ed. 2010;49:6979. doi: 10.1002/anie.201003730. [DOI] [PubMed] [Google Scholar]
- 11.a) Hansen JH, Gregg TM, Ovalles SR, Lian Y, Autschbach J, Davies HML. J. Am. Chem. Soc. 2011;133:5076. doi: 10.1021/ja111408v. [DOI] [PubMed] [Google Scholar]; b) Briones JF, Hansen J, Hardcastle KI, Autschbach J, Davies HML. J. Am. Chem. Soc. 2010;132:17211. doi: 10.1021/ja106509b. [DOI] [PubMed] [Google Scholar]; c) Lian Y, Miller LC, Born S, Sarpong R, Davies HML. J. Am. Chem. Soc. 2010;132:12422. doi: 10.1021/ja103916t. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lian Y, Davies HML. J. Am. Chem. Soc. 2010;132:440. doi: 10.1021/ja9078094. [DOI] [PubMed] [Google Scholar]; e) Li Z, Davies HML. J. Am. Chem. Soc. 2010;132:396. doi: 10.1021/ja9075293. [DOI] [PubMed] [Google Scholar]
- 12.Doyle MP, McKervey MA, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds. New York, NY: John Wiley & Sons, Inc; 1998. [Google Scholar]
- 13.The Zn(OTf)2 catalysis forms the Mukaiyama-aldol reaction product from reactions of diazoacetoacetates with α,β-unsaturated carbonyl compounds (L. Zhou, M. P. Doyle, Org. Lett. 2010, 12, 796) but the Mukaiyama-Michael reaction product is formed from reactions of vinyldiazoacetate 3-Me with α,β-unsaturated carbonyl compounds (Y. Liu, Y. Zhang, N. Jee, M. P. Doyle, Org. Lett. 2008, 10, 1605).
- 14.a) Olson JP, Davies HML. Org. Lett. 2008;10:573. doi: 10.1021/ol702844g. [DOI] [PubMed] [Google Scholar]; b) Reddy RP, Davies HML. J. Am. Chem. Soc. 2007;129:10312. doi: 10.1021/ja072936e. [DOI] [PubMed] [Google Scholar]; c) Kusama H, Onizawa Y, Iwasawa N. J. Am. Chem. Soc. 2006;128:16500. doi: 10.1021/ja0671924. [DOI] [PubMed] [Google Scholar]; d) Brummond KM, Mao S, Shinde SN, Johnston PJ, Day BW. J. Comb. Chem. 2009;11:486. doi: 10.1021/cc900024p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Reddy RP, Davies HML. J. Am. Chem. Soc. 2007;129:10312. doi: 10.1021/ja072936e. [DOI] [PubMed] [Google Scholar]; b) Schwartz BD, Denton JR, Lian Y, Davies HML. J. Am. Chem. Soc. 2009;131:8329. doi: 10.1021/ja9019484. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Miller LC, Ndungu JM, Sarpong R. Angew. Chem. Int. Ed. 2009;48:2398. doi: 10.1002/anie.200806154. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lian Y, Miller LC, Born S, Sarpong R, Davies HML. J. Am. Chem. Soc. 2010;132:12422. doi: 10.1021/ja103916t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Shimizu M, Fujimoto T, Liu X, Hiyama T. Chem. Lett. 2004;33:438. [Google Scholar]; b) Clark DL, Chou W-N, White JB. J. Org. Chem. 1990;55:3975. [Google Scholar]; c) Berson JA, Dervan PB. J. Am. Chem. Soc. 1973;95:267. [Google Scholar]
- 17.See Supporting Information.
- 18.a) Maurin P, Kim S-H, Cho SY, Cha JK. Angew. Chem. Int. Ed. 2003;42:5044. doi: 10.1002/anie.200350988. [DOI] [PubMed] [Google Scholar]; b) Danheiser RL, Gee SK, Sard H. J. Am. Chem. Soc. 1982;104:7670. [Google Scholar]; c) Gadwood RC, Lett RM. J. Org. Chem. 1982;47:2268. [Google Scholar]
- 19.See Supporting Information for X-ray structures of cis-8b and trans-10d (which has TBS removed by hydrolysis of trans-8d).
- 20.Intramolecular cyclopropantion reactions of diazo ketones or esters that form similar bicyclo[3.1.0]hexane structures are reported: Feliz M, lam EG, Llusa R, Vicent C, Stiriba S-E, Pérez-Prieto J, Barberis M. Chem. Eur. J. 2006;12:1486. doi: 10.1002/chem.200500907. Lim Y-H, McGee KF, Jr, Sieburth SM. J. Org. Chem. 2002;67:6535. doi: 10.1021/jo025909+. Pirrung MC, Liu H, Morehead AT., Jr J. Am. Chem. Soc. 2002;124:1014. doi: 10.1021/ja011599l. Barberis M, Pèrez-Prieto J, Stiriba S-E, Lahuerta P. Org. Lett. 2001;3:3317. doi: 10.1021/ol010170w.
- 21.a) Liao M, Dong S, Deng G, Wang J. Tetrahedron Lett. 2006;47:4537. [Google Scholar]; b) Doyle MP, Kundu K, Russell AE. Org. Lett. 2005;7:517. doi: 10.1021/ol052003s. [DOI] [PubMed] [Google Scholar]; c) Deng G, Tian X, Qu Z, Wang J. Angew. Chem. Int. Ed. 2002;41:2773. doi: 10.1002/1521-3773(20020802)41:15<2773::AID-ANIE2773>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]; d) Calter MA, Zhu C. J. Org. Chem. 1999;64:1415. [Google Scholar]