

Abstract
We previously identified a distinct population of human circulating hematopoietic stem and progenitor cells (CHSPCs; CD14-glyA-CD34+AC133+/-CD45dimCD31+ cells) in the peripheral blood (PB) and bone marrow, and their frequency in the PB can correlate with disease state. The proangiogenic subset (pCHSPC) play a role in regulating tumor progression, for we previously demonstrated a statistically significant increase in C32 melanoma growth in NOD.Cg-Prkdcscid (NOD/SCID) injected with human pCHSPCs (p<0.001). We now provide further evidence that pCHSPCs possess proangiogenic properties. In vitro bio-plex cytokine analyses and tube forming assays indicate that pCHSPCs secrete a proangiogenic profile and promote vessel formation respectively. We also developed a humanized bone marrow-melanoma orthotopic model to explore in vivo the biological significance of the pCHSPC population. Growth of melanoma xenografts increased more rapidly at 3-4 weeks post-tumor implantation in mice previously transplanted with human CD34+ cells compared to control mice. Increases in pCHSPCs in PB correlated with increases in tumor growth. Additionally, to determine if we could prevent the appearance of pCHSPCs in the PB, mice with humanized bone marrow-melanoma xenografts were administered Interferon α-2b, which is used clinically for treatment of melanoma. The mobilization of the pCHSPCs was decreased in the mice with the humanized bone marrow-melanoma xenografts. Taken together, these data indicate that pCHSPCs play a functional role in tumor growth. The novel in vivo model described here can be utilized to further validate pCHSPCs as a biomarker of tumor progression. The model can also be used to screen and optimize anticancer/anti-angiogenic therapies in a humanized system.
Keywords: proangiogenic, circulating hematopoietic stem and progenitor cells, biomarker, tumor growth
Introduction
Neovascularization and angiogenesis are increasingly recognized as having an important role in a wide variety of diseases. Angiogenesis has been shown to be important for tumor growth, and novel anti-angiogenic therapies are currently being tested as a means of slowing or preventing growth of tumors (as reviewed in[1]). Researchers have gained important insights on the role of purported bone marrow-derived cells of both endothelial and hematopoietic lineages in neovascularization[2-6]. Multi-parametric flow cytometry is being utilized to characterize and detect these circulating hematopoietic stem and progenitor cell subsets (CHSPCs; CD14-glyA-CD34+AC133+/-CD45dimCD31+ cells) in human peripheral blood (PB), and their presence or absence have been correlated with a variety of disease states[7-9]. We propose that CHSPCs and the mobilization of this population could be a possible target that if effectively inhibited, could lead to a restriction of vessel growth necessary for tumor progression.
The clinical utility of anti-angiogenic therapy has recently been established (as reviewed in[10-13]). Increased survival rates in phase III clinical trials were observed with the addition of vascular endothelial growth factor (VEGF) specific antibody (bevacizumab) compared to front-line therapy for colon and rectal cancer patients[14]. We previously demonstrated that the blood concentrations of CHSPCs are elevated in pediatric solid tumor cancer patients prior to treatment, which is consistent with the hypothesis that CHSPCs are an important biomarker of active tumor progression and measurement of anti-cancer treatment efficacy[9]. In this study, we demonstrated that a distinct population of CHSPCs is increased in pediatric solid tumor patients with proangiogenic potential as demonstrated previously (CD14-glyA-CD34+AC133+CD45dimCD31+ cells; pCHSPCs) compared to the non-angiogenic subset that is AC133 negative (CD14-glyA-CD34+AC133-CD45dimCD31+ cells; (nCHSPCs))[7,9]. We also believe that it is the ratio between the pCHSPCs and the nCHSPCs (pCHSPC:nCHSPC) that is the feature of significant clinical importance, and can be utilized to normalize values across a wide spectrum of clinical diseases affecting organ-specific vasculature. In the pediatric study, solid tumor patients had a pCHSPC:nCHSPC ratio of greater than 2 when diagnosed. However, following successful chemotherapeutic treatment, the pCHSPC:nCHSPC ratio decreased to be within the normal range (1.2-1.8)[7,9]. We have also previously shown that in patients with peripheral vascular disease and adolescents with type 1 diabetes, the pCHSPC:nCHSPC ratio is less than 1, which is consistent with a decrease in angiogenesis at organ-specific sites[7,15].
Developing in vivo models of human disease will be necessary for testing new anti-cancer and anti-angiogenic therapies. In our current study, we characterized and confirmed the proangiogenic properties of the pCHSPCs using human cytokine bio-plex and tube forming assays. Additionally, we utilized an integrated orthotopic humanized xenograft modeling approach in which immunodeficient mice were transplanted with human CD34+ cells, and following a reconstitution period, were also engrafted with a subcutaneous melanoma xenograft. In this model, we found that the presence of pCHSPC correlated with tumor growth and progression of disease. In addition, to determine if mobilization of pCHSPCs can be modulated in this xenograft model, interferon α-2b (an inhibitor of bone marrow mobilization) was administered to mice with humanized bone marrow-melanoma xenografts to monitor pCHSPC frequency and tumor growth. A decrease in pCHSPC frequency in the PB following interferon α-2b treatment was observed. However, it was not complete, nor sufficient to inhibit tumor growth. This model can now be used to optimize therapeutic regimens and determine if a particular pCHSPC frequency and pCHSPC:nCHSPC ratio must be obtained to block tumor progression.
Materials and Methods
Isolation of Umbilical Cord Blood CD34+ Cells
The Institutional Review Board at the Indiana University School of Medicine approved all protocols. Samples of human umbilical cord blood (UCB) were collected from normal, full term infants delivered by cesarean section and the CD34+ cells were selected using the human CD34 indirect MicroBead kit and Magnetic Cell Sorting (MACS) system (Miltenyi Biotec) exactly as directed by the manufacturer. The CD34+ fraction was subsequently isolated, with the viability of the CD34+ cells always >95%. The purity and functionality of the MACS isolated CD34+ cells was confirmed by flow cytometry analysis (>95%) and a colony forming unit-granulocyte, erythrocyte, monocyte, megakaryocyte assay (CFU-GEMM).
Colony Forming Unit Assay
CFU assays (MethoCult GF H4434, Stem Cell Technologies, Inc) were conducted using the MACS isolated CD34+ cells. The cells were seeded in 35mm dishes in triplicate at a concentration of 0.5×103 CD34+ cells per plate in order to obtain 60-70 colonies per dish per the manufacturers recommendation.
Antibodies and Staining Reagents
The following primary conjugated monoclonal antibodies were used: anti-human CD31 fluoroscein isothyocyanate (FITC, BD Pharmingen), anti-human CD34 phycoerythrin (PE, BD Pharmingen), anti-human AC133 allophycocyanin (APC, Miltenyi Biotec), anti-human CD14 PECy5.5 (Abcam), anti-human CD45 APC-AlexaFluor (AF) 750 (Invitrogen), anti-human CD235a (glyA, R&D Systems) conjugated to Pacific Blue (PacB, Invitrogen) and the amine reactive viability dye, LiveDead (Invitrogen).
In order to resolve the rare and/or dim populations of interest, specific antigen and fluorochrome conjugate coupling was optimized for the six-antibody plus viability marker staining panel described below and as previously described[16-18].
Multi-Parametric Flow Cytometry Immunostaining and Sorting
MACS isolated CD34+ cells were incubated with Fc blocking reagent (Miltenyi Biotec) and stained as previously described[7,19]. “Fluorescent minus one” (FMO) gating controls were also used to ensure proper gating[19]. Briefly, cells were incubated with antibodies for 30 minutes at 4°C, washed twice in PBS with 2% fetal bovine serum (FBS), and were run fresh on a BD Aria flow cytometer (BD, Franklin Lakes, NJ, USA) equipped with a 405nm violet laser, 488nm blue laser and 633nm red laser. Data were acquired compensated, with anti-mouse Ig BD CompBeads (BD Biosciences) stained with each of the individual test antibodies to serve as single-color compensation controls. Acquisition files were exported as FCS 3.0 files and analyzed using FlowJo software, version 8.7.3 (Tree Star, Inc). For peripheral blood analysis of the CHSPCs in the humanized mice, PB cells were collected using a red blood cell lysis and stained with the human antibodies listed above. Approximately 150,000 events per sample were collected on a BD LSRII flow cytometer. Data were acquired uncompensated and exported as FCS 3.0 files, and analyzed utilizing FlowJo software version 8.7.3.
Prior to use, each lot of antibody was individually titered, as previously described, to determine the optimal staining concentration[20].
Matrigel Tube-Forming Assay
Endothelial colony forming cells (ECFCs) were purchased from the Angiogenesis, Endothelial & Pro-Angiogenic Cell Core (AEPCC, Indiana University). Matrigel assays were conducted as previously described with slight modifications[21]. Briefly, early passage ECFCs (i.e. 2-3) were starved over night and then seeded onto a 96-well tissue culture plate coated with 45μL of Matrigel (BD Biosciences) at a cell density of 7,500 cells per well. In some experiments, 2,500 sorted pCHSPCs were added to the wells with ECFCs. In addition, 7,500 sorted pCHSPCs were plated alone to assay for tube formation. Cells were observed every 2hrs by visual microscopy with an inverted microscope at 40× magnifications for tube formation. Tube formation in each well was quantified at 8 hours post plating[21]. Assays were performed in a minimum of triplicate.
Bio-Plex Human Cytokine Assay
pCHSPCs were plated in 24-well tissue culture coated plates in 2mLs of Stemline 2 media (Sigma-Aldrich) and incubated at 37°C for 48hrs to create conditioned media. Conditioned media was assayed using the Bio-Rad Bioplex Pro-Human Cytokine 27-plex Assay (IL-1β, IL-1ra, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12, IL-13, IL-15, IL-17, basic FGF, eotaxin, G-CSF, GM-CSF, IFN-γ, IP-10, MCP-1, MIP-1α, MIP-1β, PDGF-BB, RANTES, TNF-α, and VEGF) according to the manufacturers protocol by the Bio-Plex Core of the Indiana University Simon Cancer Center.
Mice
NOD.CB17-Prkdcscid/j (NOD/SCID), NOD.Cg-PrkdcscidIL2rgtm1Wjl/Sz (NOD/SCID/γchainnull), or SCID/beige (SCID/bg) mice, 6-8 weeks old, were housed according to protocols approved by the Indiana University Laboratory Animal Research Center and adhered strictly to National Institutes of Health guidelines. All protocols were approved by the Indiana University Animal Care and Use Committee.
Transplantation of NOD/SCID, NOD/SCID/γchainnull or SCID/bg Mice
All animals were given a sub-lethal dose of 300cGy total body irradiation 4hrs before transplantation. The MACS isolated CD34+ cells (105 per mouse) were re-suspended in Dulbecco's modified eagle medium (DMEM, Gibco) and transplanted by tail vein injection. To assess human engraftment, PB samples were obtained at 4 weeks post transplantation and assayed by flow cytometry with the staining listed above.
Melanoma Xenograft Models
NOD/SCID, NOD/SCID/γchainnull, or SCID/bg mice previously transplanted with CD34+ cells were subcutaneously injected with 2×106 C32 human melanoma cells (ATCC) with tumor growth and pCHSPCs monitored over time. Tumor growth was monitored by caliper, and the volume determined by the following formula: mm3 = (width)2 × length × 0.5. The fold-increase in tumor volume was determined by comparing tumor volume over time to that of the base line tumor volume. In some experiments, PB was collected at 1, 2 and 4 weeks after implantation so as to monitor the pCHSPC release from the bone marrow into the PB using flow cytometry. Interferon α-2b (Intron A; 50,000U × 3 times a week) was given via subcutaneous injection and at rotating sites to avoid irritation of the injection sites, for 6 total doses.
At the end of the experiment, mice were euthanized; tumors, PB and bone marrow were harvested, and the weight of each tumor determined.
Statistical Analysis
Data are presented as the means±SEM. Differences in tumor volume among groups were analyzed by repeated measures two-way analysis of variance using SigmaPlot v11.2 (Systat Software, Inc.). The Holm-Sidak method was used to make post-hoc pairwise comparisons among days and treatments. Flow cytometry data were analyzed using a t-test. Tube formation data were analyzed by one-way ANOVA with Tukey's post-hoc test. A p value of ≤0.05 was considered statistically significant.
Results
pCHSPCs Secrete Proangiogenic Factors and Increase Tube Formation
To determine if pCHSPCs possess proangiogenic properties, a human bio-plex analysis and a co-culture tube-forming assay were conducted. The pCHSPCs secreted the proangiogenic cytokines VEGF and IL-1Ra, as well as IL-2, IL-9, IL-15, IL-17, Eotaxin, G-CSF, GM-CSF, INF- γ, MIP-1β, and RANTES. They did not have detectable levels of IL-1β, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12, IL-13, basic FGF, IP-10, MCP-1, MIP-1α, PDGF-BB, or TNF-α (Table 1). In addition, increased tube formation was observed when ECFCs and pCHSPCs were co-cultured compared to ECFCs alone or ECFCs co-cultured with nCHSPCs (p<0.008; Figure 1). When pCHSPCs or nCHPSCs were plated alone, no tube formation was observed. Lack of tube formation was to be expected when the pCHPSCs and nCHSPCs were plated alone since both the pCHSPCs and nCHSPCs are in fact hematopoietic, and not endothelial in origin. These data indicate that pCHSPCs could play a prominent role in promotion of angiogenesis. The pCHSPCs were found to secrete proangiogenic cytokines and functionally promote the increased vessel-like tube formation of ECFCs.
Table 1. Cytokines Assessed in pCHSPC Conditioned Media.
Conditioned media was assayed for a 27-cytokine bio-plex assay. Absolute values for the cytokines are listed as pg/mL.
| Cytokines | pCHSPC (pg/mL) |
|---|---|
| IL-1b | |
| IL-1ra | 4.86 |
| IL-2 | 4.99 |
| IL-4 | |
| IL-5 | |
| IL-6 | |
| IL-7 | |
| IL-8 | |
| IL-9 | 7.53 |
| IL-10 | |
| IL-12 (p70) | |
| IL-13 | |
| IL-15 | 3.21 |
| IL-17 | 3.16 |
| Eotaxin | 19.38 |
| FGF Basic | |
| G-CSF | 5.01 |
| GM-CSF | 14.29 |
| INF-g | 2.48 |
| IP-10 | |
| MCP-1 (MCAF) | |
| MIP-1a | |
| PDGF-bb | |
| MIP1-b | 2.07 |
| RANTES | 2.16 |
| TNF-a | |
| VEGF | 4.37 |
Figure 1. Matrigel Tube Formation with pCHSPCs.
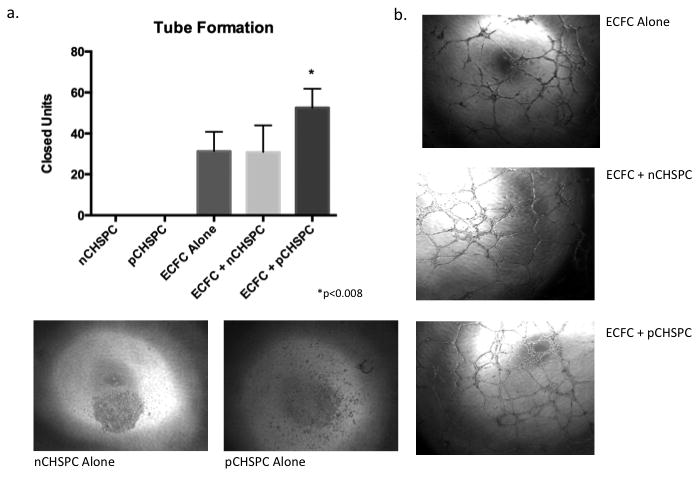
Graph of all populations tested for tube formation (a). Tube formation was increased with the addition of the pCHSPCs with no difference between ECFCs alone and ECFCs with nCHSPCs (p<0.008). Representative photomicrographs of tube formation assays comparing nCHSPCs alone, pCHSPCs alone, ECFCs alone, ECFCs with nCHSPCs, and ECFCs with pCHSPCs (b).
Monitoring Mobilization of pCHSPCs in a Humanized Bone Marrow-Melanoma Orthotopic Model
We previously demonstrated that pCHSPCs can repopulate the bone marrow following sub-lethal irradiation and transplantation into NOD/SCID mice and that C32 melanoma tumors grew more quickly in mice when pCHSPCs were infused intravenously[7]. In this current study, our goal was to develop an optimized mouse model in which to study human CHSPC function and mobilization. We first determined if NOD/SCID mice with humanized bone marrow could serve as a model to study mobilization of pCHSPCs into the PB, and whether mobilization of pCHSPCs correlated with increased tumor growth.
Three strains of immunodeficient mice were compared since these strains have been shown to promote different levels of human engraftment, and in the case of the NOD/SCIDγchainnull, enhanced differentiation of myeloid progenitors compared to SCID/bg.
Since CHSPCs have the potential to differentiate into myeloid cells, we thought it was essential to evaluate and compare CHSPC-mediated tumor progression in the SCID/bg mice versus our current model in NOD/SCID mice. Further, studies by Kirkiles-Smith et al., demonstrated that human-derived macrophages, skin, and arterial grafts actually engrafted more efficiently and consistently in SCID/bg mice compared to NOD/SCID/γchainnull mice[22]. However, NOD/SCID/γchainnull mice were better recipients for engraftment of CD34+ cells and emergence of immature and mature hematopoietic cells both in the PB and in the bone marrow compared to SCID/bg[22]. This is evidently due to differences in the innate NK responses in these mice. NOD/SCID, NOD/SCID/γchainnull, or SCID/bg mice were sub-lethally irradiated and CD34+ cells injected via the tail vein to compare the engraftment potential of all 3 strains. Engraftment was confirmed at 4 weeks post-transplantation by quantifying the number of human CD45+ cells in the PB (Figure 2). NOD/SCID and NOD/SCID/γchainnull mice had similar engraftment whether whole CD34+ cells or the pCHSPC fraction were used for transplantation (data not shown). Interestingly, SCID/bg mice failed to engraft when either whole CD34+ cells or the pCHSPC fraction was injected and were therefore not selected for further study (data not shown).
Figure 2. Schematic of C32 Melanoma Xenograft Model.
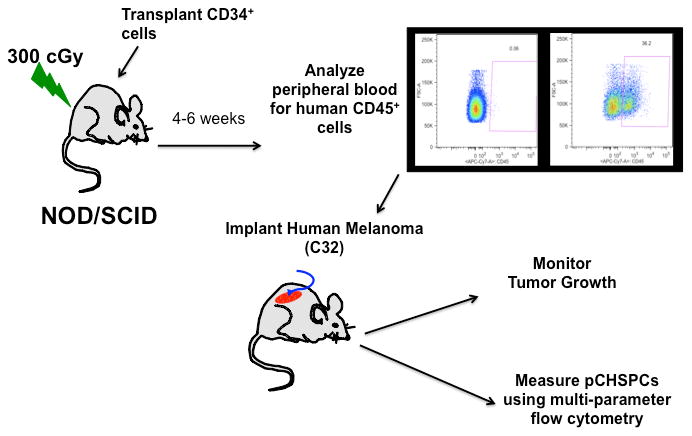
Mice were sub-lethally irradiated with 300 rads and human CD34+ cells were then transplanted into NOD/SCID mice. Following 4 weeks of engraftment, peripheral blood was analyzed for human CD45 and then C32 melanoma cells were implanted on the flanks of the humanized mice with tumor growth and CHSPCs both being monitored.
After confirming that the pCHSPCs could repopulate the bone marrow niche of both NOD/SCID and NOD/SCID/γchainnull mice, we next assessed whether pCHSPCs will mobilize into the PB in mice with humanized bone marrow and C32 melanoma xenografts. We also determined if higher levels of pCHSPCs in the PB correlated with increased tumor size. Mice were first transplanted with CD34+ cells (Figure 3a) or saline control. Following 4 weeks of recovery, engraftment was confirmed in the PB, and C32 melanoma cells were implanted on the flank. To monitor CHSPC frequency, PB was drawn each week and analyzed by flow cytometry. PB was collected from both NOD/SCID and NOD/SCID/γchainnull humanized mice and both strains had detectable levels of the parent population of the CHSPCs, but the frequency of the pCHSPCs remained low for the 5 weeks following CD34+ transplantation as well as one week following implantation of C32 melanoma (Figure 3b). While the tumors grew at a slow rate over weeks 1-2, the number of pCHSPCs remained low during this period of time. At four weeks post implantation of C32 xenografts, when the tumors started to grow rapidly, there was a statistically significant increase in the number of pCHSPCs (Figure 3c; p=<0.005; Table 2a; p=<0.005). In mice that were transplanted with CD34+ cells without C32 melanoma implantation, there was no increase in pCHSPCs compared to baseline (Figure 3d). The increase in pCHSPCs remained until harvest of the tumors at week 6 post C32 melanoma implant (Figure 3e). Interestingly, there was a specific mobilization of the pCHSPC fraction in the mice implanted with C32 melanoma cells, while the nCHSPC fraction remained at a stable level (Figure 3).
Figure 3. Mobilization of pCHSPCs (AC133+) into the Peripheral Blood in Mice Harboring C32 Melanoma Xenografts.
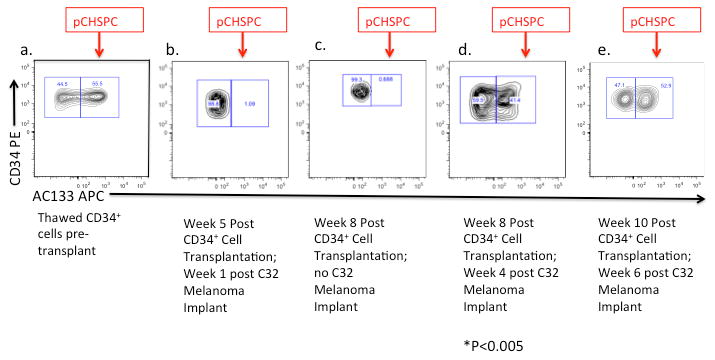
The CD34+ cells used to repopulate the marrow after sub-lethal irradiation (a). Peripheral blood, from NOD/SCID mice following transplantation with CD34+ cells was tested in mice with C32 melanoma xenografts, with representative plots at (b) 5 weeks post CD34+ transplantation/1 week post implantation of C32 cells, (c) 8 weeks post CD34+ transplantation with no C32 implantation, (d) 8 weeks post CD34+ transplantation/4 weeks post implantation of C32 cells, and (e) 10 weeks post transplantation/6 weeks post implantation of C32 cells. pCHSPCs (red arrow) are mobilized from the bone marrow into the peripheral blood following 4 weeks after C32 melanoma flank implantation (pCHSPCs at week one versus week 4 post-implantation of C32 tumor cells, p<0.005).
Table 2. Time Course of Humanized Mouse Peripheral Blood pCHSPC:nCHSPC ratios.
Peripheral blood was drawn at week 1, week 2, and week 4 following implant of C32 melanoma to monitor the pCHSPC:nCHSPC ratio in response to tumor burden. The peripheral blood pCHSPC:nCHSPC ratio increased 4 weeks post transplant of C32 melanoma when tumor volume significantly increases.
| Week 1 | Week 2 | Week 4 | |
|---|---|---|---|
| pCHSPC:nCHSPC Ratio | 0.01 | 0.02 | 0.70 |
| 0.03 | 0.01 | 0.63 | |
| 0.05 | 0.03 | 0.76 | |
| 0.02 | 0.03 | 0.93 | |
| 0.01 | 0.03 | 1.72 | |
| 0.10 | 0.02 | 0.77 | |
| 0.30 | 0.22 | 3.15 | |
| 0.44 | 0.25 | 3.67 | |
| 0.25 | 0.60 | ||
| Average: | 0.12 | 0.10 | 1.44 |
P<0.005
Tumor volumes were measured weekly for 45 days (Figure 4). Tumors in both C32 only mice and CD34+ transplanted mice grew at similar rates until about 25 days post implant. After 25 days, the CD34+ transplanted mice began to have a more rapid increase in tumor volume, which corresponds with the increase in detectable pCHSPCs in the PB (Figure 3b; Figure 4). At 45 days post tumor implant, the C32 only cohort had statistically significantly smaller tumor volumes compared to those mice that received CD34+ cells following irradiation and subsequent C32 implantation (Figure 3; p=<0.01). The in vivo model described here simulates what we have observed in patients in that increases in the pCHSPC:nCHSPC ratio correlate with increased tumor growth[9].
Figure 4. Increased Tumor Volume Following CD34+ Transplantion.
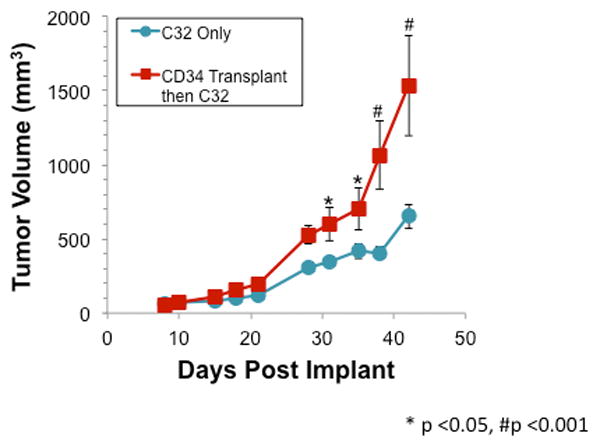
NOD/SCID mice were initially sub-lethally irradiated and then injected subcutaneously in the flank with C32 cells at 4 weeks post CD34+ transplantation. Tumors in mice transplanted with CD34+ cells were significantly larger than tumors in sham transplanted mice as indicated by a significant main effect for group in the ANOVA (F[1,60] = 11.2, p < 0.01). Post-hoc analysis indicated that tumors in CD34+ transplanted mice were significantly larger beginning on day 31 post-implant (*p<0.05, #p<0.001). Moreover, tumors in CD34+ transplanted mice grew at a significantly faster rate as indicated by a significant group-by-days interaction (F[9,69] = 6.2, p<0.001).
Quantification of pCHSPCs in a Humanized Bone Marrow Xenograft can be used as a Biomarker for Evaluation of Anticancer Therapies in Orthotopic Models of Cancer
Since it was feasible to follow the levels of the pCHSPCs in the PB in the humanized mouse model, we next determined if treatment with anticancer therapy could decrease the number of pCHSPCs in the PB, and would this also correlate with decreases in tumor size compared to control animals. NOD/SCID/γchainnull mice were irradiated and CD34+ cells were injected via tail vein, and the mice were allowed to recover for 4 weeks as previously described. Following baseline bleeds for identification of human CD45+ cells and pCHPSC:nCHSPC measurements, C32 cells were implanted in the flank of the animals.
We elected to use the chemotherapeutic agent Interferon α-2b since it is currently used for treatment of melanoma. Two weeks post-implant, treatment with the Interferon α-2b (Intron A; 50,000U × 3 times a week) was initiated and tumor size measured for 2 weeks post treatment with Interferon α-2b. Following Interferon treatment, the pCHSPC:nCHSPC ratio was significantly decreased by 15% in the transplanted mice with C32 tumors compared to transplanted mice that did not receive Interferon (p=<0.05; Figure 5b). However, the decrease in the pCHSPC:nCHSPC ratio was not sufficient to cause a change in tumor growth in the transplanted mice harboring C32 tumors. It is possible that a larger decrease in the pCHSPC:nCHSPC ratio will be required to sufficiently impact tumor growth and is the focus of ongoing studies. Additionally, the Interferon α-2b treatment did not affect the percentage of human CD45+ cells in the bone marrow in any of the three groups (see Table 3a). There was an statistically significant increased ratio of pCHSPC:nCHSPC in the mice transplanted with human CD34+ cells and then implanted with C32 melanoma regardless of Interferon treatment compared to humanized mice who did not receive a C32 implant (Table 3b).
Figure 5. Interferon α-2b-Mediated Decreases in the ratio of pCHSPC:nCHSPC in Transplanted Mice with C32 Melanoma Xenografts.
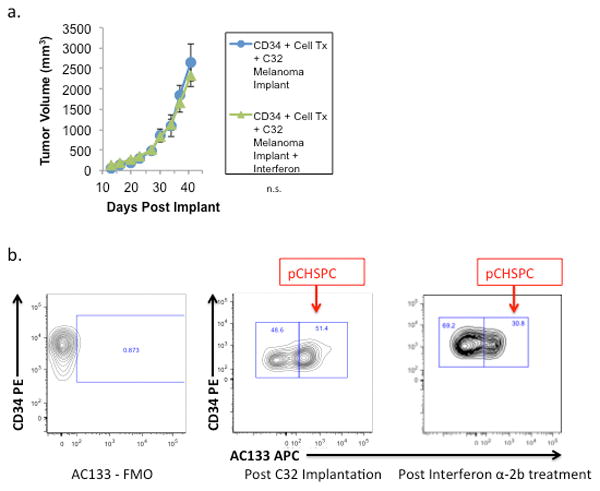
NOD/SCID/γchainnull mice were first transplanted with human CD34+ cells and then the C32 melanoma was implanted on the flank 4 weeks post-transplantation. Following two weeks of C32 melanoma unrestricted growth, some mice were treated with Interferon α-2b (Intron-A; 50,000U × 3 times per week) and tumor volume was measured via caliper over time (a). There were decreased pCHSPCs in the peripheral blood following Interferon α-2b treatment (b).
Table 3. Cell Populations in Humanized Mice From Bone Marrow.
No difference was seen between the bone marrow averages of human CD45+ cells in those mice transplanted with human CD34+ cells whether they had C32 melanoma implants (with or without treatment with Interferon) or human CD34+ cell transplantation (a). There was no difference in the bone marrow pCHSPC:nCHSPC ratios for mice transplanted with human CD34+ cells with melanoma implants (with or without treatment with Interferon), however both populations had a statistically significant bone marrow pCHSPC:nCHSPC compared to mice transplanted with CD34+ cells alone (b; p<0.01). Subpopulations (pCHSPC/nCHSPC) are the percent of the total live cells.
| a. | |||||
|---|---|---|---|---|---|
| CD45+ Percentage | CD34 BM | CD34+C32 BM | CD34+C32+lnterferon BM | No Tx BM | |
| 93.5 | 82.8 | 77.4 | 10.1 | ||
| 89.4 | 53.8 | 83.5 | 10.5 | ||
| 71.1 | 78.6 | 82.3 | 8.67 | ||
| 90.2 | 69.5 | 67.8 | 12.9 | ||
| 75.3 | 89.6 | 84.5 | 9.09 | ||
| 42.5 | 47.5 | 88.5 | 0.07 | ||
| 64.9 | 86.2 | 0.06 | |||
| 50.5 | 54.4 | ||||
| 58 | |||||
| 89.6 | |||||
| Average: | 77 | 67.15 | 77.22 | 7.34 | |
| b. | ||||
|---|---|---|---|---|
| * | * | |||
| pCHSPC:nCHSPC Ratio | CD34 BM | CD34+C32 BM | CD34+C32+lnterferon BM | |
| 9.17/13.7; 0.67 | 4.38/5.19; 0.84 | 3.21/3.79; 0.85 | ||
| 4.97/6.67; 0.75 | 2.08/2.82; 0.74 | 2.78/3.46; 0.80 | ||
| 3.49/4.98; 0.70 | 4.08/3.71; 1.1 | 3.87/4.75; 0.81 | ||
| 2.71/3.53; 0.73 | 2.9/3.45; 0.84 | 1.54/1.71; 0.90 | ||
| 3/5.22; 0.57 | 5.87/7.39; 0.79 | 3.23/3.62; 0.89 | ||
| 1.2/2.07; 0.58 | 5.48/6.86; 0.80 | 3.75/3.71; 1.0 | ||
| 4.08/5.01; 0.80 | 3.66/4.74; 0.77 | |||
| 5.22/7.720.68 | 1.26/1.46; 0.86 | |||
| 2.59/3.56; 0.73 | ||||
| 3.04/3.34; 0.91 | ||||
| Average Ratio: | 0.67 | 0.82 | 0.85 | |
P<0.01
Discussion
We describe a humanized orthotopic cancer model that can be used to track the mobilization of a potentially very powerful human cellular biomarker, the pCHSPC. The levels of these cells in the PB of pediatric patients were shown previously to correlate with progression of pediatric solid tumor malignancies[9]. Tracking of the pCHSPCs in the PB of both tumor bearing and non-tumor bearing humanized mice further validates the potential of the pCHSPC as a useful biomarker of tumor progression and in the context of anticancer therapy studies.
It is well known that hematopoietic cells participate in angiogenesis[23]. We have previously demonstrated that bone marrow-derived pCHSPCs are increased in pediatric solid tumor patients at diagnosis compared to healthy age-matched controls[9]. Monitoring these same cells in a humanized mouse model, we were able to simulate what we have observed in patients - increased pCHSPC levels correlate with tumor progression. Recently CD133, the key marker of the pCHSPC, has been shown to interact with VEGF which has pro-survival and angiogenic properties for endothelial cells and increases survival of cancer cells[24]. Further, cell growth of both endothelial and melanoma cells are also regulated through VEGF[24].
The pCHSPCs were detected at the highest levels in the peripheral blood of mice that had a humanized bone marrow and a melanoma xenograft. In addition to showing that increases in pCHSPCs correlate with increased tumor volume, the appearance of the pCHSPCs in the PB can be inhibited by the chemotherapeutic agent Interferon α-2b. We do know that the pCHSPCs do not originate from the tumor since pCHSPCs are not detected in mice that have the C32 tumors but were not transplanted with human CD34+ cells. Interferon α-2b is an adjunct therapy for patients with resected melanoma, and is thought to both alert the immune system to the presence of melanoma cells as well as suppress release of bone-marrow cells into the blood stream[25]. Since the interferon α-2b is a known modulator of bone marrow mobilization, it is most likely that the pCHPSCs do originate from the humanized bone marrow compartment. While it is most likely that the pCHSPCs are derived from the bone marrow, we cannot rule out the possibility that the cells were simply generated in the peripheral blood.
The humanized bone marrow-melanoma model was used to determine to what extent interferon α-2b could block mobilization of pCHSPCs into the PB and modulate tumor growth. A decrease in the pCHSPC:nCHSPC ratio was achieved in the PB, but was not sufficient to block tumor growth. Further studies need to be conducted to optimize the Interferon α-2b-mediated decreases in the pCHSPC:nCHSPC and to assess if a particular ratio is required to cause a block in tumor growth.
The multi-parametric flow cytometry approach employed here permits isolation and analysis of the CHSPC subsets based upon AC133 expression, and only the AC133+ pCHSPC subset possesses proangiogenic activity in promoting angiogenesis and human tumor growth in an immunodeficient mouse explant model system[7]. AC133+ cells have been associated with poor prognosis in ovarian cancer patients, including shortened overall survival time and non-response to chemotherapy[26]. Furthermore, AC133+ cells have been inversely correlated with glioblastoma, diverse gliomas, breast cancer, colon cancer, and pancreatic patient survival, thus making them an important biomarker in oncology[4,22,27-31]. In the future, identification of specific molecular pathways that promote tumor angiogenesis may be insightful and permit a better understanding of mechanisms that can block tumor angiogenesis and growth. A novel murine xenograft model is now available to investigate the recruitment of the pCHSPCs in response to many different tumor types in vivo. This will lead to a better understanding of whether the pCHSPCs can differentiate into myeloid cells with angiogenic potential, including the Tie2+ monocytes, that have been shown to actively promote tumor angiogenesis[32].
For the first time, we demonstrate the ability to detect and track human cellular subsets that may have the potential to promote angiogenesis in a humanized xenograft melanoma model. By using humanized models, a clearer grasp of how tumorigenesis occurs and the factors that are necessary for both promoting and disrupting tumor growth can be accurately studied. When human CD34+ cells were transplanted into a murine xenograft model, the pCHSPC subset was shown to mobilize in response to increased tumor growth and were subsequently decreased upon treatment with a chemotherapeutic agent (Interferon α-2b). Consistent with these findings are previously published studies by our group which demonstrated an increase in the pCHSPC:nCHSPC ratio at baseline in pediatric patients with solid tumors compared to healthy controls[9]. In addition, the pCHSPC:nCHSPC ratio was decreased following treatment with chemotherapeutic agents, thus indicating its potential use as a predictive biomarker of treatment efficacy[9].
Acknowledgments
We acknowledge the assistance and state-of-the-art facilities of the Flow Cytometry Resource Facility at the Indiana University Simon Cancer Center. We also acknowledge Barbara Bailey of the In Vivo Therapeutics Core of the Indiana University Simon Cancer Center, as well as the nursing staff and Dr. Arthur Baluyut at the St. Vincent Hospital (Indianapolis, IN) for providing some of the UCB samples used for this study. We acknowledge Hui Lin Chau and Artur Plett of the Bio-Plex Core of the Indiana University Simon Cancer Center. Finally, we acknowledge the Angiogenesis, Endothelial and Pro-Angiogenic Cell Core of the Indiana University Simon Cancer Center for providing some of the CD34+ cells and ECFCs used for this study.
Grant Support: We acknowledge the continuing support of the Riley Children's Foundation and the Indiana University Simon Cancer Center. This work was supported by the American Cancer Society IRG-84-002-25 (J.C.), Showalter Trust Fund ERA 31948 (J.C.), NIH/NIDDK P30DK090948 CEMH (J.C., J.A.M and K.E.P.), RO1 CA138798 (H.W., S.C., and K.E.P.) and the Jeff Gordon Research Foundation (H.W. and K.E.P.).
Footnotes
Conflict of Interest: All authors declare no conflict of interest.
References
- 1.Cao Y, Arbiser J, D'Amato RJ, D'Amore PA, Ingber DE, Kerbel R, Klagsbrun M, Lim S, Moses MA, Zetter B, Dvorak H, Langer R. Forty-year journey of angiogenesis translational research. Sci Transl Med. 2011;3(114):114rv113. doi: 10.1126/scitranslmed.3003149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asahara T, Kawamoto A. Endothelial progenitor cells for postnatal vasculogenesis. Am J Physiol Cell Physiol. 2004;287(3):C572–579. doi: 10.1152/ajpcell.00330.2003. 10.1152/ajpcell.00330.2003287/3/C572 [pii] [DOI] [PubMed] [Google Scholar]
- 3.Asahara T, Masuda H, Takahashi T, Kalka C, Pastore C, Silver M, Kearne M, Magner M, Isner JM. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res. 1999;85(3):221–228. doi: 10.1161/01.res.85.3.221. [DOI] [PubMed] [Google Scholar]
- 4.Rafii S, Meeus S, Dias S, Hattori K, Heissig B, Shmelkov S, Rafii D, Lyden D. Contribution of marrow-derived progenitors to vascular and cardiac regeneration. Seminars in cell & developmental biology. 2002;13(1):61–67. doi: 10.1006/scdb.2001.0285. [DOI] [PubMed] [Google Scholar]
- 5.Ingram DA, Mead LE, Tanaka H, Meade V, Fenoglio A, Mortell K, Pollok K, Ferkowicz MJ, Gilley D, Yoder MC. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood. 2004;104(9):2752–2760. doi: 10.1182/blood-2004-04-1396. 10.1182/blood-2004-04-1396-2004-04-1396 [pii] [DOI] [PubMed] [Google Scholar]
- 6.Critser PJ, Yoder MC. Endothelial colony-forming cell role in neoangiogenesis and tissue repair. Curr Opin Organ Transplant. 2010;15(1):68–72. doi: 10.1097/MOT.0b013e32833454b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Estes ML, Mund JA, Mead LE, Prater DN, Cai S, Wang H, Pollok KE, Murphy MP, An CS, Srour EF, Ingram DA, Jr, Case J. Application of polychromatic flow cytometry to identify novel subsets of circulating cells with angiogenic potential. Cytometry Part A : the journal of the International Society for Analytical Cytology. 2010;77(9):831–839. doi: 10.1002/cyto.a.20921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mund JA, Estes ML, Yoder MC, Ingram DA, Jr, Case J. Flow cytometric identification and functional characterization of immature and mature circulating endothelial cells. Arterioscler Thromb Vasc Biol. 2012;32(4):1045–1053. doi: 10.1161/ATVBAHA.111.244210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pradhan KR, Mund JA, Johnson C, Vik TA, Ingram DA, Case J. Polychromatic flow cytometry identifies novel subsets of circulating cells with angiogenic potential in pediatric solid tumors. Cytometry B Clin Cytom. 2011 doi: 10.1002/cyto.b.20602. [DOI] [PubMed] [Google Scholar]
- 10.Sarmiento R, Longo R, Gasparini G. Antiangiogenic therapy of colorectal cancer: state of the art, challenges and new approaches. The International journal of biological markers. 2012;27(4):e286–294. doi: 10.5301/JBM.2012.10441. [DOI] [PubMed] [Google Scholar]
- 11.Wilson PM, LaBonte MJ, Lenz HJ. Assessing the in vivo efficacy of biologic antiangiogenic therapies. Cancer chemotherapy and pharmacology. 2013;71(1):1–12. doi: 10.1007/s00280-012-1978-8. [DOI] [PubMed] [Google Scholar]
- 12.Zaki KA, Basu B, Corrie P. The role of angiogenesis inhibitors in the management of melanoma. Current topics in medicinal chemistry. 2012;12(1):32–49. doi: 10.2174/156802612798919240. [DOI] [PubMed] [Google Scholar]
- 13.Schneider BP, Shen F, Miller KD. Pharmacogenetic biomarkers for the prediction of response to antiangiogenic treatment. The lancet oncology. 2012;13(10):e427–436. doi: 10.1016/S1470-2045(12)70275-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Willett CG, Duda DG, di Tomaso E, Boucher Y, Ancukiewicz M, Sahani DV, Lahdenranta J, Chung DC, Fischman AJ, Lauwers GY, Shellito P, Czito BG, Wong TZ, Paulson E, Poleski M, Vujaskovic Z, Bentley R, Chen HX, Clark JW, Jain RK. Efficacy, safety, and biomarkers of neoadjuvant bevacizumab, radiation therapy, and fluorouracil in rectal cancer: a multidisciplinary phase II study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27(18):3020–3026. doi: 10.1200/JCO.2008.21.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DiMeglio LA, Tosh A, Saha C, Estes M, Mund J, Mead LE, Lien I, Ingram DA, Haneline LS. Endothelial abnormalities in adolescents with type 1 diabetes: a biomarker for vascular sequelae? The Journal of pediatrics. 2010;157(4):540–546. doi: 10.1016/j.jpeds.2010.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahnke YD, Roederer M. Optimizing a multicolor immunophenotyping assay. Clin Lab Med. 2007;27(3):469–485. doi: 10.1016/j.cll.2007.05.002. v. S0272-2712(07)00047-9 [pii]10.1016/j.cll.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baumgarth N, Roederer M. A practical approach to multicolor flow cytometry for immunophenotyping. Journal of immunological methods. 2000;243(1-2):77–97. doi: 10.1016/s0022-1759(00)00229-5. [DOI] [PubMed] [Google Scholar]
- 18.Tung JW, Heydari K, Tirouvanziam R, Sahaf B, Parks DR, Herzenberg LA, Herzenberg LA. Modern flow cytometry: a practical approach. Clinics in laboratory medicine. 2007;27(3):453–468. v. doi: 10.1016/j.cll.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Estes ML, Mund JA, Ingram DA, Case J. Identification of endothelial cells and progenitor cell subsets in human peripheral blood. In: Paul Robinson J, et al., editors. Current protocols in cytometry / editorial board. Unit 9. Chapter 9. 2010. pp. 33pp. 31–11. [DOI] [PubMed] [Google Scholar]
- 20.Herzenberg LA, Tung J, Moore WA, Parks DR. Interpreting flow cytometry data: a guide for the perplexed. Nat Immunol. 2006;7(7):681–685. doi: 10.1038/ni0706-681. ni0706-681 [pii] 10.1038/ni0706-681. [DOI] [PubMed] [Google Scholar]
- 21.Ponce ML. In vitro matrigel angiogenesis assays. Methods in molecular medicine. 2001;46:205–209. doi: 10.1385/1-59259-143-4:205. [DOI] [PubMed] [Google Scholar]
- 22.Kirkiles-Smith NC, Harding MJ, Shepherd BR, Fader SA, Yi T, Wang Y, McNiff JM, Snyder EL, Lorber MI, Tellides G, Pober JS. Development of a humanized mouse model to study the role of macrophages in allograft injury. Transplantation. 2009;87(2):189–197. doi: 10.1097/TP.0b013e318192e05d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Popa ER, Harmsen MC, Tio RA, van der Strate BW, Brouwer LA, Schipper M, Koerts J, De Jongste MJ, Hazenberg A, Hendriks M, van Luyn MJ. Circulating CD34+ progenitor cells modulate host angiogenesis and inflammation in vivo. Journal of molecular and cellular cardiology. 2006;41(1):86–96. doi: 10.1016/j.yjmcc.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 24.Adini A, Adini I, Ghosh K, Benny O, Pravda E, Hu R, Luyindula D, D'Amato RJ. The stem cell marker prominin-1/CD133 interacts with vascular endothelial growth factor and potentiates its action. Angiogenesis. 2013;16(2):405–416. doi: 10.1007/s10456-012-9323-8. [DOI] [PubMed] [Google Scholar]
- 25.Bhatia S, Tykodi SS, Thompson JA. Treatment of metastatic melanoma: an overview. Oncology (Williston Park) 2009;23(6):488–496. [PMC free article] [PubMed] [Google Scholar]
- 26.Sarkaria JN, Carlson BL, Schroeder MA, Grogan P, Brown PD, Giannini C, Ballman KV, Kitange GJ, Guha A, Pandita A, James CD. Use of an orthotopic xenograft model for assessing the effect of epidermal growth factor receptor amplification on glioblastoma radiation response. Clin Cancer Res. 2006;12(7 Pt 1):2264–2271. doi: 10.1158/1078-0432.CCR-05-2510. [DOI] [PubMed] [Google Scholar]
- 27.Giannini C, Sarkaria JN, Saito A, Uhm JH, Galanis E, Carlson BL, Schroeder MA, James CD. Patient tumor EGFR and PDGFRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme. Neuro-oncology. 2005;7(2):164–176. doi: 10.1215/S1152851704000821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan DD, Van Dyke WS, Bahls M, Connell SD, Critser P, Kelleher JE, Kramer MA, Pearce SM, Sharma S, Neu CP. Mechanostasis in apoptosis and medicine. Progress in biophysics and molecular biology. 2011;106(3):517–524. doi: 10.1016/j.pbiomolbio.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 29.Critser PJ, Voytik-Harbin SL, Yoder MC. Isolating and defining cells to engineer human blood vessels. Cell proliferation. 2011;44(Suppl 1):15–21. doi: 10.1111/j.1365-2184.2010.00719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shelley WC, Leapley AC, Huang L, Critser PJ, Zeng P, Prater D, Ingram DA, Tarantal AF, Yoder MC. Changes in the frequency and in vivo vessel-forming ability of rhesus monkey circulating endothelial colony-forming cells across the lifespan (birth to aged) Pediatric research. 2012;71(2):156–161. doi: 10.1038/pr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanz L, Cuesta AM, Salas C, Corbacho C, Bellas C, Alvarez-Vallina L. Differential transplantability of human endothelial cells in colorectal cancer and renal cell carcinoma primary xenografts. Laboratory investigation; a journal of technical methods and pathology. 2009;89(1):91–97. doi: 10.1038/labinvest.2008.108. [DOI] [PubMed] [Google Scholar]
- 32.De Palma M, Mazzieri R, Politi LS, Pucci F, Zonari E, Sitia G, Mazzoleni S, Moi D, Venneri MA, Indraccolo S, Falini A, Guidotti LG, Galli R, Naldini L. Tumor-targeted interferon-alpha delivery by Tie2-expressing monocytes inhibits tumor growth and metastasis. Cancer Cell. 2008;14(4):299–311. doi: 10.1016/j.ccr.2008.09.004. S1535-6108(08)00296-1 [pii] 10.1016/j.ccr.2008.09.004. [DOI] [PubMed] [Google Scholar]