

Abstract
KRAS codon 12 mutations are present in about 90% of ductal adenocarcinomas and in undifferentiated carcinomas of the pancreas. The role of KRAS copy number changes and resulting KRAS mutant allele-specific imbalance (MASI) in ductal adenocarcinoma (n=94), and its progression into undifferentiated carcinoma of the pancreas (n=25) was studied by direct sequencing and KRAS fluorescence in situ hybridization (FISH). Semi-quantitative evaluation of sequencing electropherograms showed KRAS MASI (ie, mutant allele peak higher than or equal to the wild-type allele peak) in 22 (18.4%) cases. KRAS FISH (performed on 45 cases) revealed a trend for more frequent KRAS amplification among cases with KRAS MASI (7/20, 35% vs 3/25, 12%, P=0.08). KRAS amplification by FISH was seen only in undifferentiated carcinomas (10/24, 42% vs 0/21 pancreatic ductal adenocarcinoma, 0%, P=0.0007). In 6 of 11 cases with both undifferentiated and well-differentiated components, transition to undifferentiated carcinoma was associated with an increase in KRAS copy number, due to amplification and/or chromosome 12 hyperploidy. Pancreatic carcinomas with KRAS MASI (compared to those without MASI) were predominantly undifferentiated (16/22, 73% vs 9/97, 9%, P<0.001), more likely to present at clinical stage IV (5/22, 23% vs 7/97, 7%, P=0.009), and were associated with shorter overall survival (9 months, 95% confidence interval, 5–13, vs 22 months, 95% confidence interval, 17–27; P=0.015) and shorter disease-free survival (5 months, 95% confidence interval, 2–8 vs 13 months, 95% confidence interval, 10–16; P=0.02). Our findings suggest that in a subset of ductal adenocarcinomas, KRAS MASI correlates with the progression to undifferentiated carcinoma of the pancreas.
Keywords: fluorescence in situ hybridization, KRAS, mutant allele-specific imbalance, pancreatic cancer, undifferentiated carcinoma
Activating KRAS codon 12 mutations are present in the majority (about 90%) of pancreatic ductal adenocarcinomas.1–3 Undifferentiated carcinomas of the pancreas, many of which contain pleomorphic large cells or osteoclast-like giant cells, are considered to be variants of pancreatic ductal adenocarcinoma and tend to have a worse prognosis than pancreatic ductal adenocarcinoma. Undifferentiated carcinomas of the pancreas also harbor KRAS mutations,4 and when accompanied by ductal adenocarcinoma, both components harbor the same KRAS mutation.5–7
Mutations in the KRAS oncogene are typically heterozygous and the ratio of the mutant allele to wild-type allele may be balanced or unbalanced.1,8,9 In rare instances, the mutant allele becomes dominant, either through deletion of the wild-type allele and/or through copy number gain of the mutant allele. This phenomenon is termed ‘mutant allele-specific imbalance’ (MASI) and reflects increased ‘dosage’ of the mutant allele.8,10,11 Cancers that exhibit KRAS MASI appear to behave aggressively, as has been reported in both lung12 and colon13 adenocarcinomas.
The mechanism and significance of KRAS MASI in human pancreatic ductal adenocarcinoma and undifferentiated carcinoma is unclear. In a mouse model of pancreatic ductal adenocarcinoma, loss of the KRAS wild-type allele was shown to be associated with a higher incidence of metastasis.14 Compared with preneoplastic changes in the pancreas, a ‘second hit’ in the KRAS gene (loss of the wild-type allele or gene amplification) may be associated with the invasive process.15 The whole-genome loss of heterozygosity study of human cell lines derived from primary and metastatic pancreatic tumors confirmed that loss of heterozygosity at KRAS locus is indeed an event associated with metastases.14 Other studies have identified amplification of the KRAS mutant allele without the loss of the wild-type allele in cell lines,16,17 mouse models9 and human pancreatic ductal adenocarcinoma,17 a feature believed to contribute to pancreatic ductal adenocarcinoma progression.18 In fact, amplification of the mutated KRAS allele in a case of pancreatic ductal adenocarcinoma metastatic to a lymph node was shown about a quarter of century ago.19 Finally, both KRAS loss and gain were reported in an array comparative genomic hybridization study of 13 pancreatic ductal adenocarcinomas. 20
Our aim was to describe the role of KRAS MASI in pancreatic tumorigenesis. Specifically, we have employed combined interpretation of KRAS sequencing and KRAS fluorescence in situ hybridization (FISH) to characterize KRAS copy number changes that are associated with the transition of pancreatic ductal adenocarcinoma into undifferentiated carcinomas. This is the first study to focus on KRAS allelic imbalance in pancreatic ductal adenocarcinoma and to directly compare conventional ductal adenocarcinoma to undifferentiated carcinoma of the pancreas.
Materials and methods
Patients
The slides and reports of all possible undifferentiated carcinomas of the pancreas in the surgical pathology files of the authors’ institutions were reviewed. All cases that fulfilled the criteria for undifferentiated carcinoma (with or without osteoclast-like giant cells) and had tissue blocks available for study were included (cases were collected from 2001 through 2011). A consecutive cohort of pancreatic ductal adenocarcinoma cases resected between 2007 and 2009 at UPMC with tissue blocks available for analysis were also studied. This study was approved by the University of Pittsburgh Institutional Review Board (# PRO11070413). Only KRAS-mutated cases were included in this study; KRAS wild-type cases were excluded (two undifferentiated carcinomas and 12 ductal adenocarcinomas were KRAS wild-type). Basic clinicopathologic data are summarized in Table 1. Clinical staging was based on 7th edition of the American Joint Committee on Cancer manual.21
Table 1.
Clinicopathologic features of patients with KRAS-mutated pancreatic ductal adenocarcinoma and undifferentiated pancreatic carcinoma, overall and by KRAS MASI
| Feature | Overall (n = 119) | No MASI (n = 97) | MASI (n = 22) | P-value |
|---|---|---|---|---|
| Sex | ||||
| Men/women | 64/55 | 52/45 | 12/10 | 1 |
| Age, average, years | 66 | 67 | 63.5 | 0.3 |
| Average tumor sizea, cm | 3.5 | 3.3 | 4.2 | 0.08 |
| Tumor site | ||||
| Head/neck | 92 | 76 | 16 | 0.6 |
| Body/tail | 27 | 21 | 6 | |
| pTa | ||||
| 1 | 6 | 4 | 2 | 0.2 |
| 2 | 9 | 6 | 3 | |
| 3 | 92 | 78 | 14 | |
| 4 | 5 | 4 | 1 | |
| pNa | ||||
| 0 | 40 | 32 | 8 | 0.8 |
| 1 | 70 | 58 | 12 | |
| Clinical stagea | ||||
| I | 9 | 5 | 4 | 0.009 |
| II | 93 | 81 | 12 | |
| III | 4 | 4 | 0 | |
| IV | 12 | 7 | 5 | |
| Histologic type | ||||
| PDA | 94 | 88 | 6 | <0.001 |
| Undifferentiated | 25 | 9 | 16 | |
| KRAS AA | ||||
| p.G12D | 63 | 52 | 11 | 0.9b |
| p.G12V | 34 | 26 | 8 | |
| p.G12R | 16 | 14 | 2 | |
| p.G12C | 3 | 3 | 0 | |
| p.G12A | 2 | 1 | 1 | |
| KRAS FISH (n = 45) | ||||
| Amplification | 10 | 3c | 7d | 0.08e |
| Hyperploidy | 7 | 4 | 3 | |
| Monosomy | 5 | 3 | 2 | |
| Normal | 23 | 15 | 8 | |
| Median overall survival, months | 20 | 22 (95 CI, 17–27) | 9 (95 CI, 5–13) | 0.015 |
| Median DFS, months | 15.7 | 13 (95 CI, 10–16) | 5 (95 CI, 2–8) | 0.02 |
Abbreviations: AA, amino acid; CI, confidence interval; DFS, disease-free survival; FISH, fluorescence in situ hybridization; MASI, mutant allele-specific imbalance; PDA, well-differentiated component.
Tumor size not available for 1 case, pT status was not available (biopsy material) for five PDAs and two undifferentiated carcinomas, pN was unknown in nine cases and clinical stage was unknown for one undifferentiated carcinoma.
Based on the distribution of three most common mutations. One case showed a complex insertion/deletion mutation, which was not included in this table.
In one case, amplification was associated with chromosome 12 monosomy (62.3% of cells) and in another case amplification was associated with chromosome 12 hyperploidy (62.9% of cells).
In two cases, amplification was accompanied by monosomy (46.7% and 56.1% of cells) and in two other cases, amplification was associated with hyperploidy (47.7% and 64.5% of cells).
Cases with KRAS/CEP12 >2 were compared with cases showing normal KRAS FISH.
KRAS Exon 2 Codons 12 and 13 Mutational Analysis
Exon 2 KRAS codon 12 and 13 mutations were assessed by direct sequencing in both directions (forward and reverse) as previously described.22 Tumor areas with >50% tumor cells (based on review of hematoxylin and eosin-stained slides) were microdissected from the 4-μm unstained formalin-fixed paraffin embedded sections.
Review of Sequencing Electropherograms and Determination of the KRAS Mutant to Wild-type Allelic Peak Height Ratio
Based on the variation of the KRAS mutant and wild-type allelic peak heights on the sequencing electropherograms, all cases were divided into two groups: (1) Tumors with mutant allele peak lower than the wild-type allele peak (Figure 1a) were considered negative for MASI; (2) Cases with mutant allele peak higher than or equal to the wild-type allele peak (Figures 1b, c, and d) were considered positive for KRAS MASI.
Figure 1.
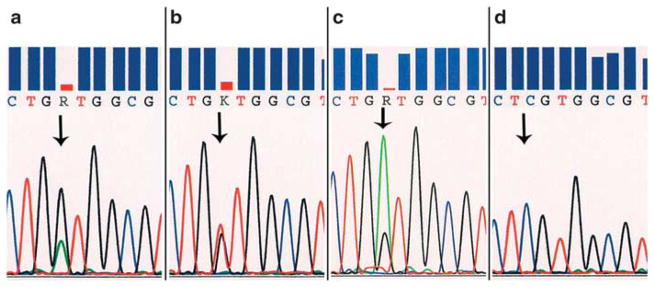
Sequencing electropherograms, comparing peak heights of the KRAS mutant allele and wild-type allele (arrows). (a) Mutant allele<wild-type allele. Cases with mutant allele peak lower than the wild-type allele peak are grouped as without mutant allele-specific imbalance (MASI); codon 12 35G>A point mutation (p.G12D). (b) Mutant allele=wild-type allele. Peaks are of about equal height; codon 12 35G>T point mutation (p.G12V). (c) Mutant allele>wild-type allele. Mutant allele peak higher than the wild-type allele peak; codon 12 35G>A point mutation (p.G12D). (d) All mutant allele. The wild-type allele is missing, suggesting complete MASI; codon 12 34 G>C point mutation (p.G12R). This was seen in two cases of undifferentiated carcinoma. Cases illustrated in panel b, c, and d are characterized as cases with MASI, partial (b, c) or complete (d).
KRAS FISH
Dual-color KRAS FISH analysis was successfully performed on 45 cases using a Spectrum Green labeled chromosome enumeration probe 12 (CEP12) (Abbott Molecular, Des Plaines, IL, USA) and a Spectrum Orange labeled KRAS locus specific (RP11-295I5, CHORI, Oakland, CA, USA) probe, as previously described.12,13,23,24 Amplification was defined as a KRAS/CEP12 ratio of >2 (Figure 2a). Chromosome 12 hyperploidy was defined as >40% of cells with >2 CEP12 signals (Figure 2b). Chromosome 12 monosomy was defined as >40% of cells with one CEP signal (Figure 2c). At least 60 cells were analyzed in each tested case. Nuclei were counterstained with DAPI, 4′,6-diamidino-2-phenylindole. The basic clinicopathologic features of cases selected for FISH were similar to the cases for which FISH analysis was not performed.
Figure 2.
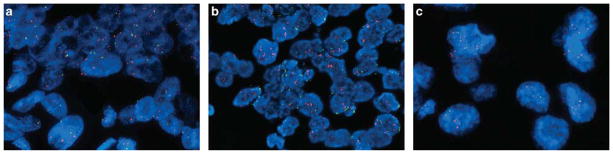
KRAS fluorescence in situ hybridization. (a) KRAS amplification; numerous KRAS (orange) signals and two CEP12 (green) signals per nucleus. (b) Example of a case with KRAS amplification and chromosome 12 hyperploidy; numerous orange and green signals in each nucleus. (c) Example of a case with KRAS amplification and chromosome 12 monosomy; numerous KRAS (orange) signals and only one CEP12 (green) signal per nucleus.
Statistical Analysis
Overall survival was assessed from the time of biopsy-confirmed diagnosis or resection to death from any cause based on review of medical record and social security death index. Living patients were censored at the time of last clinical follow-up. Disease-free survival was assessed from the time of biopsy-confirmed diagnosis to disease recurrence (eg, local recurrence, distant metastasis) or death of any cause. Follow-up for one case was not available. A t-test was used to characterize the relationship between quantitative variables, and the Fisher’s exact test was used to characterize the relationship between categorical variables. Median survival intervals with 95% confidence intervals were estimated using the Kaplan–Meier method, with statistical significance of differences between groups estimated by log-rank test. A P-value of <0.05 was defined as statistically significant. Statistical analysis was performed using SPSS 20 (Somers, NY, USA).
Results
Clinicopathologic Comparison of Ductal Adenocarcinoma with Undifferentiated Carcinoma of the Pancreas
Ninety-four conventional pancreatic ductal adenocarcinomas and 25 pancreatic undifferentiated carcinomas harboring KRAS mutations were included in this study. The ‘undifferentiated carcinoma’ variant of pancreatic carcinoma includes a spectrum of morphologies. In our study, the undifferentiated carcinomas could be divided into three subtypes (Figure 3): ten (40%) tumors were composed of two cell populations: pleomorphic round or polygonal neoplastic mononuclear cells admixed with osteoclast-like giant cells (six of these cases also contained anaplastic giant cells), 10 (40%) tumors were composed of a population of medium-to-large anaplastic epithelioid cells (anaplastic carcinoma) with variable cohesiveness, nuclear-to-cytoplasmic ratios and spindle cell components (in four of these cases, large cells contained abundant pink cytoplasm) and five (20%) tumors were composed of mixed small monotonous and anaplastic cells forming cohesive nests in a sometimes fibrous stroma.
Figure 3.
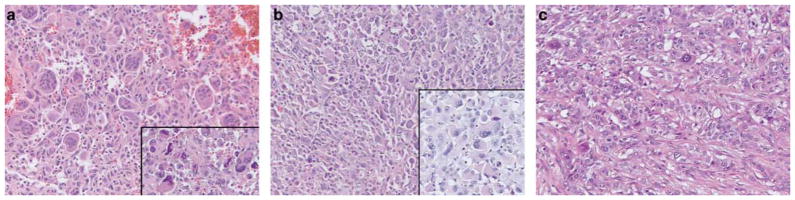
Representative examples of the undifferentiated carcinomas of the pancreas included in this study (H&E, ×200). (a) Undifferentiated carcinoma with osteoclast-like giant cells; some cases also contained anaplastic giant cells (inset) (H&E, ×200); (b) Undifferentiated carcinoma containing sheets of medium-to-large anaplastic cells (anaplastic carcinoma); in some cases, the malignant cells contained abundant pink cytoplasm creating a rhabdoid appearance (inset) (H&E, ×200). (c) Five cases contained epithelioid cells admixed with anaplastic cells that formed vague nests and trabeculae (H&E, ×200).
Sixteen (64%) of the undifferentiated carcinomas were associated with ductal adenocarcinomas; eight tumors had separate foci of pancreatic ductal adenocarcinoma comprising from 30–60% of the entire tumor mass (two of these cases also had intraductal papillary mucinous neoplasms and one had a mucinous cystic neoplasm) and eight tumors had microscopic foci of pancreatic ductal adenocarcinoma (two of these also had mucinous cystic neoplasms and one had an intraductal papillary mucinous neoplasm). Hence, there were three cases of undifferentiated carcinoma that were associated with mucinous cystic neoplasms and three that were associated with intraductal papillary mucinous neoplasms.
As a group, undifferentiated carcinomas were larger than pancreatic ductal adenocarcinomas (4.2cm vs 3.3 cm, P=0.04). Patients with undifferentiated carcinoma tended to present at younger age than patients with pancreatic ductal adenocarcinoma (average age of 63 years vs 67 years, P=0.1). Otherwise, the distribution of patients’ gender, tumor site (head/neck vs body/tail), pT, pN, clinical stage, and type of KRAS mutation were similar between these two histologically defined groups.
Overall average follow-up was 20 months. The median overall survival tended to be shorter for patients with undifferentiated carcinomas when compared with overall survival of patients with pancreatic ductal adenocarcinomas (10 months, 95% confidence interval, 6–14 vs 21 months, 95% confidence interval, 15–27, P=0.12). Similarly, disease-free survival tended to be shorter for patients with undifferentiated carcinoma (8 months, 95% confidence interval, 5–11 vs 12 months, 95% confidence interval, 9–15, P=0.4).
While histologic type (pancreatic ductal adenocarcinoma vs undifferentiated carcinoma), patients’ age, gender, and type of KRAS mutation did not correlate with overall survival or disease-free survival, clinical stage was predictive of disease-free survival and overall survival (Figure 4a). For instance, estimated median overall survival for Stage II patients was 21 months (95% confidence interval, 16–26), while it was 2 months (95% confidence interval, 0.8–3) for Stage IV patients.
Figure 4.
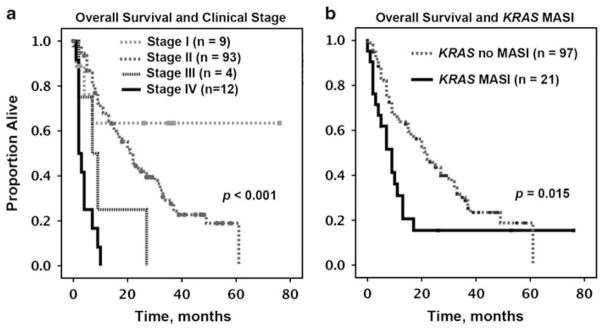
Overall survival analysis, Kaplan–Meier method, for patients with clinical follow-up. (a) Overall survival by clinical stage. (b) Overall survival by KRAS MASI (as per sequencing electropherogram).
Allelic Peak Height on Sequencing Electropherograms and KRAS Amplification by FISH
While reviewing KRAS sequencing electropherograms of all 119 pancreatic carcinomas, we noticed that 22 cases showed mutant allele peak higher or equal to the wild-type allele peak (Figure 1). In an attempt to identify the mechanism of the allelic imbalance suggested by sequencing electropherograms, KRAS FISH was performed on 45 cases, including 20 cases with mutant allele peak higher or equal to the wild-type peak and 25 cases with mutant allele peak lower than the wild-type peak (Table 1).
Chromosome 12 hyperploidy or monosomy appeared to be independent of allelic peak height on sequencing electropherograms. An equal number of cancers with chromosome 12 hyperploidy or monosomy had mutant allele peak higher/equal to the wild-type allele peak and mutant allele peak lower than the wild-type allele peak. These findings suggest that hyperploidy and monosomy may not preferentially target the chromosome harboring the KRAS mutant allele.
KRAS amplification was seen more frequently among cases with MASI compared with cases with no MASI (7/20, 35% vs 3/25, 12%, P=0.08; Table 1). Among cases with KRAS amplification, the average KRAS/CEP12 ratio was 3.6 (2.02–9.75). In six cases, KRAS amplification was associated with monosomy or hyperploidy. In four of six such cases, mutant allele peak was higher or equal to the wild-type peak.
Finally, in two undifferentiated carcinomas (one with osteoclast-like giant cells and one with anaplastic cells) sequencing electropherograms revealed mutant allele only (Figure 1d). Only one of these cancers has successful FISH testing and no FISH abnormalities were identified, suggesting that the likely mechanism of MASI in this cancer is homozygous mutation or uniparental disomy. As uniparental disomy and homozygous mutations are affecting very short DNA segments, these two mechanisms cannot be detected by FISH.
KRAS FISH: Pancreatic Ductal Adenocarcinoma vs Undifferentiated Carcinoma
KRAS FISH was performed on 21 cases of pancreatic ductal adenocarcinoma and 24 cases of undifferentiated carcinoma (Table 2). Fifteen of 21 (71%) tested pancreatic ductal adenocarcinomas showed no chromosome 12 abnormalities or KRAS copy number alteration. However, only 8 of 24 (33%) undifferentiated carcinomas showed no KRAS FISH abnormalities. Therefore, abnormal KRAS FISH result (ie, chromosome 12 hyperploidy, monosomy, or KRAS/CEP12 >2) was more common in undifferentiated carcinomas (P=0.0017) (Table 2). Most intriguingly, KRAS amplification was seen only in undifferentiated carcinomas. Ten of 24 (42%) undifferentiated carcinomas showed KRAS amplification, compared to none of the pancreatic ductal adenocarcinomas (P=0.0007) (Table 2).
Table 2.
KRAS FISH results in conventional ductal and undifferentiated pancreatic carcinomas
| KRAS FISH (n = 45) | Ductal adenocarcinomas (n = 21) | Undifferentiated carcinomas (n = 24) | P-value |
|---|---|---|---|
| Amplification | 0 | 10a | 0.0007 |
| Hyperploidy | 2 | 5 | |
| Monosomy | 4 | 1 | |
| Normal | 15 | 8 | 0.0017 |
Abbreviations: FISH, fluorescence in situ hybridization.
In three cases, amplification was associated with chromosome 12 monosomy (46.7–62.3% of cells) and in another three cases, amplification was associated with chromosome 12 hyperploidy (47.7–64.5% of cells).
The number of cases with KRAS amplification was too low for a meaningful outcome analysis; however, compared with patients without KRAS amplification, patients with carcinomas characterized by KRAS amplification showed a trend for worse median overall survival (8 months, 95% confidence interval, 0.7–15, vs 17 months, 95% confidence interval, 12–22 months, P=0.12). This is best explained by the fact that KRAS amplification was seen in undifferentiated carcinomas only.
KRAS Copy Number Alterations in Cases with Paired Ductal Adenocarcinoma and Undifferentiated Components in the Primary Tumor
In 11 cases of undifferentiated carcinoma with adjacent ductal adenocarcinoma, KRAS FISH was performed on both components (Table 3). In six of these cases (and in three additional cases without KRAS FISH data) KRAS sequencing was repeated on the well-differentiated component and in all cases the KRAS mutations were identical in both components.
Table 3.
Cases with both conventional pancreatic ductal adenocarcinoma and undifferentiated carcinoma components: results of KRAS FISH and KRAS allelic peak height evaluation on sequencing electropherogram
| Case no. | Histology | KRAS mutation | Peak height on SE |
KRAS FISH
|
||
|---|---|---|---|---|---|---|
| KRAS/CEP12 | % Of cells with hyperploidy | % Of cells with monosomy | ||||
| 1 | Undiff. | p.G12D | MA =WT | 1.1 | 41.2% | N |
| PDAa | p.G12D | MA<WT | 1.23 | N | N | |
| 2 | Undiff. | p.G12D | MA =WT | 2.02 | N | 46.7% |
| PDA | p.G12D | MA =WT | 2.43 | N | 68.3% | |
| 3 | Undiff. | p.G12V | MA =WT | 2.38 | N | N |
| PDA | n.t. | n.t. | 3.19 | N | N | |
| 4 | Undiff. | p.G12D | MA>WT | 2.51 | N | N |
| PDA | n.t. | n.t. | 1.29 | 50% | N | |
| 5 | Undiff. | p.G12V | MA>WT | 4.36 | 64.5% | N |
| PDA | n.t. | n.t. | 1.24 | N | 47.6% | |
| 6 | Undiff. | p.G12V | MA =WT | 9.75 | N | N |
| PDAb | p.G12V | MA<WT | 1.28 | N | N | |
| 7 | Undiff. | p.G12D | MA<WT | 1.02 | N | N |
| PDAa | p.G12D | MA<WT | 1.03 | N | N | |
| 8 | Undiff. | p.G12D | MA<WT | 3.16 | 62.9% | N |
| PDA | p.G12D | MA<WT | 1.75 | 88.5% | N | |
| 9 | Undiff. | p.G12D | MA<WT | 1.1 | 53.8% | N |
| PDA | p.G12D | MA<WT | 1.08 | 11.5% | N | |
| 10 | Undiff. | p.G12D | MA>WT | 1.6 | 40% | N |
| PDAb | n.t. | n.t. | 1.19 | 23.3% | N | |
| 11 | Undiff. | p.G12R | MA<WT | 1.16 | 46.3% | N |
| PDA | n.t. | n.t. | 1.1 | 56.3% | N | |
Abbreviations: FISH, fluorescence in situ hybridization; MA, mutant allele; N, normal (<40% of cells with hyperploidy or monosomy); n.t., not tested; PDA, well-differentiated component; SE, sequencing electropherogram; Undiff., undifferentiated component; WT, wild-type allele. The proportion (and size) of the PDA component ranged from 1% (0.3 cm) to 40% (3.6 cm).
The PDA component arose from a mucinous cystadenocarcinoma.
The microscopic focus of PDA arose from an intraductal papillary mucinous neoplasm.
In 6 of 11 cases, transition from the conventional ductal adenocarcinoma component to undifferentiated carcinoma was associated with an increase in KRAS copy number, either due to amplification and/or chromosome 12 hyperploidy. In three cases with KRAS sequencing performed on both components, FISH findings provided mechanistic explanation for the allelic peak height change seen on sequencing electropherograms (Table 3). For instance, in case #6, while the KRAS mutant peak was lower than the wild-type allele peak in the conventional ductal adenocarcinoma component, the height of two peaks was equal when sequencing was performed on the undifferentiated component. This change in peak height was accompanied by KRAS amplification with an increase in the KRAS/CEP12 ratio from 1.28–9.75.
Morphologic Characterization of Undifferentiated Carcinoma with KRAS MASI and KRAS FISH Abnormalities
As noted above, only undifferentiated carcinomas had KRAS amplification by FISH, and all three of the morphologic subtypes had at least one case showing amplification. Of the 10 undifferentiated carcinomas with osteoclast-like giant cells, seven (70%) had KRAS MASI. In five of these cases the likely mechanism of MASI was identified by KRAS FISH: KRAS amplification in four cases and chromosome 12 hyperploidy in one case. Of the 10 cases with predominantly anaplastic epithelioid cells (anaplastic carcinoma), five (50%) had KRAS MASI and the likely mechanism of MASI was amplification in three cases and chromosome 12 hyperploidy in one case. Four (80%) tumors with a more cohesive or nested growth pattern had KRAS MASI and the likely mechanism of MASI was amplification in one case and chromosome 12 monosomy in one case.
Clinicopathologic Correlates of KRAS MASI in Pancreatic Carcinomas
Pancreatic carcinomas with KRAS MASI (compared to those without MASI) were predominantly of undifferentiated type (16/22, 73% vs 9/97, 9%, P<0.001), more likely to present at clinical stage IV (5/22, 23% vs 7/97, 7%, P=0.009), and were associated with shorter overall survival (P=0.015) and disease-free survival (P=0.02) (Table 1 and Figure 4b). In subgroup analysis, clinical stage maintained its prognostic significance in both MASI and no MASI cases (for overall survival). Interestingly, clinical stage did not correlate with disease-free survival in the MASI group while it was significantly associated with disease-free survival in the no MASI group. The latter result allows speculating that in some cohorts the prognostic value of KRAS MASI may be independent of clinical stage.
Discussion
Undifferentiated carcinomas of the pancreas are rare and a head-to-head comparison of KRAS-mutated undifferentiated carcinomas to KRAS-mutated pancreatic ductal adenocarcinomas has not been previously reported. As KRAS mutations are so common in pancreatic cancer, we wanted to study a homogeneous group of tumors and hence only included KRAS-mutated cases. In prior studies, KRAS mutational status was employed to confirm the ductal cell origin of undifferentiated carcinomas. 4–7 Specifically, identical KRAS mutations were shown both in undifferentiated and ductal adenocarcinoma components in about 20 cases reported in the literature.5–7 Along with the cytokeratin profile, the knowledge of KRAS status was important to suggest that undifferentiated carcinomas represent variants of pancreatic ductal adenocarcinoma. The aim of our study was to characterize the role of KRAS MASI in pancreatic ductal adenocarcinoma and its progression to undifferentiated carcinoma.
The potential role of KRAS MASI in tumor progression is based on observations that KRAS MASI is associated with pancreatic ductal adenocarcinoma metastases,14 as well as aggressive behavior in lung and colon adenocarcinomas.12,13 KRAS MASI may lead to elevated KRAS mRNA levels24 and increased RAS activity in murine pancreatic adenocarcinomas and other carcinomas.8,9 In a variety of pancreatic ductal adenocarcinoma models, deletion of the wild-type allele and/or an increased copy number of the mutated allele was associated with more aggressive features. The status of the wild-type allele is also relevant, as increasing evidence indicates that the wild-type KRAS allele may act as a tumor suppressor.25–27
The possibility of the imbalance between the wild-type and mutant KRAS alleles both in pancreatic ductal adenocarcinoma and undifferentiated carcinomas was raised previously. For instance, one of the first reports on incidence of KRAS codon 12 mutations in human pancreatic ductal adenocarcinoma suggested that some pancreatic ductal adenocarcinoma cases may ‘contain increased ratios of mutant vs normal KRAS allele’.1 Similar observations were made later by others.14–16,20,28 It was also suggested that a subset of undifferentiated pancreatic carcinoma may harbor homozygous KRAS mutation.6 A more detailed discussion and comparison of KRAS MASI incidence, likely mechanism, or clinicopathologic correlates with prior studies is difficult. Other studies analyzed 12p region or KRAS locus in fewer cases, by different techniques, without clinical follow-up or detailed/explicit histologic description of cases. In fact, no other study employed FISH to characterize KRAS copy number alteration in pancreatic carcinomas.
Most prior studies reported primarily on KRAS gain/loss associated with regional lymph node or distant metastases in pancreatic ductal adenocarcinoma. For the first time, we have attempted to systematically evaluate KRAS MASI associated with transition of ductal adenocarcinomas to undifferentiated carcinoma of the pancreas. To achieve our aim, a sizable cohort of KRAS-mutated undifferentiated carcinomas of the pancreas and a cohort of pancreatic ductal adenocarcinoma were evaluated for KRAS MASI by semi-quantitative assessment of KRAS sequencing electropherograms and KRAS FISH. The semi-quantitative nature of direct sequencing was previously shown in other carcinomas, 8,13,24,29 in pancreatic ductal adenocarcinoma, 17 and was further demonstrated in this report: mutant allele peak equal to or higher than the wild-type allele peak was associated with a trend to higher incidence of amplification. In a prior report on direct KRAS sequencing in pancreatic ductal adenocarcinoma, it was shown that the KRAS allelic peak height as seen on direct sequencing was comparable to the proportion of transformed Escherichia coli colonies harboring mutant alleles.17
In our study, KRAS MASI was identified predominantly in undifferentiated carcinomas, among patients who presented at a more advance clinical stage, and correlated with worse overall survival. The difference in survival of 11 months between the KRAS MASI and no KRAS MASI groups is quite substantial for a cancer that has a median survival after resection of only 14–20 months30 and an overall (all stages) 5-year survival rate of 6%.31 We showed that KRAS MASI developed by KRAS amplification and/or chromosome 12 hyperploidy or monosomy. It appears that targeted amplification of the mutant allele may be accompanied by the loss of chromosome 12 carrying the wild-type KRAS allele or hyperploidy of the chromosome 12 harboring the mutated KRAS allele (Figure 5). Of all KRAS MASI mechanisms, KRAS amplification was the only one limited to undifferentiated pancreatic carcinomas. Pancreatic ductal adenocarcinoma develops through a multistep process that is initiated by an early activating KRAS mutation. Our findings suggest that subsequent KRAS MASI in a subset of cases correlates with the progression to undifferentiated carcinomas (Figure 5).
Figure 5.

KRAS copy number alterations and likely KRAS allelic imbalance associated with pancreatic ductal adenocarcinoma progression to undifferentiated pancreatic carcinoma. The scheme is based on previously published referenced work and a subset of cases presented in this report. Numbers refer to our cases for which both KRAS FISH and sequencing electropherograms were available. Note 1: The largest subset of pancreatic ductal adenocarcinoma (n=14) is characterized by normal results of KRAS FISH and no KRAS MASI. In these cases, the wild-type and mutated KRAS alleles are likely to be balanced and the fact that wild-type allele peak is higher than the mutant allele peak on sequencing electropherogram is best explained by the wild-type allele derived from stromal tissue and inflammatory cells. Note 2: Based on sequencing electropherograms, a subset of pancreatic ductal adenocarcinoma is characterized by KRAS MASI. KRAS FISH was performed on 5 of 6 such cases. Three cases showed normal KRAS FISH and KRAS MASI may have arisen through chromosome 12 uniparental disomy. In two cases, KRAS FISH revealed chromosome 12 monosomy in 47% and 78% of cells. As wild-type allele was predominant on sequencing electropherogram, the lost copy of chromosome 12 likely harbored the wild-type allele. Note 3: Sixteen of 25 undifferentiated carcinomas showed KRAS MASI. KRAS FISH was performed on 15 of 16 cases with KRAS MASI and amplification alone was identified in three cases. Two cases showed amplification associated with chromosome 12 monosomy, while the other two cases showed amplification associated with chromosome 12 hyperploidy.
Overall, KRAS MASI appears to represent an adverse prognostic factor. Therapeutically, there are at least two potential considerations in which KRAS MASI appears to be relevant, as well. First, it was shown that p53 restoration in a KRASG12D mouse model of non-small-cell lung carcinomas induces tumor regression only when KRAS wild-type allele was lost or the KRAS mutant allele was amplified.32,33 Second, potential targeted therapy should selectively affect the mutated KRAS gene product only, as inhibition of the wild-type KRAS gene product could mimic effects of the wild-type allele deletion.14
This study is limited by its retrospective design and focus on comparison of pancreatic ductal adenocarcinomas to undifferentiated carcinomas. Larger studies including cases of pancreatic intraepithelial neoplasia and regional lymph node and distant pancreatic ductal adenocarcinoma metastases are needed for a more granular understanding of KRAS MASI in pancreatic tumorigenesis.
In conclusion, we directly compared patients with KRAS-mutated pancreatic ductal adenocarcinomas and undifferentiated carcinomas of the pancreas and showed that KRAS MASI develops predominantly through KRAS amplification and/or chromosome 12 hyperploidy, monosomy and correlates with worse survival.
Acknowledgments
We wish to thank members of the GI Mastermind Group (2011–2012) for critical discussions, personnel in the In Situ/Developmental and Molecular Anatomic Pathology Laboratories at UPMC for excellent technical support, Jennifer Steve for assistance with the clinical database and Robyn Roche and Dee Ann Sapienza for outstanding secretarial support.
Footnotes
Disclosure/conflict of interest
The John F Fortney Charitable Pancreatic Cancer Research Group and Mellie’s Mission provided financial support. Support for advice regarding regulatory matters was provided by the National Institutes of Health through Grant Numbers UL1RR024153 and UL1TR000005.
References
- 1.Almoguera C, Shibata D, Forrester K, et al. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- 2.Smit VT, Boot AJ, Smits AM, et al. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 1988;16:7773–7782. doi: 10.1093/nar/16.16.7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hruban RH, van Mansfeld AD, Offerhaus GJ, et al. K-ras oncogene activation in adenocarcinoma of the human pancreas. A study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. Am J Pathol. 1993;143:545–554. [PMC free article] [PubMed] [Google Scholar]
- 4.Gocke CD, Dabbs DJ, Benko FA, et al. KRAS oncogene mutations suggest a common histogenetic origin for pleomorphic giant cell tumor of the pancreas, osteoclastoma of the pancreas, and pancreatic duct adenocarcinoma. Hum Pathol. 1997;28:80–83. doi: 10.1016/s0046-8177(97)90283-5. [DOI] [PubMed] [Google Scholar]
- 5.Westra WH, Sturm P, Drillenburg P, et al. K-ras oncogene mutations in osteoclast-like giant cell tumors of the pancreas and liver: genetic evidence to support origin from the duct epithelium. Am J Surg Pathol. 1998;22:1247–1254. doi: 10.1097/00000478-199810000-00010. [DOI] [PubMed] [Google Scholar]
- 6.Hoorens A, Prenzel K, Lemoine NR, et al. Undifferentiated carcinoma of the pancreas: analysis of intermediate filament profile and Ki-ras mutations provides evidence of a ductal origin. J Pathol. 1998;185:53–60. doi: 10.1002/(SICI)1096-9896(199805)185:1<53::AID-PATH45>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 7.Sedivy R, Kalipciyan M, Mazal PR, et al. Osteoclast-like giant cell tumor in mucinous cystadenocarcinoma of the pancreas: an immunohistochemical and molecular analysis. Cancer Detect Prev. 2005;29:8–14. doi: 10.1016/j.cdp.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 8.Soh J, Okumura N, Lockwood WW, et al. Oncogene mutations, copy number gains and mutant allele specific imbalance (MASI) frequently occur together in tumor cells. PLoS One. 2009;4:e7464. doi: 10.1371/journal.pone.0007464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aguirre AJ, Bardeesy N, Sinha M, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112–3126. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gandhi J, Zhang J, Xie Y, et al. Alterations in genes of the EGFR signaling pathway and their relationship to EGFR tyrosine kinase inhibitor sensitivity in lung cancer cell lines. PLoS One. 2009;4:e4576. doi: 10.1371/journal.pone.0004576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mitsudomi T, Viallet J, Mulshine JL, et al. Mutations of ras genes distinguish a subset of non-small-cell lung cancer cell lines from small-cell lung cancer cell lines. Oncogene. 1991;6:1353–1362. [PubMed] [Google Scholar]
- 12.Chiosea SI, Sherer CK, Jelic T, et al. KRAS mutant allele-specific imbalance in lung adenocarcinoma. Mod Pathol. 2011;24:1571–1577. doi: 10.1038/modpathol.2011.109. [DOI] [PubMed] [Google Scholar]
- 13.Hartman DJ, Davison JM, Foxwell TJ, et al. Mutant allele-specific imbalance modulates prognostic impact of KRAS mutations in colorectal adenocarcinoma and is associated with worse overall survival. Int J Cancer. 2012;131:1810–1817. doi: 10.1002/ijc.27461. [DOI] [PubMed] [Google Scholar]
- 14.Qiu W, Sahin F, Iacobuzio-Donahue CA, et al. Disruption of p16 and activation of Kras in pancreas increase ductal adenocarcinoma formation and metastasis in vivo. Oncotarget. 2011;2:862–873. doi: 10.18632/oncotarget.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sugio K, Molberg K, Albores-Saavedra J, et al. K-ras mutations and allelic loss at 5q and 18q in the development of human pancreatic cancers. Int J Pancreatol. 1997;21:205–217. doi: 10.1007/BF02821606. [DOI] [PubMed] [Google Scholar]
- 16.Heidenblad M, Jonson T, Mahlamaki EH, et al. Detailed genomic mapping and expression analyses of 12p amplifications in pancreatic carcinomas reveal a 3. 5-Mb target region for amplification. Genes Chromosomes Cancer. 2002;34:211–223. doi: 10.1002/gcc.10063. [DOI] [PubMed] [Google Scholar]
- 17.Aoki Y, Hosaka S, Tachibana N, et al. Reassessment of K-ras mutations at codon 12 by direct PCR and sequencing from tissue microdissection in human pancreatic adenocarcinomas. Pancreas. 2000;21:152–157. doi: 10.1097/00006676-200008000-00008. [DOI] [PubMed] [Google Scholar]
- 18.Bos JL. The ras gene family and human carcinogenesis. Mutat Res. 1988;195:255–271. doi: 10.1016/0165-1110(88)90004-8. [DOI] [PubMed] [Google Scholar]
- 19.Yamada H, Sakamoto H, Taira M, et al. Amplifications of both c-Ki-ras with a point mutation and c-myc in a primary pancreatic cancer and its metastatic tumors in lymph nodes. Jpn J Cancer Res. 1986;77:370–375. [PubMed] [Google Scholar]
- 20.Aguirre AJ, Brennan C, Bailey G, et al. High-resolution characterization of the pancreatic adenocarcinoma genome. Proc Natl Acad Sci USA. 2004;101:9067–9072. doi: 10.1073/pnas.0402932101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edge SB, Byrd DR, Compton CC, et al. AJCC Cancer Staging Manual. 7. Springer-Verlag; New York: 2010. [Google Scholar]
- 22.Chiosea S, Shuai Y, Cieply K, et al. EGFR fluorescence in situ hybridization-positive lung adenocarcinoma: incidence of coexisting KRAS and BRAF mutations. Hum Pathol. 2010;41:1053–1060. doi: 10.1016/j.humpath.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 23.Wagner PL, Perner S, Rickman DS, et al. In situ evidence of KRAS amplification and association with increased p21 levels in non-small cell lung carcinoma. Am J Clin Pathol. 2009;132:500–505. doi: 10.1309/AJCPF10ZUNSOLIFG. [DOI] [PubMed] [Google Scholar]
- 24.Modrek B, Ge L, Pandita A, et al. Oncogenic activating mutations are associated with local copy gain. Mol Cancer Res. 2009;7:1244–1252. doi: 10.1158/1541-7786.MCR-08-0532. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Z, Wang Y, Vikis HG, et al. Wildtype Kras2 can inhibit lung carcinogenesis in mice. Nat Genet. 2001;29:25–33. doi: 10.1038/ng721. [DOI] [PubMed] [Google Scholar]
- 26.Li J, Zhang Z, Dai Z, et al. LOH of chromosome 12p correlates with Kras2 mutation in non-small cell lung cancer. Oncogene. 2003;22:1243–1246. doi: 10.1038/sj.onc.1206192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, Cao HF, Wan J, et al. Growth inhibitory effect of wild-type Kras2 gene on a colonic adenocarcinoma cell line. World J Gastroenterol. 2007;13:934–938. doi: 10.3748/wjg.v13.i6.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bardeesy N, Aguirre AJ, Chu GC, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci USA. 2006;103:5947–5952. doi: 10.1073/pnas.0601273103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oakley GJ, 3rd, Chiosea SI. Higher dosage of the epidermal growth factor receptor mutant allele in lung adenocarcinoma correlates with younger age, stage IV at presentation, and poorer survival. J Thorac Oncol. 2011;6:1407–1412. doi: 10.1097/JTO.0b013e31821d41af. [DOI] [PubMed] [Google Scholar]
- 30.Bachmann J, Michalski CW, Martignoni ME, et al. Pancreatic resection for pancreatic cancer. HPB (Oxford) 2006;8:346–351. doi: 10.1080/13651820600803981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.American Cancer Society. [accessed 5 January 2013];Cancer Facts & Figures. Available from http://www.cancer.org/Research/CancerFactsFigures/index.
- 32.Feldser DM, Kostova KK, Winslow MM, et al. Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature. 2010;468:572–575. doi: 10.1038/nature09535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Junttila MR, Karnezis AN, Garcia D, et al. Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature. 2010;468:567–571. doi: 10.1038/nature09526. [DOI] [PMC free article] [PubMed] [Google Scholar]