

Abstract
Liposarcoma is the second most common soft tissue sarcoma in adults, but treatment options have been quite limited thus far. In this study, we investigated the functional and therapeutic relevance of cyclin-dependent kinase 11 (CDK11) as a putative target in liposarcoma. CDK11 knockdown by synthetic siRNA or lentiviral shRNA decreased cell proliferation, and induced apoptosis in liposarcoma cells. Moreover, CDK11 knockdown enhances the cytotoxic effect of doxorubicin to inhibit cell growth in liposarcoma cells. These findings suggest that CDK11 is critical for the growth and proliferation of liposarcoma cells. CDK11 may be a promising therapeutic target for the treatment of liposarcoma patients.
Keywords: Kinase, Liposarcoma, CDK11, Tissue microarray, Immunohistochemistry
1. Introduction
Liposarcoma is the second most common soft tissue sarcoma in adults. Histologically, liposarcoma is mainly classified into four distinct categories: well differentiated, dedifferentiated, myxoid, and pleomorphic [1,2]. Surgery serves as the primary therapy for liposarcoma. Doxorubicin alone or in combination with ifosfamide is often used as a first-line regimen in patients with high risk of recurrence or metastatic liposarcoma, and other agents are used with modest benefit, but the impact of chemotherapy in liposarcomas remains controversial [3,4]. There has been no significant progress in improving the survival rate of recurrent or metastatic liposarcoma for almost three decades. The median overall length of survival in metastatic liposarcoma is 11.5 months and the 5-year disease-specific survival rate is only 5.2% [5]. Therefore, there is need to identify novel therapeutic strategies to improve patient outcomes.
During the last decade, protein kinase targeted therapies have been shown to be effective in treating many cancers. For example in sarcomas, the small molecule inhibitor of c-Kit, imatinib, has dramatically changed the treatment and prognosis of gastrointestinal stromal tumors (GIST) [6]. Several protein kinase pathways have been found to be up-regulated or amplified in liposarcomas, including the proto-oncogenes PI3K/AKT [7] and C-Jun [8,9]. Amplification and over-expression of these kinase pathways are thought to be an early and essential part of the oncogenic program of liposarcoma. However, the roles of most human protein kinases in liposarcoma remain largely unknown.
Recently, the Cyclin-Dependent Kinases (CDKs), a family of protein kinases, have been established as master regulators of cell growth/proliferation and apoptosis, and thus inhibitors of CDKs have been explored as a novel therapeutic strategy in malignancy [10,11]. Although to this date no cell cycle protein kinase inhibitors have been approved as an anticancer drug, several CDKs inhibitors (flavopiridol and dinaciclib) have entered clinical trials in chronic lymphocytic leukemia, non-Hodgkin's lymphoma, renal, prostate, colon and gastric carcinomas patients [10,12–14]. Flavopiridol, a pan-CDK inhibitor and targeting CDK2, CDK4, CDK6 and CDK9, is associated with cell-cycle arrest and subsequent apoptosis [15,16]. More recently, a phase II study was performed to determine the safety and efficacy of CDK4 inhibitor PD0332991 in patients with advanced CDK4-amplified liposarcoma [11]. The results showed that treatment with PD0332991 displayed a favorable disease progression-free rate in liposarcoma patients. These ongoing clinical trials appeared promising and will facilitate the development of more potent CDK-based therapeutic compounds.
In previously work we screened osteosarcoma cell lines with the lentiviral shRNA kinase library, consistently identifying that knockdown of CDK11 expression led to inhibited growth and induction of apoptosis in both in vitro and in vivo models [17]. CDK11 (formerly known as PITSLRE or CDC2L1) is a member of the extended family of p34cdc2-related kinases [18]. At least 10 CDK11 isoforms have been cloned in eukaryotic cells, and the major isoforms are CDK11p110 [19,20], CDK11p58 [21,22], and CDK11p46[23]. Although no clear mechanistic functions for CDK11 are known in tumor cells, the larger CDK11p110 isoform has been reported to have effects on the regulation of transcription and RNA splicing [24–26]. CDK11p110 is ubiquitously expressed in tumor cell lines and constantly through the cell cycle [27]. Although we have observed that knockdown of CDK11 p110 decreased cell viability and increased apoptosis in osteosarcomas [17], the significance of CDK11 p110 signaling in liposarcomas is unknown. Therefore, in this study, we first comparatively examined the expression of CDK11 p110 in liposarcoma and lipoma (benign tumor) tissue microarray by Immunohistochemical analysis. Furthermore, we evaluated the function role of CDK11 p110 in the growth and proliferation of liposarcoma.
2. Materials and Methods
2.1 Tissue array and immunohistochemistry (IHC)
The human liposarcoma and lipoma tissue array was purchased from Imgenex Corporation (CA, USA). Lipomas, as the control group of liposarcomas, are the most common benign tumor and are composed of adipose tissue. Immunohistochemistry was conducted with HRP-DAB System Cell and Tissue Staining Kit (R&D Systems, CA, USA) according to the manufacturer’s protocol. Briefly, paraffin embedded slide was baked at 62°C for 60 minutes, then deparaffinized with xylenes and rehydrated. Antigen retrieval was performed using heat-induced epitope retrieval (water bath in antigen retrieval solution). The slides were incubated with peroxidase blocking reagent for 5 minutes, blocked with serum blocking reagent for 15 minutes, incubated with avidin and biotin blocking reagent for 15 minutes, and probed with rabbit anti-CDK11 (SC-928; 1:50 dilution; Santa Cruz Biotechnology, Inc., Texas, USA.) at 4°C overnight. After washing, the tissue slide was probed with biotinylated anti-rabbit secondary antibody (BA-1000; Vector Laboratories Inc., CA, USA) for 1 hour, and HSS-HRP was added for 30 minutes. DAB Chromogen Solution was added to cover the entire tissue section and incubated for 8 minutes. The tissue array was counterstained using hematoxylin QS (Vector Laboratories Inc., CA, USA), dehydrated, and mounted with Vectamount AQ (Vector Laboratories Inc., CA, USA). The slide was imaged using a Nikon Eclipse Ti-U fluorescence microscope (Nikon Corp., Melville, New York, USA) with a SPORT RT digital camera (Diagnostic Instruments Inc., MI, USA).
The degree of immunostaining of tissue array was viewed and scored separately by two independent investigators, who were blinded to the histopathological features and patient details of the samples. The proportion of positively stained tumor cells was staged as follows: 0 (no positive nuclear staining of CDK11 tumor cells), 1+ (<10% positive tumor cells), 2+ (10–25% positive tumor cells), 3+ (26–50% positive tumor cells), 4+ (51–75% positive tumor cells), 5+ (>75% positive tumor cells). A staining score of ≥ 3 was used to define tumors with high expression and a staining index ≤ 2 was used to define tumors with low expression of CDK11.
2.2 Cell lines and cell culture
The human liposarcoma cell lines, SW872 and SW982, were purchased from the American Type Culture Collection (Maryland, USA). Both the liposarcoma cell lines were cultured in DMEM (HyClone, Thermo Fisher Scientific Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (HyClone), penicillin (100 mg/ml), and streptomycin (100 mg/ml; Invitrogen, Grand Island, NY, USA). Cells were maintained in a humidified atmosphere containing 5% CO2–95% air at 37oC. Light microscopy images were documented using a Zeiss microscope from Carl Zeiss, Inc., (Oberkochen, Germany) with an attached Nikon D40 digital camera from Nikon Corp. (Melville, New York, USA).
2.3 Synthetic CDK11siRNA and transfection
CDK11 knockdown in liposarcoma cells were performed by transfection of synthetic human CDK11 siRNA (AM16708; Ambion at Applied Biosystems, Foster City, USA). The siRNA sequence targeting CDK11 corresponded to coding regions (5’-AGAUCUACAUCGUGAUGAAtt-3’, antisense 5’-UUCAUCACGAUGUAGAUCUtg-3’) of the CDK11 gene. The nonspecific siRNA oligonucleotides (AM4637; Applied Biosystems) were used as negative controls. 5,000 cells per well were seeded in 96-well plates with complete growth medium without antibiotics and transfected with nonspecific siRNA or CDK11 siRNA. Transfections were performed with LipofectamineTM RNAiMAX (Invitrogen) following the manufacturer’s instructions.
2.4 Proliferation assay
Effects of CDK11 siRNA on cellular growth and proliferation were assessed in vitro using the MTT assay. 5000 cells were seeded in 96-well plates with complete growth medium without antibiotics and transfected with non-specific siRNA or CDK11 siRNA. After 6 days of culture, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT, Sigma-Aldrich, St. Louis, MO, USA) was added to each well and absorbance was read using a SPECTRAmax Microplate Spectrophotometer (Molecular Devices, Sunnyvale, CA, USA).
2.5 Lentiviral human CDK11 shRNA and transfection
Five lentiviruses carrying different shRNA sequences targeting human CDK11 kinase genes were chosen from MISSION LentiExpress Human Kinases shRNA library (SHX001; Sigma-Aldrich, St. Louis, MO, USA). The sequences of 5 shRNA target different sites of CDK11 were TRCN0000006206 (5’-GCCGAAGAAGTAAGTGAGGAA-3’), TRCN0000006207 (5’-CGATCAGATCAACAAGGTGTT-3’), TRCN0000006208 (5’-CGGAAACGACATCGAGAAGAA-3’), TRCN0000006209 (5’-CGGCCTCAAGCATGAGTATTT-3’) and TRCN0000006210 (5’-CAGATGAAATTGTGGCTCTAA-3’). Negative control shRNA lentiviral particles include pLKO.1-puro Control (SHC001V, Sigma-Aldrich) and shRNA non-target control (SHC002V, Sigma-Aldrich). Transfection was carried out by following the manufacturer’s protocol. Briefly, 2000 cells in fresh media were added to the number of wells needed for each construct in a 96-well plate. Then Hexadimethrine bromide (final concentration 11.3 μg/ml, Sigma-Aldrich) was added to each well. Puromycin (1 μg/ml, Sigma-Aldrich) was added to the cells in fresh media to select for stable cell transfection on day 3. After 6 days, all cells were transferred to flasks to observe long-term silencing of CDK11 in cells. After establishment of stable cell transfection for 30 days, all cells were collected and total cell numbers were counted using a Bright-Line™ Hemacytometer (Cambridge Instruments, Inc., NY, USA).
2.6 Chemotherapeutic response assay
Cytotoxicity was determined by MTT assay as described above [28]. 5,000 cells were plated into each well of a 96-well plate. After 24 hours of CDK11 siRNA transfection, the cells were treated with or without doxorubicin for 6 days. MTT (Sigma-Aldrich) was added to each well and absorbance was read using a SPECTRAmax Microplate Spectrophotometer (Molecular Devices). The relative absorbance values were normalized by assigning the absorbance value of cells without CDK11siRNAand doxorubicin treatment. Experiments were performed in triplicate.
2.7 Protein preparing and western blotting
Total protein was isolated with RIPA Lysis Buffer (Upstate Biotechnology, New York, USA) 60–65 hours after CDK11 siRNA transfection. The concentration of the protein was determined by protein assay reagents (Sigma-Aldrich, St. Louis, MO, USA) with a spectrophotometer (Beckman Du-640, Beckman Instruments, Inc., Indianapolis, IN). Western blotting was performed as follows: denatured proteins were run on an SDS-PAGE gel, and then transferred to nitrocellulose membrane. Membranes were blocked in 5% nonfat milk for two hours, and probed with rabbit polyclonal antibody (SC-928) to human CDK11 (1:500 dilution) or mouse monoclonal antibody to human β-actin (A2228; Sigma-Aldrich, St. Louis, MO, USA)at 4°C overnight. Following primary antibody incubation, membranes were washed with TBST, and Goat anti-Rabbit IRDye® 800CW or Goat anti-mouse IRDye® 680LTsecondary antibody (1:2000 dilution) (926-32211 and 926-68020; LI-COR Biosciences, NE, USA) was added respectively. Bands were detected using Odyssey for Infrared Fluorescent Western Blots from LI-COR Bioscience (Lincoln, NE, USA). Quantification analysis of Western blot results was performed with Odyssey software 3.0 (LI-COR Bioscience, Lincoln, Nebraska, USA). All other antibodies used in this study were purchased from Santa Cruz Biotechnology or Cell Signaling Technologies.
2.8 Immunofluorescence
To visualize immunofluorescence of CDK11 expression, SW872 and SW982 were transfected with nonspecific siRNA or CDK11 siRNA in 12-well chambers for 60–65 hours. The samples were fixed with 4% paraformaldehyde (Sigma-Aldrich, St. Louis, MO, USA) in PBS for 15 min at room temperature. The samples were then permeabilized with ice-cold methanol (Sigma-Aldrich) for 10 min. Then samples were blocked in 1% BSA (Sigma-Aldrich) in PBST for 30 min to block unspecific binding of the antibodies. Following incubation with CDK11 primary antibody (1:50 dilution, Santa Cruz Biotechnology) or β-actin (1:200 dilution, Sigma-Aldrich) at 4°C overnight, samples were incubated with Alexa Fluor 488 (Green) conjugated goat anti-rabbit antibody (A-11034; Invitrogen, Grand Island, NY, USA) and Alexa Fluor 594 (red) goat anti-mouse antibody (A-11032; Invitrogen, Grand Island, NY, USA) for one hour. Finally, Hoechst 33342 (1μg/ml, Invitrogen) was added to counterstain the cell nucleus. Cells were then imaged on a Nikon Eclipse Ti-U fluorescence microscope (Nikon Corp., Melville, New York, USA) equipped with a SPOT RT digital camera from Diagnostic Instruments, Inc. (MI, USA).
2.9 Statistical analysis
All statistical analyses were carried out using the GraphPad PRISM 5 software from GraphPad Software, Inc (San Diego, CA, USA). The student t test was used to analyze the differences between two groups. Data were analyzed by the one-way ANOVA followed by Tukey post-hoc test. In all cases, results are expressed as mean ± SD and P<0.05 was considered statistically significant.
3. Results
3.1 CDK11 is highly expressed in liposarcoma as compared with lipoma tissues
To compare the expression of CDK11 between benign lipoma and liposarcoma tissues, CDK11 protein levels were analyzed by immunohistochemistry using lipoma and liposarcoma tissue microarray. The results showed that protein expression level of CDK11 was significantly higher in liposarcoma samples (2.6 ± 0.20) than in lipoma samples (0.56 ± 0.18) (Fig. 1A). High expression of CDK11 (CDK11 staining ≥ 3) was identified in 58.5% of liposarcoma (Table 1), whereas it was not observed in lipoma tissues, suggesting that CDK11 expression may play an important role in the growth of liposarcoma. Moreover, in the liposarcoma tissues, 38 (92.7%) of 41 samples were positive when tested for CDK11 staining, yet 5 (55.6%) of lipoma tissues were positive. These results also showed that the CDK11 protein was mainly localized in the nucleus of liposarcoma cells (Fig. 1B) which is consistent with our prior publication in osteosarcoma cells [17].
Fig 1.

CDK11 is expressed higher in liposarcoma than in lipoma tissues. A, distribution of CDK11 staining scores among the lipoma and liposarcoma tissues. The student t test was used to analyze the differences between two groups. B, representative images of different immunohistochemical staining intensities of CDK11 are shown in lipoma and liposarcoma tissues. For CDK11 immunohistochemical staining, the percentage of cells showing positive nuclear staining for CDK11 was calculated by reviewing the entire spot.
Table 1.
Clinicopathologic characteristics of studied patients and expression of CDK11 in lipoma and liposarcoma.
| NO (%) | |||||
|---|---|---|---|---|---|
| Gender | |||||
| Male | 23 (46.0) | ||||
| Female | 27 (54.0) | ||||
| Age (years) | |||||
| >=60 | 13 (26.0) | ||||
| <60 | 37 (74.0) | ||||
| Pathology | |||||
| Lipoma | 9 (18.0) | ||||
| Liposarcoma, well differntiated | 18 (36.0) | ||||
| Liposarcoma, myxoid | 15 (30.0) | ||||
| Liposarcoma, dedifferntiated | 5 (10.0) | ||||
| Liposarcoma, pleomorphic | 3 (6.0) | ||||
| Survival (n=56) | |||||
| Alive | 33 (66.0) | ||||
| Dead | 17 (34.0) | ||||
| Expression of CDK11 | |||||
| 0 | 1+ | 2+ | 3+ | 4+ | |
| Lipoma (n=9) | 4(44.4) | 5(55.6) | |||
| Liposarcoma (n=41) | 3(7.3) | 6(14.6) | 8(19.5) | 11(26.8) | 13(31.7) |
Note: 0 (no positive tumor cells); 1+ (<10% positive tumor cells); 2+ (10–25% positive tumor cells); 3+ (26–50% positive tumor cells); 4+ (51–75% positive tumor cells); 5+ (>75% positive tumor cells)
3.2 CDK11 expression is critical for liposarcoma cell growth and survival
To characterize the functional role of CDK11 in liposarcoma, synthetic human CDK11 siRNA was transfected into two liposarcoma cell lines SW872 and SW982 (Fig. 2A and 2B). The results revealed that CDK11 siRNA inhibits liposarcoma cell growth and proliferation. Meanwhile, the expression of CDK11 was measured in siRNA transfected cells. Western blot analysis suggested that down-regulated expression of CDK11 protein by CDK11 siRNA is associated with the inhibition of cell growth (Fig. 2C). SW872 and SW982 cells were also transfected with CDK11 siRNA in a dose-dependent manner. The dose-dependent inhibition of liposarcoma cell growth and survival was observed, accompanied by down-regulated expression of CDK11 protein (Fig. 3).
Fig 2.

Transfection with synthetic CDK11 siRNA decreases cell proliferation in SW872 and SW982 cells. (A) Cell growth and proliferation of SW872 and SW982 was determined by transfection with either nonspecific siRNA or CDK11 siRNA at 40nmol/L concentration. (B) Representative images of liposarcoma cells after transfection of nonspecific siRNA or CDK11 siRNA in 96-well plate. (C) Western blotting confirmed that knockdown of CDK11 by transfection with 40nmol/L siRNA significantly decreased the CDK11protein expression.
Fig 3.

Inhibition of CDK11 expression by dose-dependent siRNA in liposarcoma cell lines.A, results of synthetic siRNA against CDK11 in SW872 and SW982 cell lines.SW872 and SW982 cells were transfected with CDK siRNA in a dose-dependent manner. Proliferation was assessed by MTT as described in the Materials and Methods. C, dose-dependent CDK siRNA down-regulated the expression of CDK11. For Western blot analysis, 25 mg of total cellular proteins was used for immunoblotting with specific antibody to CDK11. Results are expressed as mean ± SD. Data were analyzed by the one-way ANOVA. Different superscripts indicate significant differences between the means as determined by the Tukey post-hoc test. ** P<0.01, *** P<0.001 vs. Cells only; #P<0.05, ##P<0.01, ### P<0.001 vs. Cells + nonspecific siRNA.
To further analyze the long-term effects of CDK11 expression knockdown on liposarcoma cell lines, five lentiviruses carrying different shRNA sequences targeting human CDK11 kinase genes were transfected into SW982 cells. After puromycin selection, lentivirus-transfected cells were collected and total cell numbers were counted. Compared to SW982 cells without puromycin and negative shRNA lentiviral controls, transfection with CDK11 shRNA lentiviruses significantly reduced cell growth and survival in SW982 cells (Fig.4A). Puromycin had little or no effect on negative shRNA lentiviral controls. The results show that CDK11shRNA (TRCN0000006208) inhibited liposarcoma cell growth by 66.7%, whereas the other four CDK11 shRNA lentiviruses significantly decreased cell growth by 90–100% (Fig. 4A).
Fig 4.

Lentiviral shRNA targeting CDK11 in SW982 cell lines. A, representative images of SW982 cells after transfection of lentiviral CDK11 shRNA and puromycin selecting for 30 days in 25 cm2 flask. B, the total cell numbers of after stable transfection of lentiviral CDK11 shRNA. All cells were continuously cultured in medium with puromycin. Note: A, cell only without puromycin; B, cell only with puromycin.
The effect of CDK11 expression on liposarcoma cell growth and the subcellular localization of CDK11 were further evaluated by immunofluorescence assay. Consistent with the results of liposarcoma tissue microarray, the immunofluorescence assay (Fig. 5) also showed that the CDK11 protein was mainly localized in the nucleus of liposarcoma cells. These data are comparable with CDK11 expression in osteosarcoma which also showed nucleus localization in previous studies. The data of CDK11 immunofluorescence analysis further confirmed that compared with cells transfected with nonspecific siRNA, SW872 and SW982 cells expressed much lower levels of CDK11 after being transfected with CDK11 siRNA.
Fig 5.
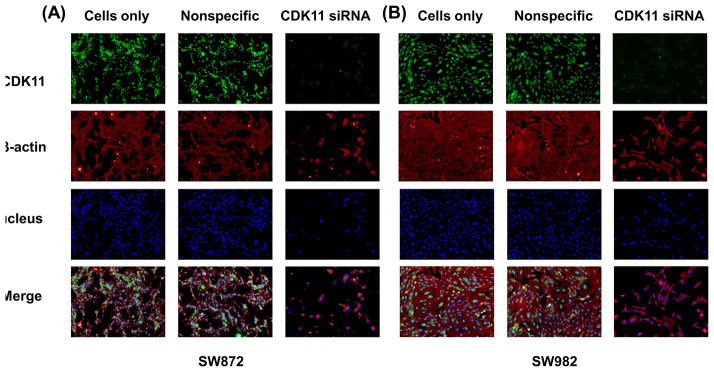
Confirmation of CDK11 knockdown induces cell death and decreases CDK11 expression in different tumor cell lines by immunofluroescence. Expression of CDK11 in SW872 and SW982 cells were assessed by immunofluresecence with antibodies to CDK11 (green) and β-actin (red). Hoechst 33342 was added to counterstain the cell nucleus (blue). Green fluorescence of CDK11 protein was mainly localized in the nucleus of liposarcoma cells.
3.3 CDK11 knockdown may induce apoptosis in liposarcoma cell lines
To investigate how CDK11 inhibited tumor cell growth and survival, western blotting was performed on several apoptotic related proteins (MCL-1, Bcl-XL, Bax, Survivin, Cytochrome C, and Cyclin D1) after CDK11 knockdown in liposarcoma cell lines SW872 and SW982(Fig. 6A). After the cells were transfected with increasing concentrations of CDK11 siRNA, the expression levels of CDK11 decreased, which was not observed with the nonspecific siRNA transfection. Two anti-apoptotic proteins (Cyclin D1 and Survivin) were highly expressed in liposarcoma, and transfection of CDK11 siRNA significantly down-regulated the expression of these anti-apoptotic proteins in both SW872 and SW982 cells (Fig. 6B).In addition, decreased expression of another anti-apoptotic protein Cytochrome C was associated with transfection of CDK11 siRNA in SW872. Thus, these results indicate that knockdown of CDK11 by siRNA could induce cell apoptosis via down-regulation of anti-apoptotic proteins Cyclin D1 and Survivin in liposarcoma cells.
Fig 6.

Synthetic siRNA targeting CDK11 induces apoptosis in liposarcoma cells.SW872 and SW982 cells were transfected with CDK siRNA as dose-dependent manner. CDK11 siRNA decreased CDK11 expression, accompanied with down-regulation of anti-apoptotic proteins expression (A). For Western blot analysis, 25 mg of total cellular proteins was subjected to immunoblotting with specificantibody to CDK11, MCL-1, BcL-XL, Bax, Survivin, Cytochrome C (Cyt C), CyclinD1, and β-actin. Western blots from A were analyzed by Odyssey software 3.0 and normalized to β-actin expression. Quantitative results for protein Cyclin D1 and Survivin were presented as relative expression (B).
3.4 CDK11 knockdown CDK11 knockdown enhances the cytotoxic effect of chemotherapeutic agent in liposarcoma cell lines
High level expression of CDK11 may contribute to the survival advantage of liposarcoma cells, in part through the induction of anti-apoptotic regulatory proteins such as Cyclin D1 and Surivivin. We hypothesized that inhibition of CDK11 pathway in liposarcoma may lower the apoptotic threshold and increase chemotherapy sensitivity. To determine whether CDK11 down-regulation influenced chemotherapeutic agents’ effects on liposarcoma cells, SW872 and SW982 cells were transfected with CDK11 siRNA and then incubated with doxorubicin, a commonly used chemotherapy drug in the treatment of liposarcomas. 5nM and 10nM of CDK11 siRNA were chosen for SW872 and SW982, respectively, which inhibited the growth of liposarcoma cells by about 30% (Fig. 7A and B). Western blotting confirmed decreased expression of CDK11 in liposarcoma cells at these concentrations of CDK11 siRNA (Fig. 7C and 7D). The combination of CDK11 siRNA and doxorubicin significantly inhibited cell growth and survival compared to those from each treatment alone in SW872 and SW982 cell lines (Fig. 7A and B). Thus, our results show that CDK11 knockdown enhances the cytotoxic effect of doxorubicin to inhibit cell growth in liposarcoma cell lines.
Fig 7.

CDK11 knockdown enhances the cytotoxic effect of doxorubicin in SW872 and SW982 cell lines. The 5 and 10 nmol/L of CDK11 siRNA were chosen for SW872 and SW982 respectively, inducing the 30% reduction of cell proliferation in liposarcoma cells (A and B). The CDK11 expression was analyzed by western blotting after tranfection with CDK11siRNA (C and D). Cells were incubated with nonspecific siRNA only, doxorubicin only, nonspecific siRNA + doxorubicin, CDK11 siRNA only, CDK11 siRNA + doxorubicin (A and B). Absorbance was determined at 6 days. Results are expressed as mean ± SD. Data were analyzed by the one-way ANOVA. Different superscripts indicate significant differences between the means as determined by the Tukey post-hoc test. *** P<0.001 vs. Cells only; ## P<0.01, ### P<0.001 vs. Cells + nonspecific siRNA; & P<0.05, &&& P<0.001 vs. Cells + doxorubicin, a P<0.05, aaa P<0.001 vs. nonspecific siRNA + doxorubicin, bb P<0.01 vs. Cells + CDK11 siRNA.
4. Discussion
This study begins by examining the expression of CDK11 in liposarcoma compared with lipoma, a common benign soft tissue tumor composed of adipose tissue. Our study shows little to no expression of CDK11 in lipoma, whereas CDK11 expression is significantly higher in liposarcoma. This suggests that CDK11 may play an important role in the proliferation of liposarcoma. Though there are currently no studies on CDK11 in liposarcoma, the function of CDK4 has been elucidated in liposarcoma [11,29–32]. CDK4, a catalytic subunit of the protein kinase complex, is important for cell cycle G1 phase progression. CDK4 is known to be amplified in well- and dedifferentiated liposarcomas specifically [29,30] and expressed up to 10–fold higher in liposarcoma than in normal fat tissues [31]. These results are consistent with our findings of increased CDK11 expression in liposarcoma. Inhibition of cell proliferation by short hairpin RNA (shRNA)-based knockdown of CDK4 has been reported in liposarcoma cell lines [32]. Our study shows that CDK11 knockdown by synthetic siRNA and lentivirus shRNA inhibited liposarcoma cell growth and survival (Fig. 3 and 4), which implies again that CDK11 plays an important role in liposarcoma cell proliferation and growth. There is currently one CDK4 inhibitor (PD0332991) has exciting results in early phase clinical trials of liposarcoma [11,33]. This has motivated the search for synthetic inhibitors of other highly expressed CDKs in cancer, such as CDK11.
Previous studies have reported that the CDK11 protein localizes to both splicing factor compartments and to the nucleoplasm [26]. We show that the location of CDK11p110 is mainly in the nucleoplasm as measured by immunofluorescence and immunohistochemical staining of CDK11 protein (Fig. 1B and 5). The results are consistent with our previous study of CDK11 in osteosarcoma cells [17].
To elucidate the molecular mechanisms underlying the effects of CDK11 in liposarcoma cells, several apoptotic related proteins were examined after induction of cell death by CDK11 knockdown in liposarcoma cell lines. Our study showed that anti-apoptotic proteins are reduced by CDK11knockdown in liposarcoma cell lines, suggesting that CDK11 is involved in apoptosis signaling. Similarly, the pro-apoptotic effect of synthetic siRNA against CDK11 was reported in osteosarcoma cell lines [17]. Many studies have also found CDK11p58 to be closely associated with apoptosis and cell cycle arrest in vitro and in vivo [34,35], and that CDK11p46 is involved in apoptotic signaling [36]. Our study suggests that CDK11p110 is also involved in apoptosis signaling in liposarcoma.
Another potential mechanism of CDK11 knockdown- induced cell growth inhibition and apoptosis in liposarcoma cells may related to the crucial role of CDK11 involved in the regulation of cellular RNA transcription and processing. CDK11 is part of large-molecular-weight complexes that also contain general pre-mRNA splicing factors, RNA polymerase II, and transcriptional elongation factors [42,43]. CDK11 may therefore couple transcription and pre-mRNA splicing by their effect(s) on certain proteins required for these processes (cell cycle progression and apoptosis) [42,43]. For example, CDK11 interacts with the general pre-mRNA splicing factors RNPS1 and 9G8, RNA polymerase II (RNAP II), CyclinL, casein kinase 2 (CK2) and checkpoint kinase 2 (CHK2)[20, 42,43].
CDK11 knockdown also enhances the cytotoxic effect of chemotherapeutic agent doxorubicin in liposarcoma cell lines, suggesting that combination therapies could be explored in liposarcoma clinical trials. These results are consistent with a recent preclinical and phase I study of the Pan-CDK inhibitor flavopiridol combined with doxorubicin in soft tissue sarcoma [37]. The results of the phase I clinical trial [37] also showed that a combination of fixed dose doxorubicin followed by escalating doses of flavopiridol is well tolerated and there were some patients with durable responses over 12 weeks. Interestingly, encouraging clinical effects can be achieved with adjuvant flavopiridol and doxorubicin treatment for pancreas and breast cancer patients [38]. Similarly, several clinical trials show that treatment with flavopiridol and other chemotherapeutic drugs (FOLFIRI, irinotecan etc.) remain a safe and effective regimen against advanced solid cancers [39–41].
In conclusion, we showed high expression of CDK11 in liposarcoma and that cell growth/proliferation of liposarcoma can be decreased by inhibiting CDK11. This effect was enhanced when CDK11 knockdown was combined with chemotherapeutic agents. These results suggest that CDK11 may be a promising therapeutic target for liposarcoma patients. Future studies on the signaling pathway and molecular mechanisms of CDK11 and cell growth have yet will be required. Elucidating these mechanisms will further help us to understand the functions of CDK11 in liposarcoma and to develop novel strategies for anti-cancer drugs.
Supplementary Material
Acknowledgments
Grant Support
This work was supported, in part, by a grant from the Jeff Guyer Fund. Support has also been provided by the Gattegno and Wechsler funds, the Kenneth Stanton Fund. Dr. Duan is supported, in part, through a grant from Sarcoma Foundation of America (SFA), a grant from Chordoma Foundation, a grant from National Cancer Institute (NCI)/National Institutes of Health (NIH), UO1, CA 151452-01, a pilot grant from Sarcoma SPORE/NIH, and a grant from an Academic Enrichment Fund of MGH Orthopaedics. Dr. Jia is supported by Scholarship from China Scholarship Council.
The authors thank Jacson Shen for assistance with editing the manuscript.
Footnotes
Conflicts of interest
None of the authors has any financial or other interest with regard to the submitted manuscript that might be constructed as a conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Matushansky I, Hernando E, Socci ND, Matos T, Mills J, Edgar MA, Schwartz GK, Singer S, Cordon-Cardo C, Maki RG. A developmental model of sarcomagenesis defines a differentiation-based classification for liposarcomas. The American journal of pathology. 2008;172:1069–1080. doi: 10.2353/ajpath.2008.070284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murphey MD. World Health Organization classification of bone and soft tissue tumors: modifications and implications for radiologists. Seminars in musculoskeletal radiology. 2007;11:201–214. doi: 10.1055/s-2008-1038310. [DOI] [PubMed] [Google Scholar]
- 3.Sleijfer S, Ouali M, van Glabbeke M, Krarup-Hansen A, Rodenhuis S, Le Cesne A, Hogendoorn PC, Verweij J, Blay JY. Prognostic and predictive factors for outcome to first-line ifosfamide-containing chemotherapy for adult patients with advanced soft tissue sarcomas: an exploratory, retrospective analysis on large series from the European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group (EORTC-STBSG) European journal of cancer. 2010;46:72–83. doi: 10.1016/j.ejca.2009.09.022. [DOI] [PubMed] [Google Scholar]
- 4.Van Glabbeke M, van Oosterom AT, Oosterhuis JW, Mouridsen H, Crowther D, Somers R, Verweij J, Santoro A, Buesa J, Tursz T. Prognostic factors for the outcome of chemotherapy in advanced soft tissue sarcoma: an analysis of 2,185 patients treated with anthracycline-containing first-line regimens--a European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1999;17:150–157. doi: 10.1200/JCO.1999.17.1.150. [DOI] [PubMed] [Google Scholar]
- 5.Ghadimi MP, Al-Zaid T, Madewell J, Peng T, Colombo C, Hoffman A, Creighton CJ, Zhang Y, Zhang A, Lazar AJ, Pollock RE, Lev D. Diagnosis, management, and outcome of patients with dedifferentiated liposarcoma systemic metastasis. Annals of surgical oncology. 2011;18:3762–3770. doi: 10.1245/s10434-011-1794-0. [DOI] [PubMed] [Google Scholar]
- 6.Lopes LF, Bacchi CE. Imatinib treatment for gastrointestinal stromal tumour (GIST) Journal of cellular and molecular medicine. 2010;14:42–50. doi: 10.1111/j.1582-4934.2009.00983.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Demicco EG, Torres KE, Ghadimi MP, Colombo C, Bolshakov S, Hoffman A, Peng T, Bovee JV, Wang WL, Lev D, Lazar AJ. Involvement of the PI3K/Akt pathway in myxoid/round cell liposarcoma. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2012;25:212–221. doi: 10.1038/modpathol.2011.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dass CR, Galloway SJ, Clark JC, Khachigian LM, Choong PF. Involvement of c-jun in human liposarcoma growth: supporting data from clinical immunohistochemistry and DNAzyme efficacy. Cancer biology & therapy. 2008;7:1297–1301. doi: 10.4161/cbt.7.8.6301. [DOI] [PubMed] [Google Scholar]
- 9.Snyder EL, Sandstrom DJ, Law K, Fiore C, Sicinska E, Brito J, Bailey D, Fletcher JA, Loda M, Rodig SJ, Dal Cin P, Fletcher CD. c-Jun amplification and overexpression are oncogenic in liposarcoma but not always sufficient to inhibit the adipocytic differentiation programme. The Journal of pathology. 2009;218:292–300. doi: 10.1002/path.2564. [DOI] [PubMed] [Google Scholar]
- 10.Blachly JS, Byrd JC. Emerging Drug Profile: Cyclin-Dependent Kinase Inhibitors. Leukemia & lymphoma. 2013 doi: 10.3109/10428194.2013.783911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dickson MA, Tap WD, Keohan ML, D'Angelo SP, Gounder MM, Antonescu CR, Landa J, Qin LX, Rathbone DD, Condy MM, Ustoyev Y, Crago AM, Singer S, Schwartz GK. Phase II Trial of the CDK4 Inhibitor PD0332991 in Patients With Advanced CDK4-Amplified Well-Differentiated or Dedifferentiated Liposarcoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013 doi: 10.1200/JCO.2012.46.5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Senderowicz AM. Flavopiridol: the first cyclin-dependent kinase inhibitor in human clinical trials. Investigational new drugs. 1999;17:313–320. doi: 10.1023/a:1006353008903. [DOI] [PubMed] [Google Scholar]
- 13.Ramaswamy B, Phelps MA, Baiocchi R, Bekaii-Saab T, Ni W, Lai JP, Wolfson A, Lustberg ME, Wei L, Wilkins D, Campbell A, Arbogast D, Doyle A, Byrd JC, Grever MR, Shah MH. A dose-finding, pharmacokinetic and pharmacodynamic study of a novel schedule of flavopiridol in patients with advanced solid tumors. Investigational new drugs. 2012;30:629–638. doi: 10.1007/s10637-010-9563-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whitlock JA, Krailo M, Reid JM, Ruben SL, Ames MM, Owen W, Reaman G S. Children's Oncology Group. Phase I clinical and pharmacokinetic study of flavopiridol in children with refractory solid tumors: a Children's Oncology Group Study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005;23:9179–9186. doi: 10.1200/JCO.2004.01.0660. [DOI] [PubMed] [Google Scholar]
- 15.Darpolor MM, Kennealey PT, Le HC, Zakian KL, Ackerstaff E, Rizwan A, Chen JH, Sambol EB, Schwartz GK, Singer S, Koutcher JA. Preclinical study of treatment response in HCT-116 cells and xenografts with (1) H-decoupled (31) P MRS. NMR in biomedicine. 2011;24:1159–1168. doi: 10.1002/nbm.1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Tanaka K, Li X, Okada T, Nakamura T, Takasaki M, Yamamoto S, Oda Y, Tsuneyoshi M, Iwamoto Y. Cyclin-dependent kinase inhibitor, flavopiridol, induces apoptosis and inhibits tumor growth in drug-resistant osteosarcoma and Ewing's family tumor cells. International journal of cancer. Journal international du cancer. 2007;121:1212–1218. doi: 10.1002/ijc.22820. [DOI] [PubMed] [Google Scholar]
- 17.Duan Z, Zhang J, Choy E, Harmon D, Liu X, Nielsen P, Mankin H, Gray NS, Hornicek FJ. Systematic kinome shRNA screening identifies CDK11 (PITSLRE) kinase expression is critical for osteosarcoma cell growth and proliferation. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:4580–4588. doi: 10.1158/1078-0432.CCR-12-1157. [DOI] [PubMed] [Google Scholar]
- 18.Shi J, Feng Y, Goulet AC, Vaillancourt RR, Sachs NA, Hershey JW, Nelson MA. The p34cdc2-related cyclin-dependent kinase 11 interacts with the p47 subunit of eukaryotic initiation factor 3 during apoptosis. The Journal of biological chemistry. 2003;278:5062–5071. doi: 10.1074/jbc.M206427200. [DOI] [PubMed] [Google Scholar]
- 19.Sachs NA, Vaillancourt RR. Cyclin-dependent kinase 11p110 and casein kinase 2 (CK2) inhibit the interaction between tyrosine hydroxylase and 14-3-3. Journal of neurochemistry. 2004;88:51–62. doi: 10.1046/j.1471-4159.2003.02119.x. [DOI] [PubMed] [Google Scholar]
- 20.Choi HH, Choi HK, Jung SY, Hyle J, Kim BJ, Yoon K, Cho EJ, Youn HD, Lahti JM, Qin J, Kim ST. CHK2 kinase promotes pre-mRNA splicing via phosphorylating CDK11(p110) Oncogene. 2012 doi: 10.1038/onc.2012.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu X, Cheng C, Shao B, Wu X, Ji Y, Lu X, Shen A. The functional interaction between CDK11p58 and beta-1,4-galactosyltransferase I involved in astrocyte activation caused by lipopolysaccharide. Inflammation. 2012;35:1365–1377. doi: 10.1007/s10753-012-9450-9. [DOI] [PubMed] [Google Scholar]
- 22.Petretti C, Savoian M, Montembault E, Glover DM, Prigent C, Giet R. The PITSLRE/CDK11p58 protein kinase promotes centrosome maturation and bipolar spindle formation. EMBO reports. 2006;7:418–424. doi: 10.1038/sj.embor.7400639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hao Y, Kong X, Ruan Y, Gan H, Chen H, Zhang C, Ren S, Gu J. CDK11p46 and RPS8 associate with each other and suppress translation in a synergistic manner. Biochemical and biophysical research communications. 2011;407:169–174. doi: 10.1016/j.bbrc.2011.02.132. [DOI] [PubMed] [Google Scholar]
- 24.Hu D, Mayeda A, Trembley JH, Lahti JM, Kidd VJ. CDK11 complexes promote pre-mRNA splicing. The Journal of biological chemistry. 2003;278:8623–8629. doi: 10.1074/jbc.M210057200. [DOI] [PubMed] [Google Scholar]
- 25.Trembley JH, Hu D, Hsu LC, Yeung CY, Slaughter C, Lahti JM, Kidd VJ. PITSLRE p110 protein kinases associate with transcription complexes and affect their activity. The Journal of biological chemistry. 2002;277:2589–2596. doi: 10.1074/jbc.M109755200. [DOI] [PubMed] [Google Scholar]
- 26.Loyer P, Trembley JH, Katona R, Kidd VJ, Lahti JM. Role of CDK/cyclin complexes in transcription and RNA splicing. Cellular signalling. 2005;17:1033–1051. doi: 10.1016/j.cellsig.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 27.Xiang J, Lahti JM, Grenet J, Easton J, Kidd VJ. Molecular cloning and expression of alternatively spliced PITSLRE protein kinase isoforms. The Journal of biological chemistry. 1994;269:15786–15794. [PubMed] [Google Scholar]
- 28.Yang W, Liu X, Choy E, Mankin H, Hornicek FJ, Duan Z. Targeting hedgehog-GLI-2 pathway in osteosarcoma. Journal of orthopaedic research : official publication of the Orthopaedic Research Society. 2013;31:502–509. doi: 10.1002/jor.22230. [DOI] [PubMed] [Google Scholar]
- 29.Binh MB, Sastre-Garau X, Guillou L, de Pinieux G, Terrier P, Lagace R, Aurias A, Hostein I, Coindre JM. MDM2 and CDK4 immunostainings are useful adjuncts in diagnosing well-differentiated and dedifferentiated liposarcoma subtypes: a comparative analysis of 559 soft tissue neoplasms with genetic data. The American journal of surgical pathology. 2005;29:1340–1347. doi: 10.1097/01.pas.0000170343.09562.39. [DOI] [PubMed] [Google Scholar]
- 30.Aleixo PB, Hartmann AA, Menezes IC, Meurer RT, Oliveira AM. Can MDM2 and CDK4 make the diagnosis of well differentiated/dedifferentiated liposarcoma? An immunohistochemical study on 129 soft tissue tumours. Journal of clinical pathology. 2009;62:1127–1135. doi: 10.1136/jcp.2009.070201. [DOI] [PubMed] [Google Scholar]
- 31.Singer S, Socci ND, Ambrosini G, Sambol E, Decarolis P, Wu Y, O'Connor R, Maki R, Viale A, Sander C, Schwartz GK, Antonescu CR. Gene expression profiling of liposarcoma identifies distinct biological types/subtypes and potential therapeutic targets in well-differentiated and dedifferentiated liposarcoma. Cancer research. 2007;67:6626–6636. doi: 10.1158/0008-5472.CAN-07-0584. [DOI] [PubMed] [Google Scholar]
- 32.Barretina J, Taylor BS, Banerji S, Ramos AH, Lagos-Quintana M, Decarolis PL, Shah K, Socci ND, Weir BA, Ho A, Chiang DY, Reva B, Mermel CH, Getz G, Antipin Y, Beroukhim R, Major JE, Hatton C, Nicoletti R, Hanna M, Sharpe T, Fennell TJ, Cibulskis K, Onofrio RC, Saito T, Shukla N, Lau C, Nelander S, Silver SJ, Sougnez C, Viale A, Winckler W, Maki RG, Garraway LA, Lash A, Greulich H, Root DE, Sellers WR, Schwartz GK, Antonescu CR, Lander ES, Varmus HE, Ladanyi M, Sander C, Meyerson M, Singer S. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nature genetics. 2010;42:715–721. doi: 10.1038/ng.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwartz GK, LoRusso PM, Dickson MA, Randolph SS, Shaik MN, Wilner KD, Courtney R, O'Dwyer PJ. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1) British journal of cancer. 2011;104:1862–1868. doi: 10.1038/bjc.2011.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lahti JM, Xiang J, Heath LS, Campana D, Kidd VJ. PITSLRE protein kinase activity is associated with apoptosis. Molecular and cellular biology. 1995;15:1–11. doi: 10.1128/mcb.15.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li T, Inoue A, Lahti JM, Kidd VJ. Failure to proliferate and mitotic arrest of CDK11(p110/p58)-null mutant mice at the blastocyst stage of embryonic cell development. Molecular and cellular biology. 2004;24:3188–3197. doi: 10.1128/MCB.24.8.3188-3197.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mikolajczyk M, Nelson MA. Regulation of stability of cyclin-dependent kinase CDK11p110 and a caspase-processed form, CDK11p46, by Hsp90. The Biochemical journal. 2004;384:461–467. doi: 10.1042/BJ20040848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luke JJ, D'Adamo DR, Dickson MA, Keohan ML, Carvajal RD, Maki RG, de Stanchina E, Musi E, Singer S, Schwartz GK. The cyclin-dependent kinase inhibitor flavopiridol potentiates doxorubicin efficacy in advanced sarcomas: preclinical investigations and results of a phase I dose-escalation clinical trial. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:2638–2647. doi: 10.1158/1078-0432.CCR-11-3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fornier MN, Rathkopf D, Shah M, Patil S, O'Reilly E, Tse AN, Hudis C, Lefkowitz R, Kelsen DP, Schwartz GK. Phase I dose-finding study of weekly docetaxel followed by flavopiridol for patients with advanced solid tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13:5841–5846. doi: 10.1158/1078-0432.CCR-07-1218. [DOI] [PubMed] [Google Scholar]
- 39.Dickson MA, Shah MA, Rathkopf D, Tse A, Carvajal RD, Wu N, Lefkowitz RA, Gonen M, Cane LM, Dials HJ, Schwartz GK. A phase I clinical trial of FOLFIRI in combination with the pan-cyclin-dependent kinase (CDK) inhibitor flavopiridol. Cancer chemotherapy and pharmacology. 2010;66:1113–1121. doi: 10.1007/s00280-010-1269-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rathkopf D, Dickson MA, Feldman DR, Carvajal RD, Shah MA, Wu N, Lefkowitz R, Gonen M, Cane LM, Dials HJ, Winkelmann JL, Bosl GJ, Schwartz GK. Phase I study of flavopiridol with oxaliplatin and fluorouracil/leucovorin in advanced solid tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:7405–7411. doi: 10.1158/1078-0432.CCR-09-1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shah MA, Kortmansky J, Motwani M, Drobnjak M, Gonen M, Yi S, Weyerbacher A, Cordon-Cardo C, Lefkowitz R, Brenner B, O'Reilly E, Saltz L, Tong W, Kelsen DP, Schwartz GK. A phase I clinical trial of the sequential combination of irinotecan followed by flavopiridol. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11:3836–3845. doi: 10.1158/1078-0432.CCR-04-2651. [DOI] [PubMed] [Google Scholar]
- 42.Drogat J, Migeot V, Mommaerts E, Mullier C, Dieu M, van Bakel H, Hermand D. Cdk11-cyclinL controls the assembly of the RNA polymerase II mediator complex. Cell reports. 2012;2:1068–1076. doi: 10.1016/j.celrep.2012.09.027. [DOI] [PubMed] [Google Scholar]
- 43.Loyer P, Trembley JH, Grenet JA, Busson A, Corlu A, Zhao W, Kocak M, Kidd VJ, Lahti JM. Characterization of cyclin L1 and L2 interactions with CDK11 and splicing factors: influence of cyclin L isoforms on splice site selection. The Journal of biological chemistry. 2008;283:7721–7732. doi: 10.1074/jbc.M708188200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.