

Abstract
Current dogma suggests that chronically demyelinated axons are at risk for degeneration, with axonal loss resulting in permanent disability in myelin disease. However, the trophic role of the myelin sheath in long-term axonal survival is incompletely understood. Previous observations of the effect of dysmyelination or demyelination on axonal survival in the myelin mutants has been limited because of their short life span. In this study, we used the Long–Evans shaker (les) rat, which can live up to 9 months, to study axonal health and survival after chronic demyelination. At 2 weeks, ∼29% of medium and ∼47% of large fiber axons are myelinated in les spinal cord. However, by 3 months, no medium and ∼<1% of large-diameter axons retain myelin. After demyelination, axons have a reduced-caliber, abnormal neurofilament distribution and an increase in mitochondrial number. However, there are no signs of axonal degeneration in les rats up to 9 months. Instead, there is a profound increase in oligodendrocytes, which were found to express BDNF, NT-3, and IGF-1. Importantly, this study provides in vivo evidence that mature glial cells produce various neurotrophic factors that may aid in the survival of axons after chronic demyelination.
Introduction
In multiple sclerosis (MS), a demyelinating disorder of the CNS, axonal loss correlates with functional impairments (De Stefano et al., 2001; Tallantyre et al., 2010) and is responsible for permanent disability (Ferguson et al., 1997). Axonal loss occurs in the early as well as the late-chronic phase of MS (De Stefano et al., 2001; Schirmer et al., 2011). Axonal degeneration during the chronic, noninflammatory phase of MS is thought to result from a loss of trophic support (Bjartmar et al., 2003; Hagemeier et al., 2012). However, the role myelin plays in the long-term survival of axons is not fully understood.
Axonal abnormalities occur in animal models of dysmyelination. For example, in the shiverer (shi) mouse, an MBP mutant that lacks compact CNS myelin (Rosenbluth, 1980; Roach et al., 1985), axons have increased slow axonal transport, an immature cytoskeleton composition (Brady et al., 1999), and metabolic abnormalities (Andrews et al., 2006). Despite these abnormalities, there is no axonal loss reported in shi (Rosenbluth, 1980; Inoue et al., 1981; Nixon, 1982; Griffiths et al., 1998). However, the short life span of shi mice (∼100 d) greatly limits these findings, although it has been predicted that shi axons may degenerate at later time points (Andrews et al., 2006). In the taiep rat, a model of demyelination, which can live up to 18–20 months (Duncan et al., 1992), axonal pathology and degeneration occur after chronic CNS demyelination (Wilkins et al., 2010). However, only small-diameter axons are affected (Song et al., 2001), and oligodendrocyte dysfunction may also play a role in degeneration (Song et al., 2003). Axonal degeneration can also be independent of myelin loss (Nave, 2010a). In mice deficient in proteolipid protein (PLP) or 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNP), axons degenerate in the presence of compact myelin (Griffiths et al., 1998; Garbern et al., 2002; Lappe-Siefke et al., 2003). Therefore, longer-lived models of demyelination are required to fully elucidate the role of the myelin sheath in axonal development and survival.
The Long–Evans shaker (les) rat (Delaney et al., 1995) lacks CNS myelin beyond early development because of a mutation in the MBP gene (O'Connor et al., 1999) and can live up to 9 months, making it an ideal model to study axonal development, integrity, and survival after chronic dysmyelination. Here, we show that, although les axons have reduced axon caliber, abnormal neurofilament distribution, and increased mitochondria after dysmyelination, there are no signs of axonal degeneration in les spinal cords up to 9 months. Instead, there is an increase in les oligodendrocytes, which were found to express neurotrophin-3 (NT-3), brain derived neurotrophic factor (BDNF), and insulin-like growth factor 1 (IGF-1) that likely aid in axonal survival after chronic demyelination. Overall, these results show that abnormal myelin development and loss alone do not directly lead to degeneration in this model. Moreover, we provide direct in vivo evidence that mature glial cells produce various trophic factors that may aid in axonal survival after myelin loss.
Materials and Methods
Experimental animals.
Control and les rats were housed and maintained according to the guidelines of the University of Wisconsin-Madison School of Veterinary Medicine. For this study, rats of either sex were killed at 2, 3, 4, and 8 weeks as well as 3, 6, and 9 months of age. Control and les rats were generated by breeding carrier pairs and identified at postnatal day 5 by the detection of the mutated MBP sequence using PCR as previously described (O'Connor et al., 1999). Control and les animals were housed in identical conditions, except with modifications found to enhance the survival of the mutant animals. les rats develop whole-body tremors at 2 weeks of age followed by seizures at 1 month. Some les rats can also develop progressive ataxia, hindlimb paresis, and incontinence (Delaney et al., 1995). Therefore, les rats were weaned a week later than control littermates to ensure proper growth and development. Once weaned, les rats were given a gelatin block of rodent diet supplemented with phenobarbital (0.175 g of phenobarbitol, 4 g of sugar, 4 g of powdered milk, 61 g of ground Harlan rodent diet) 3 times per week to control seizures. In addition, apples were placed on the floor of the cage to aid in caloric intake and hydration.
EM.
For EM, control and les rats were perfused with PBS (0.1 mm PBS, pH 7.4) followed by weak (1% PFA and 1.25% gluteraldehyde, pH 7.3) and strong (4% PFA and 5% gluteraldehyde. pH 7.3) Karnovsky's fixative or perfused with 4% PFA followed by immersion fixation in buffered 2.5% gluteraldehyde. For resin embedding, tissue was washed in phosphate buffer (0.1 mm PB) followed by fixation in 1% osmium tetraoxide. Tissue was dehydrated in a series of ethanol concentrations, and propylene oxide was used as a transitional fluid for resin infiltration. Ultrathin sections were mounted on copper grids (Electron Microscopy Sciences) and stained with uranyl acetate followed by lead citrate. Images were captured on a Hitachi 7600 transmission electron microscope housed at the Wisconsin Veterinary Diagnostics Laboratory at University of Wisconsin-Madison. Approximately 10 images were taken of each target area at an original magnification of 2500.
Immunohistochemistry.
Control and les rats were perfused with 0.1 mm PBS followed by paraformaldehyde buffered in 0.1 mm PB (4% PFA). After dissection, tissue was postfixed overnight in 4% PFA followed by sucrose cryoprotection. Tissue was cut into 20 μm free-floating sections and incubated with the following antibodies: rabbit anti-GST-π (1:30,000, MBL), mouse anti-GFAP (1:30,000, Millipore), chicken antiβ-III tubulin (1:10,000, Millipore), rabbit anti-APP (1:1000, Enzo Life Sciences), mouse anti-SMI-32 (1:1000, Covance), and rabbit anti-NF-H (1:1000, Millipore Bioscience Research Reagents). Immunolabeling was performed according to standard protocols using AlexaFlour secondary antibodies (1:2000, Invitrogen) to visualize protein expression. Images were taken on a Nikon E800 microscope equipped with a SPOT RT digital camera (Diagnostic Instruments) using MetaMorph software. Integrated densities as a relative measure of expression were calculated using ImageJ software.
In situ probe design.
Probes were designed using mRNA sequences from NCBI for IGF-1, BDNF, and NT-3. Primers were designed using Pimer3 software (Rozen and Skaletsky, 2000) within exon sequences identified using Ensembl (Hubbard et al., 2007). Primers are as follows: IGF-1 (left, TTAAGCCGCTTCCTTCACAG; right, TGCTTTGCTTTCTCCTTTCTC), BDNF (left, GGAAGCCTGAATGAATGGAC; right, CTGCCAACAAGAGACCACAG), and NT-3 (left, CCGATGATTGCAACAGACAC; right, TTGACAAGGCACACACACAG). A 5 bp leader sequence and a T7 polymerase recognition site were added to the right primer of each set: CGATG TTAATACGACTCACTATAGGG. The DNA template for each probe was amplified from genomic DNA with PCR. The appropriate band was excised from the gel using a QIAquick gel extraction kit (QIAGEN). Antisense, dioxygenin-labeled probes were generated using the dioxygenin RNA labeling kit with T7 polymerase (Roche).
In situ hybridization.
For in situ hybridization, 20 μm thoracic spinal sections were mounted on Superfrost Plus slides (Fisher) and allowed to dry. Tissue sections were washed in PBS with 0.1% Tween 20 followed by incubation in proteinase K for 6 min at room temperature. Tissue sections were then postfixed in 4% PFA, washed in PBS with 0.1% Tween 20, and incubated in acetylation solution (0.1 m triethanolamine, 0.078 m HCl, 0.25% acetic anhydride) for 10 min at room temperature. After acetylation, sections were then incubated in hybridization buffer (50% formamide, 5× SSC, 10 μg/ml tRNA, 10 μg/ml heparin, HB) for 3 h at 60.5°C and incubated with probe diluted (0.2 μg of probe/ml) in HB overnight at 60.5°C. The following day, tissue sections were washed with wash solution 1 (50% formamide, 5× SSC, 1% SDS) for 3×15 min at 60.5°C followed by a 1:1 mix with solution 2 (10 mm Tris-HCl, pH 7.5, 0.5 m NaCl, 0.1% Tween 20, 0.2 mm sodium azide) for 10 min 60.5°C. Slides were then washed in solution two for 3×5 min at room temperature followed by RNase treatment at 37°C to remove unspecific binding. The tissue sections were again washed with solution 2 1×5 min followed by solution 3 (2× SSC, 50% formamide) 2×20 min at 60.5°C. Blocking was performed with absorption buffer (1×TBS, 10% sheep serum, 1% blocking reagent, 1% BSA) for 1 h at room temperature followed by overnight incubation in antidioxygenin antibody diluted in absorption buffer (1:200). The following day, tissue was washed with Tris-buffered saline with 0.1% Tween plus 2 mm levamisole 6×5 min. Probe detection was performed by incubating sections in 50% BM Purple (Roche) in NTMT + levamisole (100 mm Tris HCl, pH 9.5, 100 mm 5 m NaCl, 50 mm MgCl2 0.2 mm sodium azide, 0.1% Tween 20, 2 mm levamisole) for up to 3 d until the color reaction was complete. After color detection, slides were washed with NTMT + levamisole at room temperature and mounted using 80% glycerol and PBS. For double labeling, immunohistochemistry was performed as described above after the in situ color reaction.
Protein extraction and Western blot.
Control and les rats (n = 3 control and les for each time point) were perfused with cold PBS. After perfusion, the spinal cord and optic nerve were dissected and immediately flash frozen in liquid nitrogen. The optic nerve was used as a more pure measure of glial cell expression. Protein extraction was performed using radio-immunoprecipitation assay buffer (20 mm Tris, 150 mm NaCl, 0.1% SDS, 1% NP40, and protease inhibiter complete; Roche). Protein samples were quantified using the Quant-iT protein detection kit and Qubit flourometer according to manufacturer's protocols (Invitrogen). A total of 20 μg of each sample was run on a 12% acrylimide gel and blotted onto Immobolin-P membranes (Millipore) using the Mini-PROTEAN electrophoresis system (Bio-Rad). After immunoblotting, membranes were blocked for 1 h in 5% BSA in 20 mm Tris-HCl buffer (TBS-T) (150 mm HCl and 0.05% Tween 20, pH 7.6). After blocking, membranes were incubated with primary antibodies: mouse anti-BDNF (1:1000, Millipore), mouse anti-IGF (1:1000, Millipore), and rabbit anti-NT-3 (1:1000, Millipore). The membranes were reprobed with β-actin (1:20,000, Sigma) to ensure loading consistency. The integrated density of each band was measured using ImageJ software. Values are expressed as relative integrated densities normalized to β-actin and averaged over three animals per time point.
Quantification.
Axon diameter, myelin thickness, axonal density, and mitochondrial number were quantified from electron micrographs of les and control spinal cord using Bioquant image analysis software. Approximately 3–5 images (∼500 axons) were measured for each animal (n = 3 for les and control animals) in the dorsal and ventral column of the thoracic spinal cord. Images were taken in the dorsal column in the gracile fasciculus and in the ventral column in the spinothalamic tract. Both areas were chosen based on the observation that each area had a variety of axon sizes. For the specific analysis of axon populations over time, axons were divided into three categories based on axon diameter: small (≤2.5 μm), medium (2.5–5 μm), and large (>5 μm). The total number of axons was reported as a proportion to account for the varying number of axons counted at each time point. Axonal density was calculated using the following calculation: total axon area/(total image area − total myelin area). Using this calculation, the differences in myelin sheath thickness for control and les animals were controlled. Axonal density was measured for each image and averaged across time points.
Statistical analysis.
An ANOVA was performed on each dataset followed by Bonferroni post hoc tests between time points using GraphPad Prism 4 software unless otherwise stated. The error bars in subsequent figures represent SEM. p values <0.05 were deemed significant.
Results
les lack myelin beyond early development
Electron micrographs of spinal cord sections demonstrated that les rats develop uncompact membrane sheaths during peak myelin development at 2 weeks as an attempt at myelination in the absence of MBP (Fig. 1B, inset). However, most of this myelin was lost by 4 weeks, and by 8 weeks little myelin was present within the spinal cord of les rats (Fig. 1A). Although this has been previously reported, the total myelin development and loss have not been quantified. To quantify myelin development and loss in les, axons were grouped by diameter into small (≤2.5 μm), medium (2.5–5 μm), and large (>5 μm) for analysis. Images were sampled in the dorsal and ventral columns for each animal. In control animals at 2 weeks, all (100 ± 0%) large-diameter axons, 77 ± 3% of medium-caliber axons, and 12 ± 3% of small-fiber axons were myelinated. In addition, the proportion of myelinated medium and small-fiber axons increased with age. In contrast, at 2 weeks 47 ± 13% of large, 28 ± 9% of medium, and no small fibers were myelinated within the spinal cord of les rats. However, at 8 weeks no medium and only 6 ± 3% of large axons retain myelin. By 3 months, <1% of large diameter axons were myelinated in les spinal cords (Fig. 1B). Therefore, the les mutant is an excellent long-lived model of chronic demyelination as les rats lack myelin beyond the initial stages of development.
Figure 1.

A, Electron micrographs of the spinal cord from 2 week, 4 week, 3 month, and 9 month control and les animals. At 2 weeks, control rats begin to develop myelin sheaths that persist as the animal ages (a–d). In contrast, at 2 weeks, les animals develop thin, uncompact myelin sheaths (e). However, most of this attempt at myelination is lost by 4 weeks (f), and at later ages there is practically no myelin in the CNS (g–h). Scale bar, 1 μm. B, Enlarged inset illustrating an example of a myelinated axon in les. Although it is uncompact, the membrane sheaths in les form an organized multilamellar structure that is characteristic of a myelin sheath. *Glial processes. Scale bar, 1 μm. The total proportion of myelinated axons were grouped by diameter into large (>5 μm; triangle), medium (2.5–5 μm; square), and small axons (≤2.5 μm; diamond) and graphed over time in les (gray) and control (black) spinal cord sections (C). All of the large-diameter axons in control spinal cords are myelinated by 2 weeks, whereas 77 ± 3% of medium and 12 ± 3% of small fibers are myelinated. In les, 43 ± 13% of large, 28 ± 9% of medium, and no small fibers are myelinated. By 3 months, <1% of large-diameter axons retain myelin in les. Error bars indicate SEM; n = 3 per time point.
Neurofilament distribution and mitochondrial number are altered in les axons
To determine whether loss of myelin had an impact on axon diameter, the total proportion of large (>5 μm), medium (2.5–5 μm), and small (≤2.5 μm) axons was quantified over time. In control animals, the number (proportion) of small diameter axons slightly decreased over time, whereas the number of medium- and large-diameter axons increased as the animal aged (Fig. 2). This trend was also observed in the les mutant. However, there was a significant increase in the proportion of small-diameter axons between 4 and 8 weeks in les compared with control animals (p < 0.01 and p < 0.001, respectively). In addition, there was a significant decrease in the proportion of medium-diameter axons between 4 and 8 weeks (p < 0.01 and p < 0.001, respectively) and large-diameter axons at 8 weeks (p < 0.01). Although there were initial differences in axonal caliber in les, from 3 to 9 months les animals had the same proportion of large-, medium-, and small-diameter axons compared with control animals (Fig. 2).
Figure 2.

The total proportion of small-diameter (≤2.5 μm; diamond), medium-diameter (2.5–5 μm; square), and large-diameter (>5 μm; triangle) axons in les (gray) and control (black) spinal cords was graphed from 2 weeks until 9 months. Between 4 and 8 weeks, there is a significant increase in the proportion of medium- and small-diameter axons. In addition, there is a significant decrease in large-diameter axons at 8 weeks. From 3 to 9 months, there is no difference in the proportion of axons grouped by diameter. Error bars indicate SEM; n = 3 per time point. *p < 0.01, compared with control of the same axon size and time point (two-way ANOVA followed by Bonferroni post hoc test). **p < 0.001, compared with control of the same axon size and time point (two-way ANOVA followed by Bonferroni post hoc test).
To determine whether les axons are structurally normal, immunohistochemical staining was performed for neurofilament heavy chain (NF-H) and dephosphorylated neurofilament (SMI-32). At early ages (2–4 weeks), the distribution of NF-H and SMI-32 was found to be similar between les and control axons. However, beginning at 3 months, there was an abnormal increase of SMI-32 expression, whereas NF-H expression was reduced in les axons compared with controls (Fig. 3).
Figure 3.
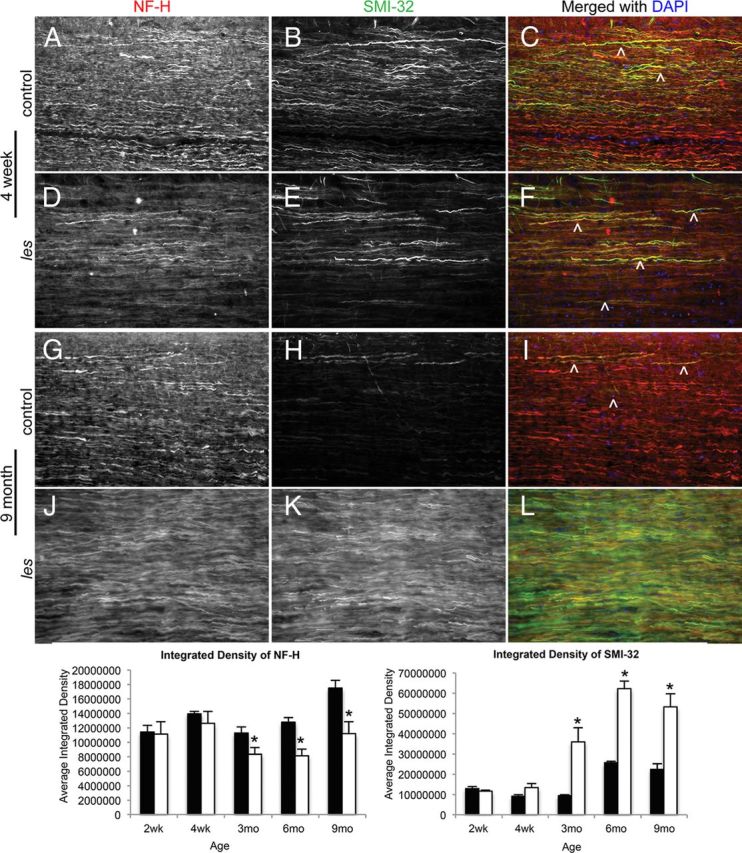
Representative images of control and les longitudinal spinal cord sections at 3 and 9 months immunostained for NF-H (red) and SMI-32 (green). A–C, H–I, In control sections, there is high NF-H expression and very little SMI-32 within axons. D–F, J–L, Between 3 and 9 months, les axons express less NF-H, whereas there is an increase in SMI-32 expression. ∧Examples of axons positive for SMI-32. Original magnifications 20×. The graphs below represent the relative integrated densities of NF-H and SMI-32 in les (white) compared with control (black). From 3 months to 9 months, there is a significant decrease in the relative expression of NF-H and a significant increase in SMI-32 in les. Error bars indicate SEM. *p < 0.05, les and control at each time point (n = 3) (two-tailed Student's t test).
It has been hypothesized that chronically demyelinated axons have an increased energy need to maintain conduction velocity in the absence of myelin (Black et al., 1990; Andrews et al., 2006). Thus, mitochondrial activity increases within demyelinated axons to fulfill this energy need. In electron micrographs of les spinal cord, it was observed that larger-diameter axons appear to have a greater number of mitochondria compared with control axons (Fig. 4A). Therefore, the total number of mitochondria was quantified for each axon and averaged across various axon sizes. It was found that there was a significant increase in mitochondria number in large-diameter axons from 4 weeks until 9 months and in medium-diameter axons from 8 weeks until 9 months (Fig. 4B).
Figure 4.

Representative electron micrographs of 9 month control and les ventral spinal cord. A, B, There is a noticeable increase in the number of mitochondria (+) in larger-diameter axons in les (A) compared with control (B). In addition, there is also an obvious increase in glial cell processes in les (*). The average number of mitochondria per axon in les and control was quantified and graphed across time points. The number of mitochondria at each time point is grouped by axon diameter (small: ≤ 2.5 μm; medium: 2.5–5 μm; and large: > 5 μm). From 4 weeks to 9 months, the number of mitochondria in large-diameter axons is significantly higher in les. In addition, the average number of mitochondria in medium axons is significantly higher from 8 weeks until 9 months in les. Error bars indicate SEM; n = 3 for each time point. *p < 0.05, compared with control of the same axon size (two-way ANOVA followed by Bonferroni post hoc test). **p < 0.01, compared with control of the same axon size (two-way ANOVA followed by Bonferroni post hoc test). ***p < 0.001, compared with control of the same axon size (two-way ANOVA followed by Bonferroni post hoc test).
Axons do not degenerate after demyelination despite abnormalities
Whole spinal cord grids were scanned for signs of axonal degeneration. However, no axonal swellings or spheroids were observed in les spinal cord sections. To confirm the lack of axonal degeneration in les rats, double immunolabeling for amyloid precursor protein (APP) and β III tubulin was performed on control and les spinal cord sections. In les axons, there was not an increase in APP expression compared with controls as late as 9 months (Fig. 5).
Figure 5.

Representative images of spinal cord sections from 9 month control and les animals. A–F, Sections are immunostained for β-III tubulin (red), APP (green), and DAPI (blue). In control spinal cord sections, APP is not highly expressed in β-III-positive axons. G–L, Although les lacks myelin by 3 months, there is no increase in APP up to 9 months as illustrated by both transverse (G–I) and longitudinal (J–L) images of les spinal cord. Original magnification 20 ×.
To determine whether les rats lose axons after demyelination, the axonal density was calculated from electron micrographs of les and control spinal cords. There was no difference in axonal density between controls and les animals at early ages (2–8 weeks). Interestingly, there was a significant increase in axonal density in les between 3 and 9 months (Fig. 6).
Figure 6.

The average axonal density of control and les electron micrographs was graphed from 2 weeks to 9 months. Quantification illustrates that there is no significant difference between the average axonal density of les spinal cord images (white) compared with controls (black) between 2 and 8 weeks. However, there is a significant increase in axonal density between 3 and 9 months in les. Error bars indicate SEM; n = 3 at each time point. *p < 0.05, compared with control at the same time point (two-way ANOVA followed by Bonferroni post hoc test). ***p < 0.001, compared with control at the same time point (two-way ANOVA followed by Bonferroni post hoc test).
Glial cells express neurotrophic factors that may aid in axonal survival
To determine whether les spinal cords exhibit signs of gliosis, immunohistochemistry was performed for oligodendrocyte marker, GST-π, and astrocyte marker, GFAP. There was an obvious increase in the expression of GFAP staining, which is probably the result of an increase in astrocyte processes. In addition, there is an increase in the number of oligodendrocytes beginning at 4 weeks in les spinal cord, and by 3 months there was a marked gliosis in the spinal cord of les mutants (Fig. 7).
Figure 7.

A–P, Representative images of immunohistochemistry for GFAP and GST-π in the ventral column of 2 week, 4 week, 3 month, and 9 month control (A–H) and les (I–P) spinal cord sections. At 4 weeks, there is an increase in glial cells in les spinal cord sections (K, L) compared with controls (C, D). By 3 months, there is a marked gliosis in les (M, N) up to 9 months (O, P). Images were taken at the same level in the thoracic spinal cord in the ventral column. Original magnification ×20. Graph of the total oligodendrocyte count (GST-π-positive cells) of whole thoracic spinal cord sections from les and control animals over time (below). les spinal cord sections have significantly more GST-π-positive oligodendrocytes than controls from 3 weeks until 9 months. Error bars indicate SEM. *p < 0.05, les versus control animals at each time point (n = 3) (two-tailed Student's t test).
In addition to producing myelin, glial cells are also thought to have a trophic role in axonal survival. Therefore, to test whether an increase in glial cells may be beneficial to les axons, in situ hybridizations were performed using antisense probes against IGF-1, BDNF, and NT-3 in les and control spinal cord sections. An increase in the number of cells expressing these neurotrophic factors was found within the white matter of les spinal cords after demyelination (Fig. 8A). In addition, immunolabeling with GST-π and GFAP illustrates that these neurotrophic factors localized within les oligodendrocyte and astrocyte processes (Fig. 8B). Western blot was used to measure the total amounts of each protein in tissue homogenates from optic nerve and spinal cords of 3 month control and les rats. Optic nerves were used to represent a higher proportion of glial cells without neuronal cell bodies. In les optic nerves, there was a significant increase in the amount of BDNF (p < 0.03), IGF-1 (p < 0.04), and NT-3 (p < 0.05) (Fig. 9). In addition, there was an increase in neurotrophic factors in les spinal cord homogenates compared with controls for NT-3 (p < 0.05) and BDNF and IGF-1 that trended toward significance (p < 0.06 and p < 0.1, respectively).
Figure 8.

Representative images of control and les ventral spinal cord in situ hybridizations for NT-3, BDNF, and IGF-1 at 3 and 9 months. From 3 to 9 months, les spinal cords show an increased expression of mRNA for NT-3 (B, D), BDNF (F, H), and IGF (I, L) compared with controls (A, D, E, G, J, K). Images were taken of the ventral column at 20×. Double labeling for GST-π (green) and GFAP (red) shows that the cells expressing mRNA for these growth factors in les spinal cords are oligodendrocytes and astrocytes (arrowheads). a–d, NT-3 mRNA is expressed in both oligodendrocytes and astrocytes at 3–6 months. e–h, Additionally, BDNF is expressed mostly in oligodendrocytes and some astrocytes at 3–9 months. i–l, IGF-1 is expressed highly in oligodendrocytes (i, k) as well as some astrocytes (j, l). Images were taken at 60×. Expression of growth factors in gray matter is similar in both control and les (data not shown).
Figure 9.

Representative image of Western blot used for quantification of NT-3, BDNF, IGF-1, and β-actin in optic nerve (ON) and spinal cord (SC) homogenates from 3-month-old control and les rats. Quantification of optical densities illustrates that homogenates from les ON (white bars) have a significant increase in BDNF, NT-3, and IGF-1 compared with controls (black bars). In addition, extracts from les SC (light gray bars) have a significant increase of NT-3 and are trending toward a significant increase of BDNF compared with controls (dark gray bars). Integrated densities were adjusted to β-actin levels as a loading control. Error bars indicate SEM. *p < 0.05, les versus control animals from the same group (n = 4) (two-tailed Student's t test). #p < 0.1, les versus control animals from the same group (n = 4) (two-tailed Student's t test).
Discussion
Here, we show that axonal degeneration does not occur in a long-lived model of dysmyelination. At 2 weeks, ∼29% of medium-diameter axons and ∼47% of large-diameter axons have myelin, yet by 3 months no medium-diameter axons and ∼<1% of large-diameter axons are myelinated in les spinal cord. Although we identified biochemical and metabolic abnormalities after demyelination, we show that axonal loss does not occur in les spinal cords up to 9 months. Finally, we illustrate an increase in mature oligodendrocytes and astrocytes that express neurotrophic factors that may promote the survival and sprouting of les axons.
Comparison of les with other myelin mutants
Few myelin mutants allow the study of chronically demyelinated or dysmyelinated axons as most die prematurely. Both the shi mouse and the les rat can live longer. However, the shi mouse only survives up to 120 d, whereas the les rat can live up to 9–12 months. Moreover, shi mice gain myelin with time hence do not provide a milieu to study the survival of chronically demyelinated axons (Rosenbluth, 1980; Inoue et al., 1983). Although the taiep rat can live up to 18–20 months, only small-diameter axons are demyelinated within the spinal cord and brain (Song et al., 2001). Although the extended life span of the les rat may not reflect the prolonged demyelination in chronic MS, the les rat is the longest-lived model of demyelination in which all axons lose myelin.
Axonal abnormalities after demyelination
At 4 weeks, les spinal cords have a significant increase in small-caliber axons. A reduction in axonal diameter has been reported in the shi mouse between 4 and 6 weeks, which was thought to be the result of a lack of compact myelin (Brady et al., 1999; Kirkpatrick et al., 2001). However, in les rats, there is not an increase in small-diameter axons from 3 to 9 months. Therefore, the increase in small-diameter axons may reflect axonal sprouting reported in the les optic nerve between 16 and 20 weeks (Phokeo et al., 2002). This is also supported by the increase in axonal density in les spinal cords from 3 to 9 months.
It is hypothesized that demyelinated axons have increased energy needs to maintain conduction velocity (Black et al., 1990; Andrews et al., 2006). In both the shi and les models, there is an increase and abnormal distribution of ion channels in demyelinated axons (Boiko et al., 2001; Eftekharpour et al., 2005). In les, a decreased amplitude and an increase in the latency of somatosensory-evoked potentials as well as a significant reduction in conduction velocity were found in 6-month-old les compared with controls (Eftekharpour et al., 2005). Mitochondrial number and activity are increased within shi axons (Andrews et al., 2006) as well as in demyelinated axons in MS lesions that correlates with increased oxidative stress (Witte et al., 2009). Thus, it was predicted that shi axons would degenerate if shi mice lived longer (Andrews et al., 2006). However, there is an increase of mitochondria in les with no signs of axonal degeneration up to 9 months.
Although β-APP accumulation, SMI-32, and mitochondrial abnormalities are often used as evidence of axonal pathology, this study illustrates that these markers are not necessarily indicative of degeneration. In response to demyelination or the accumulation of organelles in oligodendrocytes in les, there is marked microglial activation from 2 to 4 weeks and an increase in the expression of the inflammatory cytokines TNF-α, IL-1β, and iNOS in les from 3 to 8 weeks of age (Zhang et al., 2001). In this study, we detected an increase in the number of Ox-42-expressing cells in the les spinal cord up to 9 months. It has been suggested in the shi mouse that a “second hit” added to the myelin absence, such as inflammation, might result in axonal degeneration (Edgar et al., 2010). Although there was no infiltration of blood-borne cells, such as lymphocytes (Zhang et al., 2001), data from the les rat suggest that at least microglial activation does not have this effect.
Trophic role of myelin and oligodendrocytes in axonal survival
It has become increasingly apparent that oligodendrocytes and myelin play important roles in axon health and survival through glial–axonal signaling. Certain myelin proteins, such as PLP and CNP, appear critical for axon survival. Thus, in both the PLP-null mouse and in Pelizaeus–Merzbacher disease patients who lack PLP, there is a length-dependent loss of axons in the spinal cord (Griffiths et al., 1998; Garbern et al., 2002; Sima et al., 2009). The importance of PLP was confirmed when PLP-null oligodendrocytes were transplanted into the shi mouse, resulting in swellings and membranous organelle accumulation in axons with PLP-negative myelin (Edgar et al., 2004). Point mutations in the PLP1 gene result in a moderate to severe dysmyelinating phenotype and evidence of axonal degeneration, although it is not as severe as in the PLP1 knock-out mice (Dentinger et al., 1982, 1985; Duncan et al., 1995; Koeppen and Robitaille, 2002). In the CNP-null mouse, although myelin appears normal except for abnormalities in the nodal, inner-tongue processes of oligodendrocytes, there is notable axonal degeneration in early development (Lappe-Siefke et al., 2003; Edgar et al., 2009). In addition, there are abnormalities in paranodal proteins and a disruption of sodium channels before degeneration, suggesting that a lack of CNP affects axon–glial communication (Rasband et al., 2005). Recently, targeted ablation of oligodendrocytes in mice has provided interesting but variable results on axons (Traka et al., 2010; Ghosh et al., 2011; Pohl et al., 2011; Oluich et al., 2012). In one of the first studies, profound oligodendrocyte death resulted in demyelination without axonal loss (Traka et al., 2010), although the other studies reported axonal pathology. However, it will be important to differentiate between axonal damage and axon loss. It has recently been shown in cuprizone toxicity that at least some of the changes in axons, such as interruption of axonal transport and neurofilament dephosphorylation, which have been thought to predict axon loss, are indeed reversible (Xie et al., 2010). EM examination and quantification showed no significant degeneration or reduction in axonal number in the corpus callosum (Xie et al., 2010). Thus, the finding of β-APP and SMI-32-positive swellings may not always predicate axonal loss.
Although our understanding of the molecular basis for the role of oligodendrocytes in axonal survival is incomplete, recent studies show that oligodendrocytes metabolically support axons through an MTC1-dependent transport of lactate, even in the absence of mitochondrial respiration (Funfschilling et al., 2012). Moreover, disruption of this transporter results in axonal degeneration (Lee et al., 2012). les oligodendrocytes lack MBP; however, the cytoplasmic expression of other myelin proteins, including PLP and CNP, is similar to controls (O'Connor et al., 1999). In addition, there is an increase in les oligodendrocytes in response to the absence of myelin (Kwiecien et al., 1998) that also occurs in shi (Bu et al., 2004). Here, we also show evidence that les oligodendrocytes and astrocytes express mRNA for BDNF, NT-3, and IGF-1. Although oligodendrocytes have been reported to express neurotrophic factors during development (Dai et al., 2003), there is little evidence that oligodendrocytes secrete trophic factors in the mature CNS (Nave, 2010b).
The role of neurotrophins in the protection of demyelinated axons
The neurotrophic factors identified in this study are important for development (Hofer and Barde, 1988; Wozniak, 1993) and directly promote the outgrowth and survival of neurons in vitro (Nonomura et al., 1996; Wilkins et al., 2001; Hanamura et al., 2004). IGF-1 prevents neuronal death after spinal cord injury (Hollis et al., 2009), in models of amyotrophic lateral sclerosis (Kaspar et al., 2003) and protects axons from nitric oxide-mediated damage (Wilkins and Compston, 2005). IGF-1 can attenuate axonal damage by the activation of the Erk pathway and prevent apoptosis by inhibiting p38 activation as well as increasing phosphorylated Akt (Wilkins and Compston, 2005; Kaspar et al., 2003). In addition, BDNF promotes axonal survival in experimental autoimmune encephalomyelitis (Makar et al., 2009; Linker et al., 2010). BDNF and NT-3 also support axonal sprouting after spinal cord injury (von Meyenburg et al., 1998; Tobias et al., 2003). Therefore, these neurotrophic factors may promote axonal survival after demyelination through anti-inflammatory mechanisms and by preventing neuronal apoptosis as well as axonal damage. Furthermore, axonal sprouting promoted by neurotrophins may decrease the energy needs of individual axons in the absence of myelin.
In conclusion, we show that axons can survive for up to 9 months in the les rat after dysmyelination despite biochemical and metabolic abnormalities. Support from an increase in oligodendroyctes and astrocytes in les spinal cord that express BDNF, NT-3, and IGF-1 likely enhances the survival and sprouting of chronically demyelinated axons in les. Further studies are necessary to elucidate the role of each neurotrophic factor in the survival of les axons. However, there are currently very few therapies that prevent axonal loss or promote axonal regeneration in myelin disease. This work suggests that glial-derived trophic factors may prove to be an effective strategy to prevent axonal degeneration and promote regeneration after demyelination.
Footnotes
This work was supported by an NSF Graduate Research Fellowship, Training Grant T32-GM007507 from the University of Wisconsin Neuroscience Training Program, and the University of Wisconsin Graduate School Fall Competition award. We thank Drs. Ian Griffiths, John Svaren, Albee Messing, Ronald Kalil, and Zsusanna Fabry for their comments on this manuscript as well as Loklan Sherrington and Craig Radi for technical assistance with electron microscopy.
The authors declare no competing financial interests.
References
- Andrews H, White K, Thomson C, Edgar J, Bates D, Griffiths I, Turnbull D, Nichols P. Increased axonal mitochondrial activity as an adaptation to myelin deficiency in the Shiverer mouse. J Neurosci Res. 2006;83:1533–1539. doi: 10.1002/jnr.20842. [DOI] [PubMed] [Google Scholar]
- Bjartmar C, Wujek JR, Trapp BD. Axonal loss in the pathology of MS: consequences for understanding the progressive phase of the disease. J Neurol Sci. 2003;206:165–171. doi: 10.1016/s0022-510x(02)00069-2. [DOI] [PubMed] [Google Scholar]
- Black JA, Kocsis JD, Waxman SG. Ion channel organization of the myelinated fiber. Trends Neurosci. 1990;13:48–54. doi: 10.1016/0166-2236(90)90068-l. [DOI] [PubMed] [Google Scholar]
- Boiko T, Rasband MN, Levinson SR, Caldwell JH, Mandel G, Trimmer JS, Matthews G. Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron. 2001;30:91–104. doi: 10.1016/s0896-6273(01)00265-3. [DOI] [PubMed] [Google Scholar]
- Brady ST, Witt AS, Kirkpatrick LL, de Waegh SM, Readhead C, Tu PH, Lee VM. Formation of compact myelin is required for maturation of the axonal cytoskeleton. J Neurosci. 1999;19:7278–7288. doi: 10.1523/JNEUROSCI.19-17-07278.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu J, Banki A, Wu Q, Nishiyama A. Increased NG2(+) glial cell proliferation and oligodendrocyte generation in the hypomyelinating mutant shiverer. Glia. 2004;48:51–63. doi: 10.1002/glia.20055. [DOI] [PubMed] [Google Scholar]
- Dai X, Lercher LD, Clinton PM, Du Y, Livingston DL, Vieira C, Yang L, Shen MM, Dreyfus CF. The trophic role of oligodendrocytes in the basal forebrain. J Neurosci. 2003;23:5846–5853. doi: 10.1523/JNEUROSCI.23-13-05846.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney KH, Kwiecien JM, Wegiel J, Wisniewski HM, Percy DH, Fletch AL. Familial dysmyelination in a Long–Evans rat mutant. Lab Anim Sci. 1995;45:547–553. [PubMed] [Google Scholar]
- Dentinger MP, Barron KD, Csiza CK. Ultrastructure of the central nervous system in a myelin deficient rat. J Neurocytol. 1982;11:671–691. doi: 10.1007/BF01262431. [DOI] [PubMed] [Google Scholar]
- Dentinger MP, Barron KD, Csiza CK. Glial and axonal development in optic nerve of myelin deficient rat mutant. Brain Res. 1985;344:255–266. doi: 10.1016/0006-8993(85)90803-0. [DOI] [PubMed] [Google Scholar]
- De Stefano N, Narayanan S, Francis GS, Arnaoutelis R, Tartaglia MC, Antel JP, Matthews PM, Arnold DL. Evidence of axonal damage in the early stages of multiple sclerosis and its relevance to disability. Arch Neurol. 2001;58:65–70. doi: 10.1001/archneur.58.1.65. [DOI] [PubMed] [Google Scholar]
- Duncan ID, Lunn KF, Holmgren B, Urba-Holmgren R, Brignolo-Holmes L. The taiep rat: a myelin mutant with an associated oligodendrocyte microtubular defect. J Neurocytol. 1992;21:870–884. doi: 10.1007/BF01191684. [DOI] [PubMed] [Google Scholar]
- Duncan ID, Nadon NL, Hoffman RL, Lunn KF, Csiza C, Wells MR. Oligodendrocyte survival and function in the long-lived strain of the myelin deficient rat. J Neurocytol. 1995;24:745–762. doi: 10.1007/BF01191211. [DOI] [PubMed] [Google Scholar]
- Edgar JM, McLaughlin M, Yool D, Zhang SC, Fowler JH, Montague P, Barrie JA, McCulloch MC, Duncan ID, Garbern J, Nave KA, Griffiths IR. Oligodendroglial modulation of fast axonal transport in a mouse model of hereditary spastic paraplegia. J Cell Biol. 2004;166:121–131. doi: 10.1083/jcb.200312012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar JM, McLaughlin M, Werner HB, McCulloch MC, Barrie JA, Brown A, Faichney AB, Snaidero N, Nave KA, Griffiths IR. Early ultrastructural defects of axons and axon-glia junctions in mice lacking expression of Cnp1. Glia. 2009;57:1815–1824. doi: 10.1002/glia.20893. [DOI] [PubMed] [Google Scholar]
- Edgar JM, McCulloch MC, Montague P, Brown AM, Thilemann S, Pratola L, Gruenenfelder FI, Griffiths IR, Nave KA. Demyelination and axonal preservation in a transgenic mouse model of Pelizaeus-Merzbacher disease. EMBO Mol Med. 2010;2:42–50. doi: 10.1002/emmm.200900057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eftekharpour E, Karimi-Abdolrezaee S, Sinha K, Velumian AA, Kwiecien JM, Fehlings MG. Structural and functional alterations of spinal cord axons in adult Long–Evans Shaker (LES) dysmyelinated rats. Exp Neurol. 2005;193:334–349. doi: 10.1016/j.expneurol.2005.01.019. [DOI] [PubMed] [Google Scholar]
- Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain. 1997;120:393–399. doi: 10.1093/brain/120.3.393. [DOI] [PubMed] [Google Scholar]
- Fünfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, Brinkmann BG, Kassmann CM, Tzvetanova ID, Möbius W, Diaz F, Meijer D, Suter U, Hamprecht B, Sereda MW, Moraes CT, Frahm J, Goebbels S, Nave KA. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature. 2012;485:517–521. doi: 10.1038/nature11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbern JY, Yool DA, Moore GJ, Wilds IB, Faulk MW, Klugmann M, Nave KA, Sistermans EA, van der Knaap MS, Bird TD, Shy ME, Kamholz JA, Griffiths IR. Patients lacking the major CNS myelin protein, proteolipid protein 1, develop length-dependent axonal degeneration in the absence of demyelination and inflammation. Brain. 2002;125:551–561. doi: 10.1093/brain/awf043. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Manrique-Hoyos N, Voigt A, Schulz JB, Kreutzfeldt M, Merkler D, Simons M. Targeted ablation of oligodendrocytes triggers axonal damage. PLoS One. 2011;6:e22735. doi: 10.1371/journal.pone.0022735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, Schneider A, Zimmermann F, McCulloch M, Nadon N, Nave KA. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science. 1998;280:1610–1613. doi: 10.1126/science.280.5369.1610. [DOI] [PubMed] [Google Scholar]
- Hagemeier K, Brück W, Kuhlmann T. Multiple sclerosis: remyelination failure as a cause of disease progression. Histol Histopathol. 2012;27:277–287. doi: 10.14670/HH-27.277. [DOI] [PubMed] [Google Scholar]
- Hanamura K, Harada A, Katoh-Semba R, Murakami F, Yamamoto N. BDNF and NT-3 promote thalamocortical axon growth with distinct substrate and temporal dependency. Eur J Neurosci. 2004;19:1485–1493. doi: 10.1111/j.1460-9568.2004.03228.x. [DOI] [PubMed] [Google Scholar]
- Hofer MM, Barde YA. Brain-derived neurotrophic factor prevents neuronal death in vivo. Nature. 1988;331:261–262. doi: 10.1038/331261a0. [DOI] [PubMed] [Google Scholar]
- Hollis ER, 2nd, Lu P, Blesch A, Tuszynski MH. IGF-I gene delivery promotes corticospinal neuronal survival but not regeneration after adult CNS injury. Exp Neurol. 2009;215:53–59. doi: 10.1016/j.expneurol.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard TJ, Aken BL, Beal K, Ballester B, Caccamo M, Chen Y, Clarke L, Coates G, Cunningham F, Cutts T, et al. Ensembl 2007. Nucleic Acids Res. 2007;35:D610–D617. doi: 10.1093/nar/gkl996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y, Nakamura R, Mikoshiba K, Tsukada Y. Fine structure of the central myelin sheath in the myelin deficient mutant Shiverer mouse, with special reference to the pattern of myelin formation by oligodendroglia. Brain Res. 1981;219:85–94. doi: 10.1016/0006-8993(81)90269-9. [DOI] [PubMed] [Google Scholar]
- Inoue Y, Inoue K, Terashima T, Mikoshiba K, Tsukada Y. Developmental changes of oligodendroglia in the posterior funiculus of “Shiverer” mutant mouse spinal cord, with special reference to myelin formation. Anat Embryol (Berl) 1983;168:159–171. doi: 10.1007/BF00315814. [DOI] [PubMed] [Google Scholar]
- Kaspar BK, Lladó J, Sherkat N, Rothstein JD, Gage FH. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003;301:839–842. doi: 10.1126/science.1086137. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick LL, Witt AS, Payne HR, Shine HD, Brady ST. Changes in microtubule stability and density in myelin-deficient shiverer mouse CNS axons. J Neurosci. 2001;21:2288–2297. doi: 10.1523/JNEUROSCI.21-07-02288.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeppen AH, Robitaille Y. Pelizaeus-Merzbacher disease. J Neuropathol Exp Neurol. 2002;61:747–759. doi: 10.1093/jnen/61.9.747. [DOI] [PubMed] [Google Scholar]
- Kwiecien JM, O'Connor LT, Goetz BD, Delaney KH, Fletch AL, Duncan ID. Morphological and morphometric studies of the dysmyelinating mutant, the Long–Evans shaker rat. J Neurocytol. 1998;27:581–591. doi: 10.1023/a:1006922227791. [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang PW, Pellerin L, Magistretti PJ, Rothstein JD. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–448. doi: 10.1038/nature11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linker RA, Lee DH, Demir S, Wiese S, Kruse N, Siglienti I, Gerhardt E, Neumann H, Sendtner M, Lühder F, Gold R. Functional role of brain-derived neurotrophic factor in neuroprotective autoimmunity: therapeutic implications in a model of multiple sclerosis. Brain. 2010;133:2248–2263. doi: 10.1093/brain/awq179. [DOI] [PubMed] [Google Scholar]
- Makar TK, Bever CT, Singh IS, Royal W, Sahu SN, Sura TP, Sultana S, Sura KT, Patel N, Dhib-Jalbut S, Trisler D. Brain-derived neurotrophic factor gene delivery in an animal model of multiple sclerosis using bone marrow stem cells as a vehicle. J Neuroimmunol. 2009;210:40–51. doi: 10.1016/j.jneuroim.2009.02.017. [DOI] [PubMed] [Google Scholar]
- Nave KA. Myelination and support of axonal integrity by glia. Nature. 2010a;468:244–252. doi: 10.1038/nature09614. [DOI] [PubMed] [Google Scholar]
- Nave KA. Myelination and the trophic support of long axons. Nat Rev Neurosci. 2010b;11:275–283. doi: 10.1038/nrn2797. [DOI] [PubMed] [Google Scholar]
- Nixon RA. Increased axonal proteolysis in myelin-deficient mutant mice. Science. 1982;215:999–1001. doi: 10.1126/science.7156980. [DOI] [PubMed] [Google Scholar]
- Nonomura T, Kubo T, Oka T, Shimoke K, Yamada M, Enokido Y, Hatanaka H. Signaling pathways and survival effects of BDNF and NT-3 on cultured cerebellar granule cells. Brain Res Dev Brain Res. 1996;97:42–50. doi: 10.1016/s0165-3806(96)00130-7. [DOI] [PubMed] [Google Scholar]
- O'Connor LT, Goetz BD, Kwiecien JM, Delaney KH, Fletch AL, Duncan ID. Insertion of a retrotransposon in Mbp disrupts mRNA splicing and myelination in a new mutant rat. J Neurosci. 1999;19:3404–3413. doi: 10.1523/JNEUROSCI.19-09-03404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oluich LJ, Stratton JA, Lulu Xing YL, Ng SW, Cate HS, Sah P, Windels F, Kilpatrick TJ, Merson TD. Targeted ablation of oligodendrocytes induces axonal pathology independent of overt demyelination. J Neurosci. 2012;32:8317–8330. doi: 10.1523/JNEUROSCI.1053-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phokeo V, Kwiecien JM, Ball AK. Characterization of the optic nerve and retinal ganglion cell layer in the dysmyelinated adult Long–Evans Shaker rat: evidence for axonal sprouting. J Comp Neurol. 2002;451:213–224. doi: 10.1002/cne.10330. [DOI] [PubMed] [Google Scholar]
- Pohl HB, Porcheri C, Mueggler T, Bachmann LC, Martino G, Riethmacher D, Franklin RJ, Rudin M, Suter U. Genetically induced adult oligodendrocyte cell death is associated with poor myelin clearance, reduced remyelination, and axonal damage. J Neurosci. 2011;31:1069–1080. doi: 10.1523/JNEUROSCI.5035-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Tayler J, Kaga Y, Yang Y, Lappe-Siefke C, Nave KA, Bansal R. CNP is required for maintenance of axon-glia interactions at nodes of Ranvier in the CNS. Glia. 2005;50:86–90. doi: 10.1002/glia.20165. [DOI] [PubMed] [Google Scholar]
- Roach A, Takahashi N, Pravtcheva D, Ruddle F, Hood L. Chromosomal mapping of mouse myelin basic protein gene and structure and transcription of the partially deleted gene in shiverer mutant mice. Cell. 1985;42:149–155. doi: 10.1016/s0092-8674(85)80110-0. [DOI] [PubMed] [Google Scholar]
- Rosenbluth J. Central myelin in the mouse mutant shiverer. J Comp Neurol. 1980;194:639–648. doi: 10.1002/cne.901940310. [DOI] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Schirmer L, Antel JP, Brück W, Stadelmann C. Axonal loss and neurofilament phosphorylation changes accompany lesion development and clinical progression in multiple sclerosis. Brain Pathol. 2011;21:428–440. doi: 10.1111/j.1750-3639.2010.00466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sima AA, Pierson CR, Woltjer RL, Hobson GM, Golden JA, Kupsky WJ, Schauer GM, Bird TD, Skoff RP, Garbern JY. Neuronal loss in Pelizaeus–Merzbacher disease differs in various mutations of the proteolipid protein 1. Acta Neuropathol. 2009;118:531–539. doi: 10.1007/s00401-009-0562-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Goetz BD, Kirvell SL, Butt AM, Duncan ID. Selective myelin defects in the anterior medullary velum of the taiep mutant rat. Glia. 2001;33:1–11. [PubMed] [Google Scholar]
- Song J, Carson JH, Barbarese E, Li FY, Duncan ID. RNA transport in oligodendrocytes from the taiep mutant rat. Mol Cell Neurosci. 2003;24:926–938. doi: 10.1016/s1044-7431(03)00254-9. [DOI] [PubMed] [Google Scholar]
- Tallantyre EC, Bø L, Al-Rawashdeh O, Owens T, Polman CH, Lowe JS, Evangelou N. Clinico-pathological evidence that axonal loss underlies disability in progressive multiple sclerosis. Mult Scler. 2010;16:406–411. doi: 10.1177/1352458510364992. [DOI] [PubMed] [Google Scholar]
- Tobias CA, Shumsky JS, Shibata M, Tuszynski MH, Fischer I, Tessler A, Murray M. Delayed grafting of BDNF and NT-3 producing fibroblasts into the injured spinal cord stimulates sprouting, partially rescues axotomized red nucleus neurons from loss and atrophy, and provides limited regeneration. Exp Neurol. 2003;184:97–113. doi: 10.1016/S0014-4886(03)00394-7. [DOI] [PubMed] [Google Scholar]
- Traka M, Arasi K, Avila RL, Podojil JR, Christakos A, Miller SD, Soliven B, Popko B. A genetic mouse model of adult-onset, pervasive central nervous system demyelination with robust remyelination. Brain. 2010;133:3017–3029. doi: 10.1093/brain/awq247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Meyenburg J, Brösamle C, Metz GA, Schwab ME. Regeneration and sprouting of chronically injured corticospinal tract fibers in adult rats promoted by NT-3 and the mAb IN-1, which neutralizes myelin-associated neurite growth inhibitors. Exp Neurol. 1998;154:583–594. doi: 10.1006/exnr.1998.6912. [DOI] [PubMed] [Google Scholar]
- Wilkins A, Compston A. Trophic factors attenuate nitric oxide-mediated neuronal and axonal injury in vitro: roles and interactions of mitogen-activated protein kinase signalling pathways. J Neurochem. 2005;92:1487–1496. doi: 10.1111/j.1471-4159.2004.02981.x. [DOI] [PubMed] [Google Scholar]
- Wilkins A, Chandran S, Compston A. A role for oligodendrocyte-derived IGF-1 in trophic support of cortical neurons. Glia. 2001;36:48–57. doi: 10.1002/glia.1094. [DOI] [PubMed] [Google Scholar]
- Wilkins A, Kondo Y, Song J, Liu S, Compston A, Black JA, Waxman SG, Duncan ID. Slowly progressive axonal degeneration in a rat model of chronic, nonimmune-mediated demyelination. J Neuropathol Exp Neurol. 2010;69:1256–1269. doi: 10.1097/NEN.0b013e3181ffc317. [DOI] [PubMed] [Google Scholar]
- Witte ME, Bø L, Rodenburg RJ, Belien JA, Musters R, Hazes T, Wintjes LT, Smeitink JA, Geurts JJ, De Vries HE, van der Valk P, van Horssen J. Enhanced number and activity of mitochondria in multiple sclerosis lesions. J Pathol. 2009;219:193–204. doi: 10.1002/path.2582. [DOI] [PubMed] [Google Scholar]
- Wozniak W. Brain-derived neurotrophic factor (BDNF): role in neuronal development and survival. Folia Morphol (Warsz) 1993;52:173–181. [PubMed] [Google Scholar]
- Xie M, Tobin JE, Budde MD, Chen CI, Trinkaus K, Cross AH, McDaniel DP, Song SK, Armstrong RC. Rostrocaudal analysis of corpus callosum demyelination and axon damage across disease stages refines diffusion tensor imaging correlations with pathological features. J Neuropathol Exp Neurol. 2010;69:704–716. doi: 10.1097/NEN.0b013e3181e3de90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SC, Goetz BD, Carré JL, Duncan ID. Reactive microglia in dysmyelination and demyelination. Glia. 2001;34:101–109. doi: 10.1002/glia.1045. [DOI] [PubMed] [Google Scholar]