

Abstract
Multiple acquisition spectroscopy (MACSY) experiments that enable multiple free induction decays to be recorded during individual experiments are demonstrated. In particular, the experiments incorporate separated local field spectroscopy into homonuclear and heteronuclear correlation spectroscopy. The measured heteronuclear dipolar couplings are valuable in structure determination as well as in enhancing resolution by providing an additional frequency axis. In one example four different three-dimensional spectra are obtained in a single experiment, demonstrating that substantial potential saving in experimental time is available when multiple multi-dimensional spectra are required as part of solid-state NMR studies.
Keywords: MACSY, dual acquisition, dual observation, PELF, dipolar couplings, protein NMR, R-INEPT, CXCR1
Introduction
Multiple acquisition spectroscopy (MACSY) offers two universal advantages when multiple multi-dimensional spectra are required in NMR studies. First, that the overall amount of time spent signal averaging is greatly reduced, by at least a factor of 3, and potentially even larger factors in combination with non-uniform sampling techniques and further optimization. Second, that by obtaining all of the data on the same sample at the same time, accurate alignment of multiple data sets is assured. MACSY is based on the principle of acquiring multiple multidimensional data sets during a single NMR experiment where the relaxation time of at least one of the nuclei is sufficiently long, when properly handled, to maintain coherence for the duration of the experiment.
Dual acquisition was first demonstrated in 1984 for solution NMR of macromolecules by combining two-dimensional correlated spectroscopy (COSY) and nuclear Overhauser enhancement spectroscopy (NOESY) in a single experiment (COCONOSY) [1, 2]. In 2008, Fukuchi et al demonstrated applications of dual acquisition in high resolution solid-state NMR with the COCODARR experiment in which data sets for two different two-dimensional 13C/13C homonuclear correlation spectra were acquired during the course of a single experiment [3]. This was followed by the application of dual acquisition to a separated local field (SLF) spectroscopy [4] version of the experiment [5]. More recently, Gopinath et al and Lamley and Lewandowsky have built on this foundation by employing simultaneous cross-polarization (CP) to 13C and 15N to obtain two multi-dimensional spectra in a single experiment [6–9].
Here we demonstrate that there is a significant advantage to using dipolar INEPT (RINEPT) [10] for cross-polarization in dual acquisition experiments. Several additional spectroscopic enhancements, including non-uniform sampling (NUS) [11, 12], culminate in the measurement of four three-dimensional spectra in a single experiment, and multidimensional spectra of a 350-residue membrane protein in phospholipid bilayers under physiological conditions [13]. This family of experiments offers the possibility of simultaneous observation of 1H-13C and 1H-15N heteronuclear dipolar couplings in addition to various homo- and hetero- nuclear chemical shift correlations.
Heteronuclear 1H-13C and 1H-15N dipolar couplings are particularly valuable in structural studies of proteins because they provide highly reliable measurements of angles and distances. Additionally, the heteronuclear dipolar couplings can be used to measure order parameters that quantify the local and global dynamics of peptides and proteins. In these experiments the use of proton evolved local field spectroscopy (PELF) [14] has several advantages over the original versions of separated local field spectroscopy. In particular, PELF has better sensitivity compared to constant time conventional separated local field experiments because of the absence of the signal-depleting extra delay. Also, it gives simple Pake powder pattern spectra for all sites of interest in protein studies, including CH2, and CH3, also in contrast to the original version of SLF spectroscopy [15].
In these experiments, the one-bond heteronuclear dipolar couplings are correlated with chemical shift frequencies in a site-specific manner that can be either intra- or inter- residue in polypeptides; this is valuable in the resonance assignment process. Moreover, in rotationally aligned samples of membrane proteins in phospholipid bilayers, the wide range of heteronuclear dipolar coupling frequencies, which have uniform values in static polycrystalline samples, add another frequency dimension for resolution of signals that have the same chemical shift frequencies; this too is valuable in the resonance assignment process [16].
Experimental
The experiments were performed on spectrometers with 1H resonance frequencies of 750 MHz and 700 MHz. The 750 MHz spectrometer was equipped with a Bruker Avance console and a Bruker 3.2 mm Efree 1H/13C/15N triple-resonance MAS probe (www.bruker.com). The 700 MHz spectrometer was equipped with a Bruker Avance II console and a home-built 3.2 mm 1H/13C/15N triple-resonance MAS probe incorporating Revolution (http://www.revolutionnmr.com) spinning hardware. The spinning rate was controlled at 10.000 kHz ± 2 Hz. The 1H resonance frequency of water was used to monitor the temperature of the protein-containing phospholipid bilayer sample. It also served as an internal chemical shift reference frequency at 4.8 ppm at 20 °C. The 13C chemical shift frequencies of the polycrystalline samples were referenced externally to solid samples with the methylene 13C resonance of adamantane at 38.48 ppm and the 15N resonance of ammonium sulfate at 26.8 ppm [17–19].
The experimental data were acquired using the pulse sequences diagrammed in Figure 1. In all of the experiments, swept frequency two-pulse phase modulation (SWf-TPPM) [20] with 90 kHz radio frequency (RF) field strength was used to provide 1H decoupling. 50 kHz, 62 kHz and 90 kHz RF field strength pulses were applied at the resonance frequencies for the 15N, 13C, and 1H nuclei, respectively. Double cross-polarization (DCP) from 15N to 13C was accomplished using spectrally induced filtering in combination with cross-polarization (SPECIFIC-CP) [21] and proton assisted insensitive nuclei cross-polarization (PAIN-CP) [22, 23]. 10% ramped amplitude pulses at the 13C resonance frequencies were optimized for maximum polarization transfer in the applications of SPECIFIC-CP. Typical RF field strengths for SPECIFIC-CP were 27 kHz for 15N, 17 kHz for 13CA and 37 kHz for 13CO. During PAIN-CP ~50 kHz RF fields were applied synchronously to the 1H, 13C and 15N nuclei, and their amplitudes were adjusted for maximum PAIN-CP efficiency. Experiments were optimized with 2 ms and 3 ms heteronuclear mixing for PAIN and SPECIFIC-CP. Homonuclear 13C/13C spin-exchange was effected by proton driven spin diffusion (PDSD) [24], dipolar assisted rotational resonance (DARR) [25], and proton assisted recoupling techniques [23, 26, 27]. One to three bond correlations among carbon nuclei were optimized using 20 ms mixing under PDSD and DARR. Long-range correlation experiments were carried out using 2 ms PAR and up to 100 ms DARR mixing.
Figure 1.

Diagrams of MACSY pulse sequences that integrate separated local field spectroscopy and chemical shift correlation. A. Single acquisition, dual observation (SADO). B. Dual acquisition, dual observation (DADO). C. Dual acquisition, multiple observation (DAMO). D. Triple acquisition, multiple observation (TAMO). Thin and thick vertical lines represent 90° and 180° pulses, respectively. The thinner lines in Panel B represents small flip angle (35°) pulses. The phase cycles are as follows; ϕ1= 1, ϕ2= 1133, ϕ3= 11113333, ϕ4= 0022, ϕ5=3311, ϕ6= 1133. ϕ(N)(DCP)= 0022, ϕ(C)(DCP)= 00002222, ϕ(rec)= 02202002
Recoupling of the hetero-nuclear dipolar coupling frequencies and cross-polarization in MAS experiments utilized a symmetry-based R1871 scheme [28]. A pair of 180° pulses with 70° phase modulation of (π70π-70) was employed in the R1871 scheme. The scaling factors for the pulse sequences were measured experimentally with 13C and 15N detection using a uniformly 13C, 15N labeled sample of polycrystalline N-acetyl leucine (NAL). The measured dipolar splitting of 6.8 kHz for 1H-13C and 3.6 kHz for 1H-15N correspond to a scaling factor of 0.18. Two- and three-dimensional separated local field experiments were performed using direct 13C-detection with or without 15N editing.
Three-dimensional data were collected with 2 ms dipolar evolution, 3 ms to 5 ms 13C and 15N chemical shift evolution in indirect dimensions, and 10 ms direct acquisition. All of the experiments were performed with a 2 s recycle delay. A total number of 16 scans were co-added for the MLF sample, 4 scans for the NAL sample, and 512–1024 scans for the protein sample. The experimental data were processed in NMRPipe [29] and visualized using SPARKY (University of California, San Francisco). Equal numbers of data points were linear predicted for the indirect dimensions prior to Fourier transformation. Sine bell window functions shifted by 30° or 60° were used in the direct and indirect dimensions to process the multidimensional datasets, except for the NUS data. The NUS protein data in Figure 5 were processed with 0.5 ppm exponential line broadening in the direct dimension and sine bell functions shifted by 30° in the indirect dimensions. The NUS scheduling was optimized using parameters from Bruker’s TOPSPIN 3.1 program. A J coupling of 55 Hz and a T2 relaxation time of 30 ms were used to determine the optimal selection of 50% of the complete set of data points. The NUS data were processed and visualized using the same program.
Figure 5.
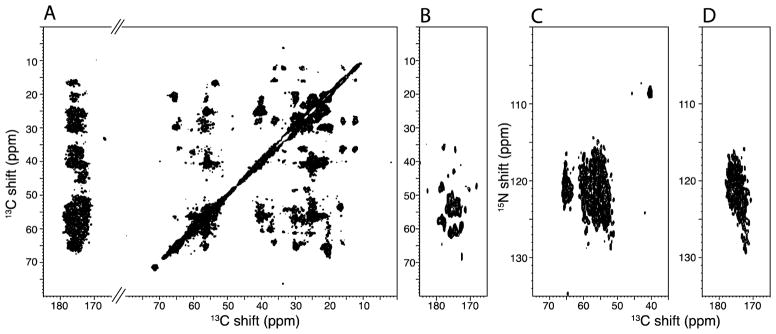
Two-dimensional correlation spectra obtained from a uniformly 13C,15N labeled sample of the 350-residue membrane protein CXCR1 in phospholipid bilayers at 750 MHz. It was obtained with 50% data point coverage NUS using the pulse sequence diagramed in Figure 1D without the dipolar frequency evolution in the third dimension. A. and B. Sections of the same 13C/13C homonuclear correlation spectrum with 13C chemical shift frequency axes. C. and D. Section of 15N/13C heteronuclear correlation spectra with 13C chemical shift and 15N chemical shift axes.
Results
The pulse sequences utilized in this study are diagrammed in Figure 1. They are named following their coherence transfer pathways. The pulse sequence in Figure 1A is referred to as single acquisition, dual observation (SADO) in which 1H-13C and 1H-15N dipolar frequencies are encoded in the indirect dimensions followed by simultaneous coherence transfer from 1H to 13C and 15N. Spin diffusion among 13C nuclei and heteronuclear mixing of 13C and 15N magnetization is carried out using PAIN [22] and PAR cross-polarization [27]. This class of experiments correlates polarization transfer between nuclei separated by relatively large distances. The pulse sequence in Figure 1B is referred to as dual acquisition, dual observation (DADO); it is the same as the pulse sequence shown in Figure 1A except that the amide and aliphatic 1H resonance frequencies are evolved simultaneously followed by the selective 15N magnetization transfer to either 13Cα(13CA) or 13C′ (13CO) resonances within the same or preceding residue in a polypeptide, respectively. Additionally, amide 1HN chemical shift frequencies are correlated with the 13CA resonances. The pulse sequence in Figure 1C is referred to as dual acquisition, multiple observation (DAMO); here 1H-13C and 1H-15N dipolar frequencies are correlated with the 13C and 15N chemical shift frequencies of the same or preceding residues. The experiments are either carried out with same dwell time for 13C (t1) and 15N evolution (t1′) or by increasing the 15N dwell time. The acquisition of 15N edited data with a longer dwell time was carried out using the method described by Gopinath et al [7, 8]. 1HA-13CA dipolar frequencies in the backbone of a peptide plane are correlated to the side chain chemical shifts separated by multiple bonds within the same amino acid; the same is true for correlation of 1H-13C dipolar frequencies in side chains to the backbone nuclei (13CA and 13CO) and can potentially be extended to long-range correlation depending on the details of the spin diffusion mixing. In addition, 1H-15N dipolar frequencies are correlated to the 13C shifts of backbone and side chain sites. The pulse sequence in Figure 2D is referred to as triple acquisition, multiple observations (TAMO). Triple acquisition provides the simplest method for transfer of magnetization among homo nuclei or from 15N to 13C. Here, 15N magnetization is transferred to 13CA chemical shift frequencies prior to the second acquisition, and the remaining magnetization is transferred to the 13CO chemical shift frequencies prior to the third acquisition. The pulse sequences diagrammed in Figure 1 have many features in common, in particular the strategy of using RINEPT for highly selective one-bond cross-polarization from the abundant 1H to the 13C and 15N nuclei in isotopically labeled peptides and proteins. This is also easier to implement than conventional Hartmann-Hahn cross-polarization. And the experiments are fully compatible with non-uniform sampling.
Figure 2.

Four different three-dimensional spectra obtained from a single DAMO experiment at 700 MHz. The three-dimensional spectra are from a uniformly 13C, 15N labeled sample of polycrystalline methionine-leucine-phenylalanine (MLF) obtained using the pulse sequence diagrammed in Figure 1C. A. 1H-15N/N(CA)CX spectrum with 13C chemical shift, 15N chemical shift, and 1H-15N heteronuclear dipolar coupling frequency dimensions. B.1H-13C/CXCY spectrum with 13C chemical shift, 13C chemical shift, and 1H-13C dipolar coupling frequency axes. C. 1H-15N/NCO spectrum with 13C chemical shift, 15N chemical shift, and 1H-15N dipolar coupling axes. D. 1HA-13CA/CA(N)CO spectrum with 13C chemical shift, 13C chemical shift, and 1H-13C dipolar coupling frequency axes.
The four three-dimensional spectra shown in Figure 2 were obtained from a polycrystalline sample of uniformly 13C, 15N labeled Met-Leu-Phe (MLF) using the DAMO pulse sequence diagrammed in Figure 1C. 1H magnetization was transferred to 13C and 15N simultaneously during a period corresponding to two rotor cycles with RINEPT. 90° pulses were then applied to flip the magnetization to the z-axis of the laboratory frame, followed by a z-filter period corresponding to four rotor cycles. Following the 90° flip-back pulses, 1H decoupled 13C and 15N chemical shift frequencies evolved. A bidirectional coherence transfer between 13CA and 15N was accomplished under SPECIFIC-CP conditions followed by two 90° pulses. The magnetization was stored along the laboratory frame z-axis. Homonuclear 13C/13C spin diffusion with 20 ms DARR mixing followed by a 90° pulse on 13C enabled the first free induction decay (FID) to be acquired. The first FID (t3) encodes two three-dimensional data sets, 1H-15N/N(CA)CX and 1H-13C/CXCY. After the first acquisition period, a 90° pulse on 15N followed by SPECIFIC-CP pulses enabled the acquisition of the second FID. During the second CP period the 13C carrier frequency was set to the middle of the 13CO spectral region (175 ppm). The second FID also encodes two three-dimensional data sets, 1H-13C/CA(N)CO and 1H-15N/NCO. Phase sensitive chemical shifts were obtained by incrementing the phases ϕ2 and ϕ3 in the States mode [30]. Two independent data sets were obtained by 180° phase alternation of ϕ3. Addition and subtraction of the first FID yield the spectra in Panel A (1H-15N/N(CA)CX) and Panel B (1H-13C/CXCY), respectively. In a similar manner, the three-dimensional spectra shown in Panel C (1H-15N/NCO) and Panel D (1H-13C/CA(N)CO) were obtained from the second FID.
In Panel A, the CO region (170 ppm – 180 ppm) shows three resolved N-H dipolar couplings. These have peak-to-peak frequency separations of 10 kHz for the rigid lattice since they represent the perpendicular discontinuities of the Pake doublets [31]. Significantly, these values vary over the full range in rotationally aligned membrane proteins due to motional averaging resulting from rotational diffusion about the bilayer normal [16]. The resolved CO signals can be directly correlated to the CA and aliphatic side chain resonances (CX). Notably, all of the side chain signals appear as simple doublets, regardless of the number of bonded hydrogens, due to the use of PELF, and all of the expected side chain resonances are observed due to the ability to establish long-range correlations. Panel C is an NCO inter-residue correlation spectrum. Panel B shows the CA and side chain resonances correlated to CO resonances. The high field resonance from the methionine methyl group has a small dipolar coupling due to motional averaging of the side chain. Panel D correlates CAi-HAi to COi-1 and is 15N edited. This is in contrast to the original DAAP experiment [16] with Ni-Hi to CAi to COi-1.
The spectra in Figure 3 were obtained from the same tripeptide sample used for the experimental results shown in Figure 2. The data in Figure 3 were obtained using the pulse sequence diagrammed in Figure 1A. The coherence transfer scheme is similar to that described above for Figure 1C. The two-dimensional 15N/13C heteronuclear correlation spectrum in Panel A was obtained using PAIN cross-polarization [22], and the two-dimensional 13C/13C homonuclear correlation spectrum in Panel B was obtained using PAR cross-polarization [27]. In Panels C and D, the two-dimensional planes were obtained from a three-dimensional data set at a 15N shift of 128 ppm and a 13C shift of 52 ppm, respectively. Two three-dimensional data sets were obtained independently by 180° phase alternation of ϕ3. Addition of two data sets yields the three-dimensional spectrum correlating 1H-13C dipolar coupling frequencies with 13C chemical shifts (1H-13C/CXCY). Subtraction of the two data sets yields the three-dimensional spectrum correlating 1H-15N dipolar coupling frequencies with 15N and 13C isotropic chemical shifts.
Figure 3.

Two-dimensional SLF and HETCOR planes from three-dimensional spectra of a uniformly 13C, 15N labeled sample of polycrystalline methionine-leucine-phenylalanine obtained using the pulse sequence diagrammed in Figure 1A (SADO) at 700 MHz. A. 15N/13C two-dimensional heteronuclear correlation spectrum with 13C chemical shift and 15N chemical shift frequency axes. B. 13C/13C two-dimensional homonuclear correlation spectrum with 13C chemical shift frequency axes. C. 1H-15N/13C two-dimensional separated local field spectrum with 13C chemical shift and 1H-15N heteronuclear dipolar coupling frequency axes. (It corresponds to the 15N chemical shift frequency at 128 ppm marked with an arrow in Panel A.) D. 1H-13C/13C two-dimensional separated local field spectrum with 13C chemical shift and 1H-13C heteronuclear dipolar coupling frequency axes. (It corresponds to the methionine 13C chemical shift frequency at 52 ppm marked with an arrow in Panel B.)
The spectral planes from three-dimensional spectra shown in Figure 4 were obtained from a polycrystalline sample of uniformly 13C, 15N labeled N-acetyl leucine using the pulse sequence diagrammed in Figure 1B. In this experiment, simultaneous evolution of heteronuclear dipolar frequencies followed by 1H chemical shift evolution was accomplished in a time-shared manner. After RINEPT, the 15N magnetization was stored along the z-axis in the laboratory frame followed by acquisition of the first FID. The second FID was acquired in a similar manner, as described above with heteronuclear mixing using SPECIFIC-CP. Panel A is a 15N-edited two-dimensional plane that correlates 1H and 13C chemical shifts. Panel B is a two-dimensional plane that correlates amide 1H and 13CA chemical shifts with the chemical shifts of side chain 13C resonances. Panel C is a 1H-13C dipolar coupling/13C chemical shift plane corresponding to a 1H chemical shift of 8 ppm. Panel D is a 1H-15N dipolar coupling/13CA chemical shift plane corresponding to a 1H chemical shift of 4 ppm. The one-dimensional dipolar slices obtained from the two-dimensional planes were taken at the positions marked with arrows. The 1H chemical shift dimensions were obtained using the States mode [30] by incrementing the 1H 90° pulse phase (ϕ(s)). The results highlight the one-bond selectivity that results from using RINEPT for cross-polarization.
Figure 4.

Two-dimensional SLF and HETCOR planes from three-dimensional spectra of a uniformly 13C, 15N labeled sample of polycrystalline N-acetyl leucine obtained using the pulse sequence diagrammed in Figure 1B (DADO) at 700 MHz. A. and B. 1H/13C two-dimensional heteronuclear correlation spectrum with 13C chemical shift and 1H chemical shift frequency axes. C. 1H-15N/13C two-dimensional separated local field spectrum with 13C chemical shift and 1H-15N heteronuclear dipolar coupling frequency axes. (It corresponds to the signal marked with an arrow in Panel A.) D. 1H-13C/13C two-dimensional separated local field spectrum with 13C chemical shift and 1H-13C heteronuclear dipolar coupling frequency axes. (It corresponds to the signal marked with an arrow in Panel B.)
Figure 5 contains two-dimensional correlation spectra obtained from a uniformly 13C, 15N labeled sample of CXCR1 in phospholipid bilayers. ~3.5 mg of protein was reconstituted in dimyristoylphosphatidylcholine (DMPC) liposomes and the experiments were carried out at 15° C in a “low-E” probe that resulted in minimal sample heating at 750 MHz. At this temperature the protein is not undergoing rotational diffusion about the bilayer normal on the relevant NMR timescales [32]. The spectra were obtained using the pulse sequence diagrammed in Figure 1D without the dipolar frequency evolution in the third dimension. All spectra were acquired with 50% NUS in the indirect dimensions, except for that in Panel A, which is a uniformly sampled 13C/13C homonuclear correlation spectrum obtained from the first FID (t2, t1). Panel C is a 15N/13C heteronuclear correlation spectrum obtained from the second FID (t2′, t1′). Panel D is a 15N/13CO heteronuclear correlation spectrum obtained from the third FID (t2″, t1′). Panel B is an inter-residue CA(N)CO correlation spectrum obtained from the second FID of Figure 1C.
Discussion
Here we introduce SLF versions of MACSY. PELF was utilized for several reasons, chief among them the simplifications of the resonances in two-dimensional planes of the heteronuclear dipolar couplings to carbons that are bonded to more than one hydrogen, e.g., CH2 and CH3. The results serve to reinforce several of the general spectroscopic advantages of MACSY. Foremost among these is the improvement in efficiency in obtaining the data. In Figure 2, we demonstrate that it is feasible to obtain four different three-dimensional spectra from a single experiment. The total expected gain in saving of experimental time by a factor of four is reduced in practice by 30%–40%. This is in consistent with the earlier results of Gopinath et al [7, 8]. It is anticipated that the efficiency can be improved with further development, although depending upon the sample some of the loss is due to relaxation effects. Short T1ρ values in membrane proteins undergoing global rotational diffusion are the main limitation. The experiments whose pulse sequences are diagrammed in Figure 1 provide only a sample of the wide variety of multiple two- and three- dimensional spectra that can be obtained from single experiments. For example, the pulse sequence in Figure 1C enables four different three-dimensional spectra to be obtained from a single experiment. In particular, these experiments enable the simultaneous observation of 1H-13C and 1H-15N heteronuclear dipolar couplings. And the dipolar couplings are correlated with the chemical shifts in a sequential manner. Notably, the experiments presented here can readily be extended to higher dimensions by adding 1H shift coherences.
The use of RINEPT is highly selective for one-bond transfers, providing high- resolution spectra in combination with a super cycled frequency switched Lee-Goldburg pulses. The 1H shift line widths are <0.3 ppm on the peptide crystals and ~0.35 ppm (after scaling) on the membrane protein in phospholipid bilayers. These sequences were developed with protein structure determination in mind. In particular, in order to measure 1H-13C and 1H-15N heteronuclear dipolar couplings that vary widely in rotationally aligned protein samples, previously described experiments utilized the variation in the magnitudes of the dipolar couplings to resolve signals with the same chemical shift(s) in the dipolar assisted assignment protocol (DAAP). In combination, these experiments provide much-needed efficiency and flexibility for studies of large membrane proteins in phospholipid bilayers under physiological conditions.
Highlights.
Consecutive acquisition of two and three FIDS in a single experiment.
Four three-dimensional spectra from a single experiment.
Multiple acquisition (MACSY) experiments that include SLF dimensions.
Acknowledgments
We thank Albert Wu and Chris Grant for their assistance with the instrumentation and Joachim Struppe of Bruker Bio-Spin for loaning us the MLF sample used to obtain the data in Figures 2 and 3. The research was supported by grant P41EB002031, R01EB005161, R01GM099986, R01GM066978, and P01AI074805 from the National Institutes of Health. It utilized the Biotechnology BTRC for NMR Molecular Imaging of Proteins at the University of California, San Diego.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Haasnoot CAG, Van De Ven FJM, Hilbers CW. COCONOSY. Combination of 2D correlated and 2D nuclear Overhauser enhancement spectroscopy in a single experiment. J Magn Reson. 1984;56:343–349. [Google Scholar]
- 2.Gurevich AZ, Barsukov IL, Arseniev AS, Bystrov VF. Combined COSY-NOESY experiment. J Magn Reson. 1984;56:471–478. [Google Scholar]
- 3.Fukuchi M, Takegoshi K. Combination of 13C-13C COSY and DARR (COCODARR) in solid-state NMR. Solid State Nucl Magn Reson. 2008;34:151–153. doi: 10.1016/j.ssnmr.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 4.Waugh JS. Uncoupling of local field spectra in nuclear magnetic resonance: Determination of atomic position in solids. Proceedings of the National Academy of Sciences of the United States of America. 1976;73:1394–1397. doi: 10.1073/pnas.73.5.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fukuchi M, Inukai M, Takeda K, Takegoshi K. Double-acquisition: Utilization of discarded coherences in a 2D separation experiment using the States method. J Magn Reson. 2008;194:300–302. doi: 10.1016/j.jmr.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 6.Gopinath T, Veglia G. Orphan spin operators enable the acquisition of multiple 2D and 3D magic angle spinning solid-state NMR spectra. The Journal of chemical physics. 2013;138:184201. doi: 10.1063/1.4803126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gopinath T, Veglia G. 3D DUMAS: Simultaneous acquisition of three-dimensional magic angle spinning solid-state NMR experiments of proteins. J Magn Reson. 2012;220:79–84. doi: 10.1016/j.jmr.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gopinath T, Veglia G. Dual acquisition magic-angle spinning solid-state NMR-spectroscopy: simultaneous acquisition of multidimensional spectra of biomacromolecules. Angew Chem Int Ed Engl. 2012;51:2731–2735. doi: 10.1002/anie.201108132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lamley JM, Lewandowski JR. Simultaneous acquisition of homonuclear and heteronuclear long-distance contacts with time-shared third spin assisted recoupling. Journal of Magnetic Resonance. 2012;218:30–34. doi: 10.1016/j.jmr.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 10.Zhao X, Hoffbauer W, Schmedt auf der Günne J, Levitt MH. Heteronuclear polarization transfer by symmetry-based recoupling sequences in solid-state NMR. Solid state nuclear magnetic resonance. 2004;26:57–64. doi: 10.1016/j.ssnmr.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 11.Hyberts SG, Arthanari H, Robson SA, Wagner G. Perspectives in magnetic resonance: NMR in the post-FFT era. Journal of Magnetic Resonance. 2014;241:60–73. doi: 10.1016/j.jmr.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin EC, Opella SJ. Sampling scheme and compressed sensing applied to solidstate NMR spectroscopy. Journal of Magnetic Resonance. 2013;237:40–48. doi: 10.1016/j.jmr.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park SH, Das BB, Casagrande F, Tian Y, Nothnagel HJ, Chu M, Kiefer H, Maier K, De Angelis AA, Marassi FM, Opella SJ. Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature. 2012;7426:779–783. doi: 10.1038/nature11580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dvinskikh SV, Zimmermann H, Maliniak A, Sandström D. Measurements of motionally averaged heteronuclear dipolar couplings in MAS NMR using R-type recoupling. Journal of Magnetic Resonance. 2004;168:194–201. doi: 10.1016/j.jmr.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 15.Rybaczewski EF, Neff BL, Waugh JS, Sherfinski JS. High resolution 13C NMR in solids: 13C local fields of CH, CH2, and CH3. The Journal of chemical physics. 1977;67:1231–1236. [Google Scholar]
- 16.Das BB, Zhang H, Opella SJ. Dipolar assisted asignment protocol (DAAP) for MAS sold-state NMR of rotationally aligned membrane proteins in phospholipid bilayers. J Magn Reson. 2014;242:224–232. doi: 10.1016/j.jmr.2014.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morcombe CR, Zilm KW. Chemical shift referencing in MAS solid state NMR. Journal of Magnetic Resonance. 2003;162:479–486. doi: 10.1016/s1090-7807(03)00082-x. [DOI] [PubMed] [Google Scholar]
- 18.Wishart D, Bigam C, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley J, Sykes B. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. Journal of biomolecular NMR. 1995;6:135–140. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- 19.De Angelis AA, Howell SC, Nevzorov AA, Opella SJ. Structure determination of a membrane protein with two trans-membrane helices in aligned phospholipid bicelles by solid-state NMR spectroscopy. J Am Chem Soc. 2006;128:12256–12267. doi: 10.1021/ja063640w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thaukur RS, Kurur ND, Madhu PK. Swept-frequency two-pulse phase odulation for heteronuclear dipolar decoupling in solid-state NMR. Chem Phys Lett. 2006;426:459–463. [Google Scholar]
- 21.Baldus M, PETKOVA AT, HERZFELD J, GRIFFIN RG. Cross polarization in the tilted frame: assignment and spectral simplification in heteronuclear spin systems. Molecular Physics. 1998;95:1197–1207. [Google Scholar]
- 22.Lewandowski JR, De Paëpe G, Griffin RG. Proton Assisted Insensitive Nuclei Cross Polarization. Journal of the American Chemical Society. 2007;129:728–729. doi: 10.1021/ja0650394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Paëpe G, Lewandowski JR, Loquet A, Böckmann A, Griffin RG. Proton assisted recoupling and protein structure determination. The Journal of chemical physics. 2008;129:245101. doi: 10.1063/1.3036928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suter D, Ernst R. Spin diffusion in resolved solid-state NMR spectra. Physical Review B. 1985;32:5608. doi: 10.1103/physrevb.32.5608. [DOI] [PubMed] [Google Scholar]
- 25.Takegoshi K, Nakamura S, Terao T. 13C-1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chemical Physics Letters. 2001;344:631–637. [Google Scholar]
- 26.Lewandowski JR, De Paëpe G, Eddy MT, Struppe J, Maas W, Griffin RG. Proton Assisted Recoupling at High Spinning Frequencies. The Journal of Physical Chemistry B. 2009;113:9062–9069. doi: 10.1021/jp810280t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lewandowski JR, Paëpe GD, Eddy MT, Griffin RG. 15N–15N Proton Assisted Recoupling in Magic Angle Spinning NMR. Journal of the American Chemical Society. 2009;131:5769–5776. doi: 10.1021/ja806578y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao X, Eden M, Levitt MH. Recoupling of heteronuclear dipolar interactions in solid-state NMR using symmetry-based pulse sequences. Chemical Physics Letters. 2001;342:353–361. [Google Scholar]
- 29.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. Journal of biomolecular NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 30.States DJ, Haberkorn RA, Ruben DJ. A two-dimensional nuclear overhauser experiment with pure absorption phase in four quadrants. Journal of Magnetic Resonance (1969) 1982;48:286–292. [Google Scholar]
- 31.Pake GE. Nuclear Resonance Absorption in Hydrated Crystals: Fine Structure of the Proton Line. The Journal of chemical physics. 1948;16:327–336. [Google Scholar]
- 32.Park SH, Casagrande F, Das BB, Albrecht L, Chu M, Opella SJ. Local and global dynamics of the G protein-coupled receptor CXCR1. Biochemistry. 2011;50:2371–2380. doi: 10.1021/bi101568j. [DOI] [PMC free article] [PubMed] [Google Scholar]