

Abstract
From the subventricular zone (SVZ), neuronal precursor cells (NPCs), called neuroblasts, migrate through the rostral migratory stream (RMS) to become interneurons in the olfactory bulb (OB). Ion channels regulate neuronal migration during development, yet their role in migration through the adult RMS is unknown. To address this question, we utilized Nestin-CreERT2/R26R-YFP mice to fluorescently label neuroblasts in the adult. Patch-clamp recordings from neuroblasts reveal K+ currents that are sensitive to intracellular Ca2+ levels and blocked by clotrimazole and TRAM-34, inhibitors of intermediate conductance Ca2+-activated K+ (KCa3.1) channels. Immunolabeling and electrophysiology show KCa3.1 expression restricted to neuroblasts in the SVZ and RMS, but absent in OB neurons. Time-lapse confocal microscopy in situ showed inhibiting KCa3.1 prolonged the stationary phase of neuroblasts' saltatory migration, reducing migration speed by over 50%. Both migration and KCa3.1 currents could also be inhibited by blocking Ca2+ influx via transient receptor potential (TRP) channels, which, together with positive immunostaining for transient receptor potential canonical 1 (TRPC1), suggest that TRP channels are an important Ca2+ source modulating KCa3.1 activity. Finally, injecting TRAM-34 into Nestin-CreERT2/R26R-YFP mice significantly reduced the number of neuroblasts that reached the OB, suggesting an important role for KCa3.1 in vivo. These studies describe a previously unrecognized protein in migration of adult NPCs.
Keywords: KCa channels, KCa3.1, migration, rostral migratory stream, TRP channels
Introduction
Life-long continuous neurogenesis has been demonstrated in essentially all mammalian species (Eriksson et al. 1998) through bromodeoxyuridine labeling. In the adult brain, 2 sites of neurogenesis have been particularly well studied, namely the dentate gyrus of the hippocampus and the subventricular zone (SVZ; Doetsch et al. 1999; Alvarez-Buylla and Garcia-Verdugo 2002). The latter gives rise to neuroblasts that migrate long distances along the rostral migratory stream (RMS) to become interneurons in the olfactory bulb (OB; Doetsch et al. 1999; Alvarez-Buylla and Garcia-Verdugo 2002; Zhao et al. 2008). The role of these adult neural precursor cells (NPCs) is under considerable debate; particularly in light of the recent finding that neurogenesis decreases progressively during the first 18 months of life in humans (Sanai et al. 2011). Studies in rodents suggest that NPCs are important sources to replenish neurons following acute injury or chronic neurodegeneration (Saha et al. 2012) and participate in learning and odor discrimination (Prickaerts et al. 2004; Bardy and Pallotto 2010). There appears to be significant consensus that the SVZ may be an important source for the generation of primary brain tumors in humans (Quinones-Hinojosa and Chaichana 2007; Jackson and Alvarez-Buylla 2008; Soroceanu et al. 2008). Similar to neuroblasts in the SVZ, these tumors show an unusual propensity to migrate over long distances as they invade the brain. In light of this commonality, studies of migratory NPCs may provide insights into migratory mechanisms shared during the neural development and malignant migration of primary brain tumors.
Before arriving in the OB, neuroblasts must migrate a considerable distance from their site of birth. Signals that promote and guide neuroblast migration are not well understood, although successful migration appears to require exogenous factors, including growth factors such as EGF and FGF2, signaling molecules such as Reelin and Shh, and cytokines such as CXCR4 (Saha et al. 2012). These factors activate downstream pathways that are poorly understood; however, several studies indicate that elevations in intracellular Ca2+ regulate cellular migration in fibroblasts, HEK293 cells, cortical neurons, glial cells, glioblastoma, and other cancerous and noncancerous cells (Schwab et al. 2007). In the nervous system, the most well-studied migratory cells are cerebellar granule neurons, in which entry of Ca2+ through the n-methyl-d-aspartic acid receptor causes oscillatory Ca2+ changes that correlate with the velocity of cell movement (Mori et al. 1993; Komuro and Rakic 1996). Similarly, in glioma cells, Ca2+ oscillations induced either by ACh or by Glu are required for coordinated cell movement (Bordey et al. 2000; Lyons et al. 2007). These Ca2+ changes are likely to alter actin–myosin interactions and focal cell adhesion sites to promote motility (Giannone et al. 2002; Martini and Valdeolmillos 2010). In addition, Ca2+ changes can engage a number of Ca2+-activated ion channels, and it has recently been suggested that activation of Ca2+-activated K+ (KCa) and Cl− channels causes hydrodynamic changes in cell shape and volume, which are required for cell movement through tissue (Schwab et al. 2007; Becchetti 2011; Watkins and Sontheimer 2011).
KCa channels encompass 3 gene families classified by their conductance: Small (SK, KCa2), intermediate (IK, KCa3.1), and big conductance (BK, KCa1.1) K+ channels (Sah and Faber 2002). Their role in embryonic development and adult neurogenesis is entirely unknown, albeit their presence in the early neonatal and adult SVZ has been indirectly documented by showing K+ currents responsive to changes in extracellular Ca2+ (Stewart et al. 1999; Wang et al. 2003).
In this study, we investigated the role of KCa channels in neuroblast migration. We elected to study the largest and most well-understood site of adult neurogenesis, the SVZ. From here, neuroblasts migrate through the RMS to the OB where they differentiate into olfactory interneurons (Doetsch et al. 1999; Alvarez-Buylla and Garcia-Verdugo 2002; Zhao et al. 2008).
We found selective expression of KCa3.1 channels in migratory neuroblasts of the SVZ and RMS, but an absence in OB neurons that have completed migration. Pharmacological blockade of KCa3.1 significantly reduced overall migration of neuroblasts through the RMS in situ by increasing the stationary phase of saltatory migration. Most importantly, chronic treatment of mice with the specific KCa3.1 inhibitor, TRAM-34, significantly reduced the number of neuroblasts that successfully migrated to the OB in vivo. These studies suggest an important modulatory role for KCa3.1 in adult neuroblast migration that could be capitalized on in future attempts to alter the intrinsic repair response of the brain using NPCs.
Materials and Methods
Animals
All methods are in accordance with the University of Alabama Institutional Animal Care and Use Committee. Nestin-CreERT2/R26R-YFP mice were a generous gift from Dr Amelia Eisch, and as previously described (Lagace et al. 2007). Unless otherwise stated, mice of both sexes were injected with tamoxifen once daily for 5 days after the age of postnatal day 21 (p21). Mice were allowed another 5 or more days for cells to migrate through the RMS before use.
Drugs and Solutions
The inhibitors such as clotrimazole (10 μM), Benidipine HCl (10 μM), ω-conotoxin MVIIC (100 nM), SKF96365 (25 μM), 2-aminoethoxydiphenyl borate (2-APB) (100 μM), apamin (300 nM), and tamoxifen (180 mg/kg/day) were obtained from Sigma (Miwaukee, WI, USA). tetrodotoxin (TTX) from Tocris Biosciences (Minneapolis, MN, USA) was used at 1 μM. TRAM-34 ([1-(2-chlorophenyl)diphenylmethyl]-1H-pyrazole) (Tocris Biosciences) was used at 1 μM in situ and 20–40 mg/kg/day in vivo. For in vivo injections of tamoxifen and TRAM-34, both were dissolved in 10% ethanol and 90% sunflower oil.
Acute slices for electrophysiology and in situ migration were cut using the following solution (in mM): 135 n-methyl-d-glucamine (NMDG), 1.5 KCl, 1.5 KH2PO4, 23 Choline-Bicarbonate, 25 d-glucose, and 0.4 ascorbic Acid. After NMDG was added, pH was adjusted to 7.4 with concentrated HCl. Both 0.35 CaCl2 and 1.75 MgCl2 were added the day of cutting. The solution was bubbled with 95%O2/5%CO2 to achieve a pH of 7.4. The following artificial cerebrospinal fluid (ACSF) solution was used during electrophysiology and in situ migration (in mM): 125 NaCl, 3 KCl, 1.25 NaH2PO4, 25 NaHCO3, and 25 d-glucose. Both 2 CaCl2 and 1 MgCl2 were added the day of recording. For a pH of 7.4, the solution was bubbled with 95% O2/5% CO2.
Unless otherwise stated, the internal pipette solution used during recording contained (in mM): 120 potassium gluconate, 15 KCl, 4 MgCl2, 10 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 4 Mg-ATP, 0.3 Na-GTP, 0.1 ethylene glycol tetraacetic acid (EGTA), and 7 phosphocreatine. The day of recording, 1 mM CaCl2, was added to achieve free calcium levels of 500 nM. Tris-base was used to pH the solution to 7.3. The high 1,2-bis(o-aminophenoxy)ethane-n,n,n',n'-tetraacetic acid (BAPTA) internal pipette solution used contains (in mM): 115 potassium gluconate, 15 KCl, 4 MgCl2, 10 HEPES, 4 Mg-ATP, 0.3 Na-GTP, 10 BAPTA, and 7 phosphocreatine. Tris-base was used to achieve a pH of 7.3.
Electrophysiology
From the transgenic mice, brains were sliced in ice cold cutting solution and bubbled with 95%O2/5%CO2. Two hundred micrometer sagittal slices were cut and allowed to recover in ACSF for 45 min at 37°C. Whole-cell recordings were made using an Axopatch200B amplifier (Axon Instruments, Foster City, CA, USA) with standard recording techniques. pClamp 8.2 software (Axon Instruments) was used to acquire data. Glass pipette tips were pulled from thin borosilicate glass (World Precision Instruments, Sarasota, FL, USA) to reach 5–7 MΩ. Series resistance was compensated to 80%, and cells were omitted if they resealed during recording.
Immunohistochemistry
Brains of transgenic or wild-type animals were fixed in 4% paraformaldehyde (PFA) and slices cut at 100 μm. Free-floating coronal slices were stained for Ki67 and cell death, and sagittal slices were made for all other staining. For KCa3.1, slices were stained using the TSA™ Plus Cyanine 3 System (Perkin-Elmer, Waltham, MA, USA) following their suggested methods. For all other antibodies, slices were blocked for >1 h at room temperature with 10% goat serum and 0.5% Triton X-100 in PBS. The primary antibody was applied overnight at 4°C. The next day, slices were washed and the secondary antibody was applied for >1 h. Slices were washed again and mounted on glass slides using polyvinyl alcohol - 1,4 diazabicyclo [2.2.2]octane (Sigma).
To enhance the yellow fluorescent protein (YFP) stain, a green fluorescent protein (GFP) antibody (Aves, Tigard, OR, USA) was used at 1:1000. Doublecortin (DCX) (1:1000) and Ki67 (1:500) were obtained for Abcam. Transient receptor potential canonical 1(TRPC1) (1:50) was obtained from Alomone (Jerusalem, Israel, USA). Goat antirabbit Alexa 488 and 633, goat antichicken Alexa 488, and goat anti-mouse Alexa 546 (Invitrogen, Grand Island, NY, USA) were secondary antibodies used at 1:500. The KCa3.1 antibody (Alomone) was used at 1:500 with the TSA™ Plus Cyanine 3 System (Perkin-Elmer) with a horseradish peroxidase-conjugated secondary antibody at 1:150 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). An In Situ Cell Death Detection Kit, TMR Red (Roche Applied Sciences), was used as recommended to detect cell death. Images were taken using the Olympus Fluoview FV1000 laser scanning microscope with 60× water or 40× air objectives.
Data Analysis
Origin 7.5 was used to analyze the data. Analysis of variance or Student's t-tests were calculated using the GraphPad software in order to test significance.
In Situ Migration
From the transgenic mice, the brains were cut in ice cold cutting solution and bubbled with 95%O2/5%CO2. Two hundred micrometer sagittal slices were cut and allowed to recover in ACSF for 45 min at 37°C. Using the Olympus Fluoview FV1000 laser scanning microscope on acute slices constantly perfused with ACSF, z-stacks 2 μm apart were acquired under a 40× water-immersion lens.
Two different experimental setups were used:
(1) Pictures were taken every 20 min for an 80-min baseline. Drug or vehicle was then washed on for 80–160 min under the same conditions. Images were analyzed using the National Institute of Health ImageJ software. X/Y plane drift was minimized using StackReg. The Manual Tracking tool was used to quantify the migration of all cells that stayed within the field of view for the entire experiment. Migration was quantified by tracking nuclear translocation. Using the Chemotaxis Tool, the distance each cell migrated was calculated during baseline and after drug application. The speed of each cell and the change in speed after drug application were also determined. Only cells that migrated at speeds over 0.1 μm/min were used in analysis, as cells with lower speeds were defined to be stationary. Using these data, the directionality was calculated for all cells by dividing the total tracked migration distance by the “Euclidean” distance (the distance between the cells' start and end position).
(2) Pictures were taken every 5 min for a 40-min baseline, and drug or vehicle was applied for 45 min. The migration distance was determined using similar methods. To determine the migration speed, every 5-min interval in which migration occurred was averaged. Every 5-min interval in which no migration occurred was used to determine the average time spent migrating.
In Vivo Migration
At postnatal day 28 (p28), transgenic mice of both sexes were given a TRAM-34 or vehicle control pretreatment by intraperitoneal (i.p.) injection for 5 days. On day 6, mice were injected with tamoxifen and TRAM-34 or vehicle. Mice were sacrificed on day 8, and brains were fixed in PFA. One hundred micrometer sagittal slices were cut from one hemisphere and stained for DCX and YFP. After being blinded to the treatment, images of YFP+ cells along the RMS were taken using the Olympus Fluoview FV1000 laser scanning microscope under a 40× objective. The number of cells per image was calculated using the National Institute of Health ImageJ software Cell Counter plugin, and the number of cells for each area was averaged.
To assess changes in proliferation and cell death in the SVZ, the remaining hemispheres of the same mice were cut for 100 μm coronal slices and stained for Ki67 or with the in situ cell death detection kit, TMR red (Roche). DNAse was added to some slices as a positive control for cell death. Some hemispheres were also stained with KCa3.1 in order to visualize possible down regulation in the RMS after treatment. Pictures of the SVZ were taken and analyzed using the same methods as described above for YFP+ cells in the RMS.
Results
Visualization of Neuroblasts in the Rostral Migratory Stream
To visualize the relatively narrow RMS, we used Nestin-CreERT2/R26R-YFP transgenic mice to fluorescently label neuroblasts (Lagace et al. 2007; Supplementary Fig. 1). These carry a modified Cre recombinase expressed under the control of 5.8 kB of the Nestin promoter and exons 1–3 of the Nestin gene. Tamoxifen injection yields selective YFP expression in NPCs (Supplementary Fig. 1). Mice were injected daily with 180 mg/kg tamoxifen beginning at p21 for 5 consecutive days, and allowed at least an additional 5 days before experimental use to allow cells to migrate from the SVZ into the RMS.
Expression of KCa3.1 Channels in the RMS
To study KCa channel expression in neuroblasts of the RMS, we prepared acute brain slices from CreERT2/R26R-YFP mice. Although relatively small, the RMS can be readily recognized by the YFP fluorescence of the neuroblasts (Fig. 1A). We obtained whole-cell patch-clamp recordings from individually identified YFP-expressing neuroblasts at different locations along the RMS, including the SVZ and OB as indicated in Figure 1. To ensure the activation of Ca2+-dependent K+ currents, the pipette solution contained 500 nM free Ca2+, a concentration sufficient to activate KCa channels (Vergara et al. 1998). Neuroblasts were held at −50 mV and stepped from −160 to +100 mV in 20 mV increments. Representative current traces are shown in Figure 1B for recordings made in the SVZ, RMS, and OB. Cells recorded differed significantly in their resting membrane potential, with average values of −24.5 (standard error of the mean, SEM = 9.0; n = 8) for the SVZ, −24.4 (SEM = 1.6; n = 39) for the RMS, and −52 mV (SEM = 2.5, n = 15) for the OB. In light of the more hyperpolarized potential of OB neurons, these were held at −70 mV, but identical voltage steps were used for current activation. Furthermore, to maximize our ability to isolate even relatively small KCa3.1 currents in the OB, we perfused slices with 1 μM TTX to suppress action potentials after mature neurons were identified. The input resistance (1–5 GΩ) and average membrane capacitance (5.7 pF) of neuroblasts were similar to those reported previously for RMS neuroblasts (Lacar et al. 2010). To isolate currents mediated by KCa channels, we used a number of channel-specific inhibitors, including paxilline, apamin, and TRAM-34. Drug-sensitive currents were isolated by subtraction of currents before and after drug application. Of the inhibitors investigated, the specific KCa3.1 inhibitor TRAM-34 (1 μM) (Wulff et al. 2000) consistently inhibited outward K+ currents in neuroblasts of the SVZ and RMS (Fig. 1B). However, no TRAM-34-sensitive current was found in neurons that had already differentiated in the OB (Fig. 1B).
Figure 1.
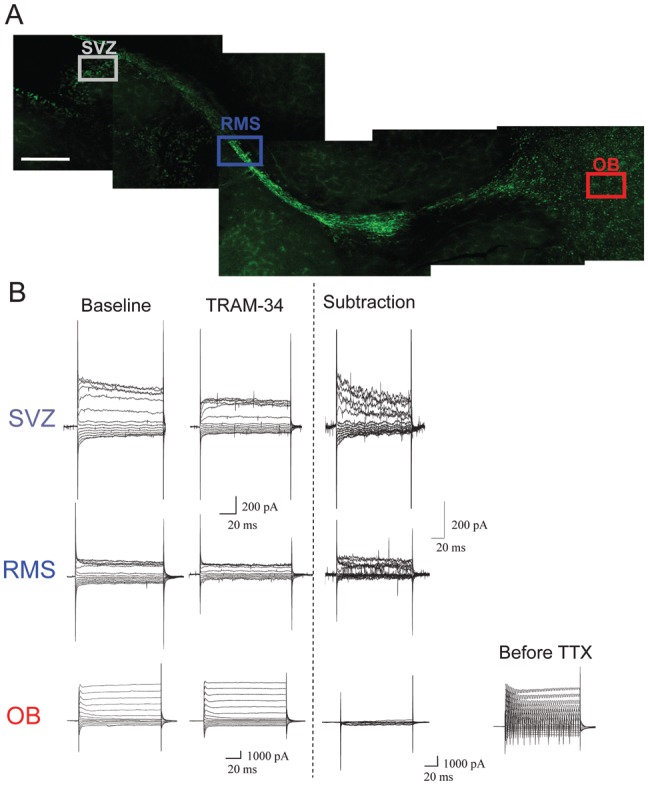
KCa3.1 channel expression along the RMS. (A) A sagittal section demonstrating RMS visualization in Nestin-CreERT2/R26R-YFP mice and highlighting the areas of interest: SVZ, RMS, and OB. Scale bar 250 μm. (B) Representative traces of whole-cell patch-clamp recordings of neuroblasts in the SVZ (n = 4) and RMS (n = 5), and of neurons in the OB (n = 7). The traces were compared before and after application of the KCa3.1 channel inhibitor, TRAM-34, and a subtraction is shown. In the OB, 1 μM TTX was also added to the bath solution to inhibit action potentials in order to visualize the currents. The current before TTX application is also presented. There is a TRAM-34-sensitive current in the SVZ and RMS, but not in the OB.
Next, we further characterized the TRAM-34-sensitive current in neuroblasts. To compare current amplitudes among cells of potentially different sizes, we normalized the recorded currents for each cell to the membrane capacitance and plotted the current density as a function of voltage, as shown in Figure 2B. The resulting I–V curve suggests that the drug-sensitive current reverses close to the K+ equilibrium potential (Fig. 2B). At very positive voltages, currents showed the characteristic inactivation due to blockade by intracellular divalent cations as previously described for KCa3.1 currents (Stocker 2004). In addition to TRAM-34, currents were also blocked by the less-specific KCa3.1 inhibitor, clotrimazole (10 μM); whereas addition of vehicle alone induced no measurable current changes (Fig. 2). Representative currents are shown in Figure 2A. These data suggest that neuroblasts in the SVZ and RMS express TRAM-34-sensitive KCa3.1 channels that are absent in the OB.
Figure 2.

Validation of KCa3.1 channel currents in the RMS. (A) Representative traces of whole-cell patch-clamp recordings of neuroblasts in the RMS before and after addition of vehicle or the KCa3.1 channel inhibitors such as TRAM-34 and clotrimazole. As shown in the subtraction, addition of TRAM-34 or clotrimazole elicited a similar current, but no current was found on vehicle addition. The application of TRAM-34 using an internal pipette solution with high 1,2-bis(o-aminophenoxy)ethane-n,n,n',n'-tetraacetic acid (BAPTA) to chelate the calcium produced no change in current, similar to the application of vehicle. (B) An I–V plot was produced from the averaged subtractions, demonstrating the presence of TRAM-34 and clotrimazole-sensitive currents in RMS neuroblasts.
To further substantiate that the TRAM-34-sensitive current is mediated by KCa3.1, recordings from YFP+ neuroblasts in the RMS were taken using high BAPTA (10 mM) in the internal solution in order to chelate the free internal Ca2+. Because KCa3.1 requires Ca2+ for activation, we hypothesized that this solution would reduce the TRAM-34-sensitive current. Indeed, the current subtraction after addition of TRAM-34 was similar to vehicle (Fig. 2). This provides further evidence that the TRAM-34-sensitive current is mediated by Ca2+-sensitive channels, namely KCa3.1.
To gain a more refined picture of the spatial expression profile of KCa3.1 throughout the RMS, we used immunostaining on fixed slices with a KCa3.1 antibody. The YFP+ cells were also located to identify neuroblasts in the RMS and newly integrated neurons in the OB. Because much of the YFP fluorescence was lost after fixation with PFA, neuroblasts were stained with an anti-GFP antibody to enhance the YFP signal. In the RMS, cells that were YFP+ were also consistently immunoreactive for KCa3.1 antibodies (Fig. 3B). Likewise, in the SVZ, there was colocalization between YFP and KCa3.1 staining (Fig. 3A). In contrast, and in agreement with our biophysical data, neurons in the OB did not show KCa3.1 immunoreactivity (Fig. 3E). Note that YFP is not expressed in every neuroblast, hence there was not complete colocalization between YFP and KCa3.1. These experiments were repeated by staining neuroblasts with DCX, instead of YFP. With this approach, there was almost complete colocalization between KCa3.1 and DCX in the RMS (Supplementary Fig. 2). Together, these results suggest that KCa3.1 channels are expressed in immature cells of the SVZ and RMS, but are absent in cells that have reached the OB. We assume that KCa3.1 expression is specifically lost once cells exit the RMS and begin to mature in the OB. In agreement with this assumption, KCa3.1 was still highly expressed in the RMS elbow (Fig. 3C); however, at the entry to the OB, KCa3.1 expression was sparse (Fig. 3D), suggesting that neuroblasts lose KCa3.1 as they enter the OB.
Figure 3.

Expression of KCa3.1 channels in the RMS. (A) Image of KCa3.1 channel staining (red) in the SVZ of Nestin-CreERT2/R26R-YFP mice, with some expression in YFP+ cells (green). Scale bar 50 μm. (B) In the RMS, immunostaining of YFP+ neuroblasts (green) show colocalization with KCa3.1 channel expression (red). Scale bar 20 μm. (C) YFP+ neuroblasts (green) in the RMS elbow (El) continue to express high levels of KCa3.1 (red). Scale bar 50 μm. (D) As YFP+ neuroblasts (green) enter the OB, they begin to lose KCa3.1 expression (red). Scale bar 50 μm. (E) YFP+ neurons in the OB (green) have no KCa3.1 channel expression (red). Dotted line indicates the RMS and OB areas. Scale bar 50 μm.
Disruption of KCa3.1 Decreases Neuroblast Migration Along the RMS
The spatial expression profile of KCa3.1 suggests that they are restricted to immature neuroblasts that are actively migrating. KCa3.1 channels have been shown to facilitate the migration of cancerous cells, such as melanoma and glioblastoma, yet little is known about their expression and function in neuroblasts (Schwab et al. 2007).
To examine a putative involvement of KCa3.1 in cell migration through the RMS, neuroblast migration was studied in acute sagittal slices in situ. Nestin-CreERT2/R26R-YFP transgenic mice were used to locate the RMS and to identify the migrating neuroblasts. Time-lapse images were acquired on a Fluoview 1000 confocal microscope equipped with water-immersion optics allowing us to obtain z-stacks containing 10–20 optical sections 2 μm apart every 20 min. Slices were maintained in an environmental control chamber providing a stable temperature, pH, and O2 environment. For each cell, an 80-min baseline was acquired to determine each cells' baseline migration speed. Next, we added either vehicle or drug and imaged cell migration for an additional 80–160 min. Overall migration speeds were quantified by measuring the accumulated distance of cell movement over the total time of capture. Analysis was performed offline using the National Institute of Health ImageJ Manual Tracking and Chemotaxis Tool plug-ins. We included only cells that displayed a baseline migration speed of >0.1 μm/min for further analysis as cells with lower speeds were defined to be stationary. The mean overall speeds for migrating cells in a slice were quantified, and then the overall migration speeds were averaged for each slice.
As seen in Figure 4, in the absence of drug, cells migrated at an average speed of 10.2 μm/h (SEM = 0.48; 5 slices, 64 cells), which was not significantly changed after application of a vehicle control for an additional 80–160 min (Fig. 4A, 9.41 μm/h, SEM = 0.57; 5 slices, 64 cells; P = 0.302). In contrast, overall migration speed was reduced by over 50% after addition of TRAM-34 from a baseline value of 10.1 μm/h (SEM = 0.43; 5 slices, 71 cells) to 4.51 μm/h (SEM = 0.55; 5 slices, 71 cells; P < 0.05; Fig. 4B). To compare cell migration under different treatment conditions, we normalized the migration speed for each cell to its own baseline, acquired during the initial 80 min of each recording. Then, the overall change in migration speed was determined for cells in a given slice, and all slice data were averaged. With this quantification, the change in overall migration speed on application of TRAM-34 yielded a mean of 48.1% of control (SEM = 0.067; 5 slices, 65 cells), less than half that of vehicle (normalized at 100%; SEM = 0.082; 5 slices, 59 cells; P < 0.01; Fig. 4C). Supplementary Figure 3A presents a representative series of time-lapse images of cells undergoing migration, and then slowing after addition of TRAM-34, as indicated by an asterisk. The experiments were repeated with clotrimazole, the less-specific KCa3.1 inhibitor, with similar results (Fig. 4C). Here, the overall change in migration speed decreased significantly to 40.1% of its baseline (SEM = 0.047; 3 slices, 30 cells; P < 0.01compared with vehicle). These results indicate that inhibition of KCa3.1 decreases neuroblast migration along the RMS.
Figure 4.

Application of KCa3.1 channel inhibitors slows neuroblast migration in situ by decreasing the time spent migrating. (A) The average overall speed of in situ migration of RMS neuroblasts was unchanged after addition of vehicle for control. (B) After application of the KCa3.1 channel inhibitor, TRAM-34, the average overall migration speed decreased by >50% of baseline. *P < 0.05. (C) The average change in overall migration of individual cells over their own baseline is quantified for vehicle and drug treatment. Both KCa3.1 channel inhibitors, TRAM-34 and clotrimazole, decrease the migration by over half compared with vehicle. *P < 0.05. (D) The percentage of time cells spent migrating under vehicle or TRAM-34 was normalized to baseline. The time spent migrating was significantly decreased upon TRAM-34 application. *P < 0.05. (E) The percentage of migratory cells for each 5-min time point during baseline, and after TRAM-34 or vehicle addition. Arrow indicates addition of TRAM-34 or vehicle.
As a control, we also applied the KCa2 inhibitor, apamin. KCa2 is a calcium-activated potassium channel similar to KCa3.1; however, the channels respond to different inhibitors. After application of apamin, there was no significant decrease in migration below baseline (90.0%; SEM = 0.148; 4 slices, 18cells) compared with vehicle (Supplementary Fig. 3C). This experiment confirms the specificity of KCa3.1 inhibition.
We also assessed changes in directionality after TRAM-34 application. Using all cells in the same set of images, we determined the total tracked distance that the cell traveled and the Euclidean distance, or the distance between the beginning and end position of the cell. By dividing the Euclidean distance by the tracked distance, we determined whether inhibition of KCa3.1 alters chemotactic migration, with the assumption that the closer the directionality value is to 1, the more directional the migration. As seen in Supplementary Figure 3B, there was no change in the directionality of the migration from baseline after vehicle or TRAM-34 application (vehicle baseline: Mean = 0.54, SEM = 0.049, n = 5 slices, 108 cells; vehicle: Mean = 0.45, SEM = 0.041, n = 5 slices, 108 cells; TRAM-34 baseline: Mean = 0.58, SEM = 0.049, n = 5 slices, 149 cells; and TRAM-34: Mean = 0.42, SEM = 0.049, n = 5 slices, 149 cells). Although there was no significant difference, it is likely that continuous perfusion of ACSF over brain slices washes out endogenous chemoattractants.
KCa3.1 Disruption Inhibits Migration by Increasing the Stationary Phase During Saltatory Migration
Neuroblasts undergo saltatory chain migration during which there are both “migratory” and “stationary” phases. To further elucidate the mechanism of KCa3.1 on migration, we investigated whether inhibition affects the speed of cell migration or the amount of time the cells spend migrating. Because the migratory and stationary phases last only a few minutes, we repeated the confocal time-lapse experiments, but obtained images for shorter time periods, every 5 min during a 40-min baseline and a 45-min drug application. We then averaged the migration speed for all 5-min intervals in which migration occurred, normalized to baseline migration, and compared the normalized speed between vehicle and drug treatment (vehicle: Mean = 0.99, SEM = 0.15, n = 5 slices, 70 cells; TRAM-34: Mean = 0.87, SEM = 0.07, n = 5 slices, 73 cells). There was no significant difference between the normalized migration speeds after vehicle and TRAM-34 application (Supplementary Fig. 4A,B). We next assessed the differences in the amount of time the cells spent migrating after drug application. Using the same data set, we quantified the percentage of time the cells were in the “migratory” or “stationary” phase (Fig. 4D) and found that, after TRAM-34 application, cells spent significantly less time migrating (vehicle: Mean = 76.6%, SEM = 5.10, n = 5 slices, 70 cells; TRAM-34: Mean = 49.0%, SEM = 4.00, n = 5 slices, 73 cells). This can also be visualized by graphing the percentage of cells that are migrating during any 5-min time point, as seen in Figure 4E. These data suggest that TRAM-34 primarily inhibits the initiation of cell migration rather than affecting the speed of nuclear translocation.
Calcium Source for KCa3.1 Modulation of Migration
KCa3.1 is primarily activated in response to changes in intracellular Ca2+. Therefore, any disruption of the Ca2+ source that engages KCa3.1 should also disrupt KCa3.1 activity and hence cell migration. Putative Ca2+ sources include intracellular inositol trisphosphate receptor (IP3R)-regulated stores, voltage-gated Ca2+ channels (VGCCs), and transient receptor potential (TRP) channels. As a first step toward identifying the source of calcium for KCa3.1 activation in the context of RMS migration, we examined the effect of inhibitors for these different calcium release or entry pathways in acute slices and assessed the resulting change in overall migration speed. Taking baseline and drug application images at 20-min intervals, the overall change in migration speed normalized to baseline was compared with that of TRAM-34, which directly inhibits KCa3.1. The application of a cocktail of the VGCC inhibitors 10 μM Benidipine HCl and 100 nM ω-conotoxin MVIIC decreased migration speed to 72.3% of baseline (SEM = 0.026; 5 slices, 71 cells; Fig. 5A). While this decrease was significantly different from vehicle-treated controls (P < 0.05), it was significantly less than TRAM-34 (P < 0.05). Hence, we next assessed overall migration speed after blocking IP3R on internal calcium stores and various TRP channels through addition of 2-APB (100 μM). This yielded a significant reduction in migration (60.5% of initial migration speed; SEM = 0.095; 4 slices, 61 cells; P < 0.05) comparable in effect size to TRAM-34 (Fig. 5B). We also utilized SKF96365 (25 μM), which nonspecifically targets TRP channels, but also blocks some VGCCs, and the store-operated calcium entry channel STIM1. SKF96365 significantly decreased migration compared with vehicle (P < 0.05), similar to 2-APB and TRAM-34 (58.1%; SEM = 0.105; 4 slices, 38 cells; Fig. 5B).
Figure 5.

Calcium source for KCa3.1 channel modulation of migration. (A) The normalized change in overall migration was decreased upon application of a cocktail of VGCC inhibitors, but to a lesser extent than TRAM-34 application. *P < 0.05. (B) The application of the calcium channel inhibitors, SKF96365 or 2-APB, caused a decrease in overall migration, similar in amount to TRAM-34. *P < 0.05. (C and D) The application of TRAM-34 and the calcium channel inhibitors such as 2-APB (C) or SKF96365 (D) together does not cause an additive decrease in migration over that of TRAM-34, 2-APB, or SKF96365 alone. *P < 0.05. (E) Immunostaining in the RMS shows expression of transient receptor potential canonical 1 (TRPC1) (blue) around the RMS, but also colocalized with YFP+ neuroblasts (green) in the RMS, along with KCa3.1 channel expression (red). Scale bar 100 μm. (F) A higher magnification confocal image of TRPC1 (white) and YFP (green) colocalization on neuroblasts in the RMS. Scale bar 20 μm.
Because these drugs are somewhat nonspecific and therefore affect multiple calcium sources, we used additive application of drugs to further narrow the identity of the KCa calcium source. We applied both 2-APB and TRAM-34 together and compared the change in migration speed with that of TRAM-34 alone. Overall migration speed was changed to a similar degree (58.5%; SEM = 0.118; 5 slices, 32 cells) as TRAM-34 alone and was significantly different than vehicle (P < 0.05; Fig. 4C). Likewise, addition of SKF96365 together with TRAM-34 produced similar changes in overall migration speed (67.2%; SEM = 0.088; 5 slices, 44 cells; P < 0.05 compared with vehicle; Fig. 4D). This additive application of drugs shows that there is a maximal decrease in migration with the calcium inhibitors, and no further change in migration upon inhibition of KCa3.1. This suggests that KCa3.1 activation is dependent on calcium influx through channels inhibited by both 2-APB and SKF96365. Because TRP channels are inhibited by both 2-APB and SKF96365, they appear to be the most likely candidate to act as a calcium source for KCa3.1 activation in RMS neuroblasts.
TRP Channels are Required for KCa3.1 Activation
We next confirmed the coupling of TRP-mediated Ca2+ entry to KCa3.1 activation using whole-cell patch-clamp electrophysiology. For recording from RMS neuroblasts identified in Nestin-CreERT2/R26R-YFP acute slices, we used an internal solution with no additional calcium and low levels (0.1 mM) of EGTA. With this solution, there should be minimal chelation of internal calcium, especially if activation of KCa3.1 occurs in microdomains. Indeed, in control experiments, TRAM-34-sensitive currents were reliably activated (n = 4), albeit of smaller amplitude than were recorded with high intracellular calcium (Fig. 6A,B). For these recordings, neuroblasts were held at −50 mV, and stepped from −140 to +80 mV in 20 mV increments. Representative current traces are shown in Figure 6A along with the drug-sensitive subtractions. Normalized I–V plots are shown in Figure 6B.
Figure 6.

Transient receptor potential (TRP) channels are required for KCa3.1 channel activation. (A) Representative traces and subtractions of patch-clamp recordings of neuroblasts in the RMS before and after addition of vehicle or calcium channel inhibitors include SKF96365 or 2-APB, and then addition of TRAM-34. As shown in the subtraction, addition of SKF96365 or 2-APB, but not vehicle, produced a drug-sensitive current. The subsequent application of TRAM-34 produced a change in current after vehicle application, but not in SKF96365 or 2-APB application, as seen in the current subtractions. (B–D) I–V plots were produced from the averaged currents elicited at baseline; after vehicle (B), SKF96365 (C), or 2-APB (D) application; and after TRAM-34 application. TRAM-34 only produced a change in current after vehicle application, but not after inhibiting the calcium source with SKF96365 or 2-APB.
To determine the calcium source for KCa3.1 activation, we first applied the calcium inhibitors SKF96365 or 2-APB. These produced a similar drug-sensitive current, as seen in representative traces in Figure 6A and I–V plots in Figure 6C,D (n = 4 and 3, respectively). With the subsequent application of TRAM-34, there was no further decrease in current (Fig. 6A,C,D). Therefore, SKF96365 and 2-APB block the calcium source activating KCa3.1, likely TRP channels, a finding in agreement with our studies on cell migration above. Supplementary Figure 5 provides the current subtractions on an I–V plot to visualize the difference between vehicle and calcium channel inhibitors.
There are no specific inhibitors to further delineate the type of TRP channel providing the calcium influx. Because TRPC1 has been implicated in chemotactic migration in gliomas (Bomben et al. 2011) and neuroblast growth cone turning during development (Li et al. 2005; Wang and Poo 2005), this appeared to be a logical candidate. Consistent with this assumption, confocal imaging on slices from Nestin-CreERT2/R26R-YFP mice shows TRPC1 expression in the YFP+ cells in the RMS (Fig. 5E). Although TRPC1 is also found outside the neuroblasts, likely on astrocytes in the glial tube, there are clear points of colocalization in YFP+ neuroblasts, as highlighted in a more magnified image in Figure 5F. Hence, TRPC1 is a potential mediator of calcium influx activating KCa3.1.
KCa3.1 Inhibition Reduces Successful Neuroblast Migration to the OB In Vivo
If KCa3.1 channels support the migration of neuroblasts in vivo, it would be expected that chronic inhibition of KCa3.1 should slow or impair the migration of neuroblasts to the OB. This question was examined in the Nestin-CreERT2/R26R-YFP mice. Since the Cre recombinase is expressed under the nestin gene, Nestin-expressing cells in the SVZ will become fluorescent on tamoxifen injection. Because Nestin is expressed by cells in the SVZ, but is lost as cells become neuroblasts (Ming and Song 2011), we can assume that any YFP+ cells found in the RMS started in or near the SVZ. By timing the injections of TRAM-34 and tamoxifen accordingly, we can track the number of YFP+ cells that have reached specific locations along the RMS, and can determine whether TRAM-34 slows RMS neuroblast migration.
Transgenic mice were injected once daily i.p. with TRAM-34 or vehicle for 5 days of pretreatment, starting at p28. The blood–brain barrier permeability of TRAM-34 has been previously demonstrated, showing that i.p. injection of TRAM-34 can produce μM levels in the rat brain (Chen et al. 2011). After the pretreatment, mice were injected with TRAM-34 or vehicle along with 180 mg/kg tamoxifen for 2 days. We performed the pretreatment to ensure that neuroblasts in the SVZ had been exposed to TRAM-34 before application of tamoxifen to fluorescently label the cells. Therefore, all fluorescent cells in the RMS at the conclusion of the experiment started in the SVZ and were exposed to inhibitory levels of TRAM-34 before moving through the RMS. Animals were sacrificed, and slices were made from fixed tissue and then stained both for DCX and YFP. The presence of DCX+ cells was used to locate slices containing the RMS and to specify the areas for analysis. After being blinded to the treatment, confocal images were taken in 4 defined areas along the RMS: Exiting the SVZ, early along the RMS, the RMS elbow, and the entry to the OB, as illustrated in Supplementary Figure 6A. Using the National Institute of Health ImageJ software Cell Counter plug-in, we determined the number of YFP+ cells in each area.
The results show that, upon administration of TRAM-34, the number of cells that reached the end of the RMS was less than half that of vehicle. Specifically, with TRAM-34 treatment, the number of cells in the RMS elbow (mean = 6.82; SEM = 1.28; n = 11) and the entry to the OB (mean = 2.27; SEM = 0.456; n = 11) was significantly less than that of vehicle (RMS El: Mean = 16.99; SEM = 4.16; n = 9; P < 0.01; OB entry: Mean = 5.94; SEM = 1.11; n = 9; P < 0.05). The number of cells exiting the SVZ (TRAM-34: Mean = 10.98; SEM = 1.51; n = 11; vehicle: Mean = 17.54; SEM = 3.63; n = 9) and in the beginning of the RMS (TRAM-34: Mean = 5.75; SEM = 0.797; n = 9; vehicle: Mean = 10.69; SEM = 2.46; n = 9) were not significantly different between treatment groups (Fig. 7A). Representative images comparing vehicle and TRAM-34 injection in the RMS elbow and OB entry are presented in Figure 7B.
Figure 7.

In vivo injection of TRAM-34 slows migration through the RMS. (A) The numbers of YFP+ cells in each area are quantified after injection of vehicle or TRAM-34. There is no significant difference in the number of cells at the exit of the SVZ or in the beginning of the RMS between treatment groups. In the later areas of the RMS, the RMS El and OB entry, there are significantly less cells in TRAM-34-treated animals. *P < 0.05, **P < 0.01. (B) Representative images of quantified sections are presented for vehicle control and TRAM-34 injection in vivo in the RMS elbow and OB entry. Scale bar 100 μm.
To assess whether the decrease in migration upon injection of TRAM-34 was caused by the drug acutely inhibiting KCa3.1, or by down regulation of the channel after chronic inhibition, we stained slices taken from the remaining hemisphere of previously analyzed animals. We found no difference in KCa3.1 expression in the RMS of vehicle and TRAM-34-injected animals, indicating that the drug acts by acutely inhibiting KCa3.1. Representative images are presented in Supplementary Figure 6D. To ensure that the change cell number was not due to alteration in death or proliferation of SVZ cells caused by injection of TRAM-34, we stained slices from the remaining hemisphere of the injected animals that were previously analyzed. We used the in situ cell death kit, TMR red, to label apoptotic cells and Ki67 to label proliferating cells. After being blinded to the treatment, images of TMR red fluorescence or Ki67+ cells in the SVZ were analyzed using the National Institute of Health ImageJ software Cell Counter. There were very few apoptotic cells overall in the SVZ of vehicle control slices [mean = 0.50, standard deviation (SD) = 0.76, n = 20 slices], and the number did not increase with TRAM-34 injection (mean = 0.40, SD = 0.60, n = 20 slices; Supplementary Fig. 6C). DNAse was applied to slices as a positive control for the cell death kit. Similarly, there was no change in proliferation after TRAM-34 injection (vehicle: Mean = 91.6, SD = 26.2, n = 40 slices; TRAM-34: Mean = 96.2, SD = 28.8, n = 32 slices; Supplementary Fig. 6B). Together, these data suggest that KCa3.1 channels are important modulators of neuroblast migration in the RMS in vivo.
Discussion
In this study, we identified a novel and hitherto unrecognized regulator of neural cell migration, the Ca2+-activated K+ channel KCa3.1. Using transgenic animals, we demonstrate that neuroblasts born in the SVZ and destined to migrate along the RMS transiently express KCa3.1, but lose this channel upon differentiation in the OB. Inhibition of KCa3.1 with the specific inhibitor, TRAM-34, or blockade of Ca2+ influx that regulates the channels slowed migration of neuroblasts by over 50% by decreasing the time the cell spent migrating. Expression of KCa3.1 correlates positively with a migratory phenotype, as channel expression was exclusively observed in motile neuroblasts. Given that KCa3.1 is Ca2+ sensitive, we screened for potential Ca2+ sources and found TRPC1 channels as a likely upstream modulator of KCa3.1. Most importantly, chronic inhibition of KCa3.1 in vivo through i.p. injection of TRAM-34 resulted in a significant reduction in neuroblasts reaching the OB.
Characterization of KCa3.1 Channels
Several lines of evidence point to the presence of KCa3.1 in migrating neuroblasts of the RMS. Whole-cell patch-clamp studies in situ revealed a current that is sensitive to TRAM-34, a specific blocker of KCa3.1. TRAM-34 is a derivative of clotrimazole, which is also a potent KCa3.1 channel blocker (Wulff et al. 2000). We found that clotrimazole inhibited the same current as TRAM-34, further indicating the presence of KCa3.1 channels (Fig. 2). Another well-described property of KCa3.1 is their Ca2+-dependence, due to Ca2+ binding by constitutively bound calmodulin in the C terminus of the protein (Sah and Faber 2002; Stocker 2004). We found that chelating intracellular Ca2+ with BAPTA led to complete inhibition of KCa3.1 (Fig. 2). Further immunohistochemical staining showed KCa3.1 channel protein in migratory neuroblasts along the RMS (Fig. 3B), yet a complete absence of the channels in differentiated neurons of the OB (Fig. 3E).
Calcium Sources for KCa3.1 Channels
KCa channels localize to microdomains and can be activated by adjacent Ca2+-permeable channels (Hosli and Hosli 1990; Vergara et al. 1998; Stocker 2004; Kim et al. 2009). Therefore, after establishing the Ca2+ dependence of KCa3.1 through whole-cell patch clamping, we used neuroblast migration to screen for upstream Ca2+ sources. We found that inhibition of VGCCs decreased neuroblast migration. This is in contrast to a recent study of neuroblasts, demonstrating that L-type calcium channels did not have a significant effect on neuroblast migration (Darcy and Isaacson 2009). The discrepancy between this study and ours may be due to differences in cerebral localization. We studied tangential migration, focusing on the early RMS, and the former study examined radial migration, which occurs after entry into the OB when neuroblasts disperse and begin to differentiate (Sun et al. 2010; Yasuda and Adams 2010). Furthermore, we found that KCa3.1 expression decreases after OB entry, implicating different mechanisms of migration in the 2 distinct areas. Despite a potential role for VGCCs in migration, our studies suggest that VGCCs are unlikely candidates for KCa3.1 activation, because inhibition of VGCCs did not inhibit neuroblast migration to the same magnitude as inhibition of KCa3.1 (Fig. 5A).
However, both 2-APB and SKF96365 slowed migration by an equal and nonadditive amount to KCa3.1 inhibition (Fig. 5B–D). 2-APB inhibits IP3R and TRP channels (Maruyama et al. 1997; Sekaran et al. 2007), and SKF96365 inhibits TRP channels, VGCCs, and STIM1 (Varnai et al. 2009). Although these drugs are not specific, the fact that both affect TRP channels and slowed migration to the same extent as KCa3.1 inhibition strongly implicates TRP channels as the Ca2+ source. This was confirmed by patch-clamp electrophysiology, where application of either 2-APB or SKF96365 markedly decreased TRAM-34-sensitive current (Fig. 6). Several studies have demonstrated that TRP channels specifically colocalize with KCa channels (Hosli and Hosli 1990; Pearce et al. 1990; Kim et al. 2009). For example, TRPC3 and TRPC6 bind to BK channels and act as a calcium source in cultured podocytes (Kim et al. 2009). In addition, in vascular smooth muscle cells, TRPC1 colocalizes with and activates BK channels, inducing membrane hyperpolarization (Hosli and Hosli 1990). We did not attempt to discern the molecular identity of the TRP channel, due to the lack of specific inhibitors. However, we observed TRPC1 expression in the RMS (Fig. 5E), and others have found TRPC1 transcripts in adult NPCs (Kong et al. 2008), making it a likely candidate worthy of further testing.
Interestingly, TRP channels themselves are known to affect migration in various systems. The most well-known studies include TRPC channel regulation of growth cone turning toward chemoattractants in developing axons. Here, inhibition of TRPC1 abolishes the calcium changes and growth cone turning in response to netrin-1 (Wang and Poo 2005). Similarly, responses to brain-derived neurotrophic factor are inhibited by expression of a dominant negative form of TRPC3 and 6 (Li et al. 2005). In glioma cells, TRPC1 is involved in chemotactic migration toward EGF (Bomben et al. 2011). From these studies, it is possible to hypothesize a scenario in which TRP channels respond to a growth factor or ligand with increasing intracellular calcium, which in turn activates KCa3.1 to facilitate migration.
To determine the relationship between KCa3.1 and calcium channels during RMS neuroblast migration, it is of interest to directly observe basal calcium changes and the effect of these inhibitors using a calcium sensor, similar to previous studies (Komuro and Rakic 1998). In situ, we bath applied a variety of calcium indicator dyes in a range of excitation/emission wavelengths, including Fura-2AM, Fluo-2 HighAff AM, Fluo-4 AM, Oregon Green 488 BAPTA-1 AM, Asante Calcium Red, and Rhod-2. While we were able to visualize the surrounding cells, the neuroblasts in the RMS were not visibly fluorescent over the background. Therefore, future observation of calcium changes in RMS neuroblasts will require manipulations through in vivo or in situ insertion of a viral construct containing a calcium sensor.
Mechanism of KCa3.1 Control of Migration
KCa3.1 has been implicated in the migration of cancerous cell types, including melanoma, pancreatic cancer, breast cancer, and prostate cancer. Nonmalignant cells that have been shown to utilize KCa3.1 in migration include fibroblasts, microglia, and leukocytes (Schwab et al. 2007). The function and localization of KCa3.1 has been studied in depth in Madin-Darby canine kidney (MDCK) cells, where it was found that either blockade or continual activation of KCa3.1 slows migration. Therefore, it is presumed that fluctuations in KCa3.1 activity, caused by calcium oscillations, are necessary for its function in migration (Schwab et al. 2007).
Here, we demonstrate that disruption of KCa3.1 suppressed neuroblast migration along the RMS. Inhibition of the channels increases the amount of time spent in the stationary phase of their saltatory migration, implicating that KCa3.1 facilitates the transition to the migrating phase. KCa3.1 could support migration through at least 2 possible mechanisms: (1) KCa3.1 may be activated to serve as a signal to initiate migration. (2) KCa3.1 may be required to retract the trailing edge of the cell to allow it to move forward.
In the RMS, neuroblasts undergo saltatory migration; however, the signals regulating the “stationary” and “migration” phases are not well studied. Based on our data, it is possible to hypothesize that cells receive a signal from a chemoattractant or growth factor, which causes a rise in calcium and subsequent activation of KCa3.1 to signal the initiation of the migratory phase. KCa3.1 could transduce the migration signal in multiple ways, but likely through changes in resting membrane potential (Catacuzzeno et al. 2012) or cell volume (Schwab et al. 2007; Becchetti 2011).
Activation of KCa3.1 leads to membrane hyperpolarization, which can affect downstream channels or other processes. For example, changes in resting membrane potential have been shown to affect voltage-sensitive potassium channels that are located near integrins that directly affect migration (Schwab et al. 2008). It is also possible that hyperpolarization leads to inactivation of VGCCs, which have also been shown to affect migration. The Ca2+ changes are thought to affect the cytoskeleton and the cell's attachment to substrates via integrins (Komuro and Rakic 1998). L-type calcium channels have been shown to be involved in the regulation of growth cone and neurite extension (Tang et al. 2003). It is possible that inhibiting KCa3.1 reduces the likelihood for hyperpolarization and, therefore, the cell is unable to complete the downstream signaling that allows the cell to move forward.
KCa3.1 could also affect volume changes necessary for migration in tissue. Dynamic regulation of cell shape and volume is necessary as cells navigate through densely populated tissue. In gliomas, K+ and Cl− ions act as osmolytes, and through the obligated movement of cytoplasmic water, the movement of these ions causes coordinated “hydrodynamic” changes in cell volume (Becchetti 2011; Watkins and Sontheimer 2011). In other cell types, KCa3.1 has been shown to specifically regulate the retraction of the trailing edge of the cell. In these cells, KCa3.1 is expressed ubiquitously on the membrane, but a gradient of calcium activates KCa3.1 only on the trailing edge. This causes an outflow of water only at the end of the cell, which causes shrinkage and allows for movement (Schwab et al. 2007; Sciaccaluga et al. 2010; Catacuzzeno et al. 2011). Retraction of the trailing edge may be necessary before the cell continues to move forward, and hindering retraction could prolong the stationary phase, as seen in KCa3.1 inhibition.
To observe whether KCa3.1 could be activated downstream of a chemoattractant source, we attempted to assess whether inhibition with TRAM-34 produced a change in directionality of cell movement. While there was no change after drug application, we also observed that the movement was not highly directional at baseline (Supplementary Fig. 3B). This is likely due to our in situ system, in which a constant perfusion of ACSF may wash out ligand or growth factor gradients and affect directional movement.
Disruption of KCa3.1 Inhibits Migration of Neuroblasts In Vivo
Neuroblasts move quickly, explaining how they cover the distance from the SVZ to the OB in just a few days (Martinez-Molina et al. 2011). Our finding that chronic inhibition of KCa3.1 in vivo decreased the speed of neuroblast migration and resulted in a reduction in the number of cells that reached the OB provides an important extension for our in situ findings, clearly ascribing an important role for the channels in the maintenance of the OB in vivo.
Although this study examined only the behavior of neuroblasts in the adult RMS, the implications of our findings are possibly more far-reaching. KCa3.1 is known to be involved in the migration of a number of cell types and, in some, the channel has been suggested to be a primary element of the migration machinery (Schwab et al. 2007). However, one major area in which KCa 3.1 has not been studied is development. In the present study, we provide evidence to support a role for KCa3.1 during adult neuroblast migration. Due to the similarities in signaling pathways between adult neurogenesis and embryonic development (Ming and Song 2011), it is possible that these findings hold true in embryonic development as well.
Adult NPCs have been suggested as one potential cell of origin for primary brain tumors, gliomas (Quinones-Hinojosa and Chaichana 2007; Zong et al. 2012). Globus and Kuhlenbeck (1944) proposed that the human brain contains a distinct cell layer lining the ventricles, the subependymal cell matrix, and containing multipotent cells that can give rise to paraventricular neoplasms, subsequently proposed to represent the possible cells of origin for a variety of gliomas (Lewis 1968). Genetic mouse models support this view (Holland et al. 1998) and explain why primary brain tumors and SVZ precursor cells share many similar characteristics. These include the expression of similar intermediate filament proteins such as Nestin and GFAP, a shared responsiveness to growth factors such as EGF and PDGF, and the activation of the pathways regulating growth and survival including Notch, PTEN, and Wnt (reviewed in Quinones-Hinojosa and Chaichana 2007). Importantly, both show a common potential to proliferate and migrate over long distances. Most pertinent to the present study, gliomas also express KCa3.1 and utilize it during migration (Sciaccaluga et al. 2010; Catacuzzeno et al. 2012) and, therefore, KCa3.1 modulators may be an opportune target for glioma therapy (Catacuzzeno et al. 2012). KCa3.1 inhibitors are currently being explored as treatment for sickle cell anemia, arthritis, immune diseases, and ischemic brain injury (Wulff et al. 2000, 2007). Clearly much more work is needed before considering KCa3.1 as a viable therapeutic target for neurological disease, yet the above described reasoning may serve as “food for thought” to consider manipulating migration in a context specific manner.
Supplementary Material
Supplementary material can be found at: http://www.cercor.oxfordjournals.org/.
Funding
This work was supported by NIH (RO1-NS031234, RO1NS-036692, and T32 GM008111).
Supplementary Material
Notes
Transgenic mice were received as a generous gift from Dr Amelia Eisch. Special thanks to Vishnu A. Cuddapah for critical reading of the manuscript. Conflict of Interest: None declared.
References
- Alvarez-Buylla A, Garcia-Verdugo JM. Neurogenesis in adult subventricular zone. J Neurosci. 2002;22:629–634. doi: 10.1523/JNEUROSCI.22-03-00629.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardy C, Pallotto M. What happens to olfaction without adult neurogenesis? Front Neurosci. 2010;4:24. doi: 10.3389/fnnes.2010.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becchetti A. Ion channels and transporters in cancer. 1. Ion channels and cell proliferation in cancer. Am J Physiol Cell Physiol. 2011;301:C255–C265. doi: 10.1152/ajpcell.00047.2011. [DOI] [PubMed] [Google Scholar]
- Bomben VC, Turner KL, Barclay TT, Sontheimer H. Transient receptor potential canonical channels are essential for chemotactic migration of human malignant gliomas. J Cell Physiol. 2011;226:1879–1888. doi: 10.1002/jcp.22518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordey A, Sontheimer H, Trouslard J. Muscarinic activation of BK channels induces membrane oscillations in glioma cells and leads to inhibition of cell migration. J Mem Biol. 2000;176:31–40. doi: 10.1007/s002320001073. [DOI] [PubMed] [Google Scholar]
- Catacuzzeno L, Aiello F, Fioretti B, Sforna L, Castigli E, Ruggieri P, Tata AM, Calogero A, Franciolini F. Serum-activated K and Cl currents underlay U87-MG glioblastoma cell migration. J Cell Physiol. 2011;226:1926–1933. doi: 10.1002/jcp.22523. [DOI] [PubMed] [Google Scholar]
- Catacuzzeno L, Fioretti B, Franciolini F. Expression and role of the intermediate-conductance calcium-activated potassium channel KCa3.1 in glioblastoma. J Signal Transduct. 2012;2012:421564. doi: 10.1155/2012/421564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YJ, Raman G, Bodendiek S, O'Donnell ME, Wulff H. The KCa3.1 blocker TRAM-34 reduces infarction and neurological deficit in a rat model of ischemia/reperfusion stroke. J Cereb Blood Flow Metab. 2011;31:2363–2374. doi: 10.1038/jcbfm.2011.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darcy DP, Isaacson JS. L-type calcium channels govern calcium signaling in migrating newborn neurons in the postnatal olfactory bulb. J Neurosci. 2009;29:2510–2518. doi: 10.1523/JNEUROSCI.5333-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/S0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH. Neurogenesis in the adult human hippocampus. Nat Med. 1998;4:1313–1317. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- Giannone G, Ronde P, Gaire M, Haiech J, Takeda K. Calcium oscillations trigger focal adhesion disassembly in human U87 astrocytoma cells. J Biol Chem. 2002;277:26364–26371. doi: 10.1074/jbc.M203952200. [DOI] [PubMed] [Google Scholar]
- Globus JH, Kuhlenbeck H. The subependymal cell plate (matrix) and its relationship to brain tumors of the ependymal type. J Neuropathol Exp Neurol. 1944;3:1–35. doi: 10.1097/00005072-194401000-00001. [DOI] [Google Scholar]
- Holland EC, Hively WP, DePinho RA, Varmus HE. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev. 1998;12:3675–3685. doi: 10.1101/gad.12.23.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosli E, Hosli L. Immunohistochemical studies on the cellular localization of GABAA-receptors in explant cultures of rat central nervous system using a monoclonal antibody. Exp Brain Res. 1990;82:667–671. doi: 10.1007/BF00228810. [DOI] [PubMed] [Google Scholar]
- Jackson EL, Alvarez-Buylla A. Characterization of adult neural stem cells and their relation to brain tumors. Cells Tissues Organs. 2008;188:212–224. doi: 10.1159/000114541. [DOI] [PubMed] [Google Scholar]
- Kim EY, Alvarez-Baron CP, Dryer SE. Canonical transient receptor potential channel (TRPC)3 and TRPC6 associate with large-conductance Ca2+-activated K+ (BKCa) channels: role in BKCa trafficking to the surface of cultured podocytes. Mol Pharmacol. 2009;75:466–477. doi: 10.1124/mol.108.051912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komuro H, Rakic P. Intracellular Ca2+ fluctuations modulate the rate of neuronal migration. Neuron. 1996;17:275–285. doi: 10.1016/S0896-6273(00)80159-2. [DOI] [PubMed] [Google Scholar]
- Komuro H, Rakic P. Orchestration of neuronal migration by activity of ion channels, neurotransmitter receptors, and intracellular Ca2+ fluctuations. J Neurobiol. 1998;37:110–130. doi: 10.1002/(SICI)1097-4695(199810)37:1<110::AID-NEU9>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Kong H, Fan Y, Xie J, Ding J, Sha L, Shi X, Sun X, Hu G. AQP4 knockout impairs proliferation, migration and neuronal differentiation of adult neural stem cells. J Cell Sci. 2008;121:24–36. doi: 10.1242/jcs.035758. [DOI] [PubMed] [Google Scholar]
- Lacar B, Young SZ, Platel JC, Bordey A. Imaging and recording subventricular zone progenitor cells in live tissue of postnatal mice. Front Neurosci. 2010;4:1–16. doi: 10.3389/fnins.2010.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagace DC, Whitman MC, Noonan MA, Ables JL, DeCarolis NA, Arguello AA, Donovan MH, Fischer SJ, Farnbauch LA, Beech RD, et al. Dynamic contribution of nestin-expressing stem cells to adult neurogenesis. J Neurosci. 2007;27:12623–12629. doi: 10.1523/JNEUROSCI.3812-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PD. Mitotic activity in the primate subependymal layer and the genesis of gliomas. Nature. 1968;217:974–975. doi: 10.1038/217974a0. [DOI] [PubMed] [Google Scholar]
- Li Y, Jia YC, Cui K, Li N, Zheng ZY, Wang YZ, Yuan XB. Essential role of TRPC channels in the guidance of nerve growth cones by brain-derived neurotrophic factor. Nature. 2005;434:894–898. doi: 10.1038/nature03477. [DOI] [PubMed] [Google Scholar]
- Lyons SA, Chung WJ, Weaver AK, Ogunrinu T, Sontheimer H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 2007;67:9463–9471. doi: 10.1158/0008-5472.CAN-07-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Molina N, Kim Y, Hockberger P, Szele FG. Rostral migratory stream neuroblasts turn and change directions in stereotypic patterns. Cell Adhes Migr. 2011;5:83–95. doi: 10.4161/cam.5.1.13788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini FJ, Valdeolmillos M. Actomyosin contraction at the cell rear drives nuclear translocation in migrating cortical interneurons. J Neurosci. 2010;30:8660–8670. doi: 10.1523/JNEUROSCI.1962-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama T, Kanaji T, Nakade S, Kanno T, Mikoshiba K. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J Biochem. 1997;122:498–505. doi: 10.1093/oxfordjournals.jbchem.a021780. [DOI] [PubMed] [Google Scholar]
- Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70:687–702. doi: 10.1016/j.neuron.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori Y, Matsubara H, Folco E, Siegel A, Koren G. The transcription of a mammalian voltage-gated potassium channel is regulated by cAMP in a cell-specific manner. J Biol Chem. 1993;268:26482–26493. [PubMed] [Google Scholar]
- Pearce B, Morrow C, Murphy S. Further characterisation of excitatory amino acid receptors coupled to phosphoinositide metabolism in astrocytes. Neurosci Lett. 1990;113:298–303. doi: 10.1016/0304-3940(90)90601-5. [DOI] [PubMed] [Google Scholar]
- Prickaerts J, Koopmans G, Blokland A, Scheepens A. Learning and adult neurogenesis: survival with or without proliferation? Neurobiol Learn Mem. 2004;81:1–11. doi: 10.1016/j.nlm.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Quinones-Hinojosa A, Chaichana K. The human subventricular zone: a source of new cells and a potential source of brain tumors. Exp Neurol. 2007;205:313–324. doi: 10.1016/j.expneurol.2007.03.016. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ES. Channels underlying neuronal calcium-activated potassium currents. Prog Neurobiol. 2002;66:345–353. doi: 10.1016/S0301-0082(02)00004-7. [DOI] [PubMed] [Google Scholar]
- Saha B, Jaber M, Gaillard A. Potentials of endogenous neural stem cells in cortical repair. Front Cell Neurosci. 2012;6:14. doi: 10.3389/fncel.2012.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanai N, Nguyen T, Ihrie RA, Mirzadeh Z, Tsai HH, Wong M, Gupta N, Berger MS, Huang E, Garcia-Verdugo JM, et al. Corridors of migrating neurons in the human brain and their decline during infancy. Nature. 2011;478:382–386. doi: 10.1038/nature10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab A, Hanley P, Fabian A, Stock C. Potassium channels keep mobile cells on the go. Physiology (Bethesda) 2008;23:212–220. doi: 10.1152/physiol.00003.2008. [DOI] [PubMed] [Google Scholar]
- Schwab A, Nechyporuk-Zloy V, Fabian A, Stock C. Cells move when ions and water flow. Pflugers Arch. 2007;453:421–432. doi: 10.1007/s00424-006-0138-6. [DOI] [PubMed] [Google Scholar]
- Sciaccaluga M, Fioretti B, Catacuzzeno L, Pagani F, Bertollini C, Rosito M, Catalano M, D'Alessandro G, Santoro A, Cantore G, et al. CXCL12-induced glioblastoma cell migration requires intermediate conductance Ca2+-activated K+ channel activity. Am J Physiol Cell Physiol. 2010;299:C175–C184. doi: 10.1152/ajpcell.00344.2009. [DOI] [PubMed] [Google Scholar]
- Sekaran S, Lall GS, Ralphs KL, Wolstenholme AJ, Lucas RJ, Foster RG, Hankins MW. 2-Aminoethoxydiphenylborane is an acute inhibitor of directly photosensitive retinal ganglion cell activity in vitro and in vivo. J Neurosci. 2007;27:3981–3986. doi: 10.1523/JNEUROSCI.4716-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soroceanu L, Akhavan A, Cobbs CS. Platelet-derived growth factor-alpha receptor activation is required for human cytomegalovirus infection. Nature. 2008;455:391–395. doi: 10.1038/nature07209. [DOI] [PubMed] [Google Scholar]
- Stewart RR, Zigova T, Luskin MB. Potassium currents in precursor cells isolated from the anterior subventricular zone of the neonatal rat forebrain. J Neurophysiol. 1999;81:95–102. doi: 10.1152/jn.1999.81.1.95. [DOI] [PubMed] [Google Scholar]
- Stocker M. Ca(2+)-activated K+ channels: molecular determinants and function of the SK family. Nat Rev Neurosci. 2004;5:758–770. doi: 10.1038/nrn1516. [DOI] [PubMed] [Google Scholar]
- Sun W, Kim H, Moon Y. Control of neuronal migration through rostral migration stream in mice. Anat Cell Biol. 2010;43:269–279. doi: 10.5115/acb.2010.43.4.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, Dent EW, Kalil K. Spontaneous calcium transients in developing cortical neurons regulate axon outgrowth. J Neurosci. 2003;23:927–936. doi: 10.1523/JNEUROSCI.23-03-00927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnai P, Hunyady L, Balla T. STIM and Orai: the long-awaited constituents of store-operated calcium entry. Trends Pharmacol Sci. 2009;30:118–128. doi: 10.1016/j.tips.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergara C, Latorre R, Marrion NV, Adelman JP. Calcium-activated potassium channels 39. Curr Opin Neurobiol. 1998;8:321–329. doi: 10.1016/S0959-4388(98)80056-1. [DOI] [PubMed] [Google Scholar]
- Wang DD, Krueger DD, Bordey A. Biophysical properties and ionic signature of neuronal progenitors of the postnatal subventricular zone in situ. J Neurophysiol. 2003;90:2291–2302. doi: 10.1152/jn.01116.2002. [DOI] [PubMed] [Google Scholar]
- Wang GX, Poo MM. Requirement of TRPC channels in netrin-1-induced chemotropic turning of nerve growth cones. Nature. 2005;434:898–904. doi: 10.1038/nature03478. [DOI] [PubMed] [Google Scholar]
- Watkins S, Sontheimer H. Hydrodynamic cellular volume changes enable glioma cell invasion. J Neurosci. 2011;31:17250–17259. doi: 10.1523/JNEUROSCI.3938-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff H, Kolski-Andreaco A, Sankaranarayanan A, Sabatier JM, Shakkottai V. Modulators of small- and intermediate-conductance calcium-activated potassium channels and their therapeutic indications. Curr Med Chem. 2007;14:1437–1457. doi: 10.2174/092986707780831186. [DOI] [PubMed] [Google Scholar]
- Wulff H, Miller MJ, Hansel W, Grissmer S, Cahalan MD, Chandy KG. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: a potential immunosuppressant. Proc Natl Acad Sci USA. 2000;97:8151–8156. doi: 10.1073/pnas.97.14.8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda T, Adams DJ. Physiological roles of ion channels in adult neural stem cells and their progeny. J Neurochem. 2010;114:946–959. doi: 10.1111/j.1471-4159.2010.06822.x. [DOI] [PubMed] [Google Scholar]
- Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132:645–660. doi: 10.1016/j.cell.2008.01.033. [DOI] [PubMed] [Google Scholar]
- Zong H, Verhaak RG, Canoll P. The cellular origin for malignant glioma and prospects for clinical advancements. Expert Rev Mol Diagn. 2012;12:383–394. doi: 10.1586/erm.12.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.