

Abstract
The applications of ethylenediaminetetraacetic acid (EDTA) have been expanded from the treatment of heavy metal poisoning to chelation therapies for atherosclerosis, heart disease, and cancers, in which EDTA reduces morbidity and mortality by chelating toxic metal ions. In this study, EDTA was used in a drug delivery system by adopting an NH4EDTA gradient method to load doxorubicin into liposomes with the goal of increasing therapeutic effects and decreasing drug-related cytotoxicity. The particle size of the optimum NH4EDTA gradient liposomes was 79.4±1.87 nm, and the entrapment efficiency was 95.54%±0.59%. In vitro studies revealed that liposomes prepared using an NH4EDTA gradient possessed long-term stability and delayed drug release. The in vivo studies also showed the superiority of the new doxorubicin formulation. Compared with an equivalent drug dose (5 mg/kg) prepared by (NH4)2SO4 gradient, NH4EDTA gradient liposomes showed no significant differences in tumor inhibition ratio, but cardiotoxicity and liposome-related immune organ damage were lower, and no drug-related deaths were observed. These results show that use of the NH4EDTA gradient method to load doxorubicin into liposomes could significantly reduce drug toxicity without influencing antitumor activity.
Keywords: NH4EDTA, liposome, doxorubicin, ion gradient, antitumor activity, toxicity
Introduction
Ethylenediaminetetraacetic acid (EDTA) is a general chelating agent that was approved by the US Food and Drug Administration for the treatment of heavy metal poisoning in the early 1950s, and remains the first choice for such treatment.1–6 From the earliest clinical trials, EDTA-mediated chelation therapy has consistently demonstrated a remarkable ability to cleanse the organism of metals and other deposits that are responsible for atherosclerosis, heart disease, and cancers.7–12 It has been reported that EDTA can reduce radical reactions and oxidation processes by chelating toxic metals that are responsible for cell membrane injury.13 EDTA and its analogs are inhibitors of calcium/calmodulin-dependent protein kinase II and protein kinase C, which play important roles in the occurrence and development of numerous diseases.14 EDTA can mitigate hypercalcinemia, soften arteries, and reduce the incidence of osteoporosis by chelating calcium ions,15,16 and it can also reduce cancer mortality by inhibiting suppression of the immune system.10,17,18 Furthermore, EDTA can improve the stability of preparations by inhibiting bacterial contamination via chelating divalent metal ions, which are essential for germs.19
Doxorubicin (DOX) is one of the best-known members of the anthracycline family of antibiotics. It was first introduced in the 1960s and was widely utilized against hematological and solid tumors.20 However, the antitumor effects of DOX were often followed by numerous undesired side effects, in particular serious cardiotoxicity,21 which led to research devoted to investigating the mechanisms of its antitumor and cardiotoxic effects, and the relationship between them. Although the mechanisms might overlap to some extent, it is generally thought that DOX exerts its antitumor effects and cardiotoxicity by distinct mechanisms: the former is associated with DNA intercalation, topoisomerase-II inhibition, and apoptosis, while the latter is mainly mediated by oxidative stress.22 Trace element research also showed that the antitumor effects of DOX generally do not require the participation of transition metal ions, while its cardiotoxic effects do.23–25 Therefore, it is believed that the antitumor effects of DOX will not be influenced by a decreased concentration of transition metal ions in vivo, but that cardiotoxicity will be weakened by the reduction in reactive oxygen species (ROS).26,27
Thus far, a common approach to prevent DOX-related cardiotoxicity has been to employ drug carriers that utilize an enhanced permeation and retention effect to alter tissue distribution and pharmacokinetics of the drug.28–30 The loading of DOX into liposomes by the application of a transmembrane NH4EDTA gradient method should maintain the advantages of liposomes and also afford several other known and potential benefits. First, intraliposomal EDTA should increase the stability of liposomal formulations by inhibiting bacterial contamination and the catalytic effect of metal ions on lipids.19 Second, EDTA reduces the generation of ROS by chelating transition metal ions, which should decrease damage to the cardiomyocyte membrane and reduce the risk of DOX-related cardiomyopathy.13 Previous studies have shown that liposomes prepared by a transmembrane NH4EDTA gradient method can significantly increase drug retention and decrease liposome-related damage to the immune system compared with liposomes prepared by other gradient methods.31,32 Finally, EDTA binds to calcium, which should lead to a decreased incidence rate of hypercalcinemia and improve the quality of life of advanced cancer patients.16
In this paper, EDTA was used in a drug delivery system to encapsulate doxorubicin into liposomes using a transmembrane NH4EDTA gradient. Through comparison with the classic ammonium sulfate gradient method, it was investigated whether liposomes prepared via an NH4EDTA gradient show superior therapeutic efficacy and decreased toxicity as well as the potential advantages of EDTA mentioned above.
Materials and methods
Materials
DOX hydrochloride was provided by Huafeng Lianbo Technology Co. Ltd. (Beijing, People’s Republic of China). Hydrogenated soy phosphatidylcholine (HSPC; MW 785) was obtained from Lucas Meyer GmbH (Düsseldorf, Germany). Cholesterol (CH) was supplied by Nanjing Xinbai Pharmaceutical Co. Ltd. (Nanjing, People’s Republic of China). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-(methoxy[polyethylene glycol]-2000) (mPEG2000-DSPE) was purchased from J&K Scientific (Beijing, People’s Republic of China). Sephadex® G-50 (medium) was obtained from Sigma-Aldrich (St Louis, MO, USA). All other reagents were of analytical grade.
Animals
Male Kunming mice weighing 18–22 g were purchased from the Experimental Animal Center of Shenyang Pharmaceutical University (Shenyang, People’s Republic of China). All mice had free access to food and water and all animal experiments were in accordance with the guidelines for the Care and Use of Laboratory Animals at Shenyang Pharmaceutical University.
Preparation of liposomes
Liposomes were prepared using a modified ethanol injection method. Briefly, a lipid mixture containing HSPC/CH/mPEG2000-DSPE (3:1:1, w/w/w) was dissolved in ethanol at 65°C, and the solvent was subsequently removed by stirring. The resulting lipid film was hydrated in 200 mM NH4EDTA solution (pH 5.5) under magnetic agitation at an appropriate rate. Liposomes containing an inner aqueous medium of 200 mM (NH4)2SO4 (pH 5.5) were also prepared for the in vitro release assays, long-term stability tests, and animal experiments.
The initially produced bulk liposome suspension was transformed into small unilamellar vesicles by sonication using a JY92-2D Vibra-cell probe sonicator (Ningbo Xinzhi Biological Technology Co. Ltd., Zhejiang, People’s Republic of China) for an initial 6-minute cycle (400 W; 3 seconds on, 3 seconds off) followed by a 2-minute cycle (200 W). The resulting vesicles were extruded through polycarbonate membranes with gradually decreasing pore sizes (0.8, 0.45, and 0.22 μm) to remove large particles.
The mean diameters and size distributions of the liposomes were estimated using a dynamic laser light scattering instrument (Nicomp™ 380 Submicron Particle Sizer; Particle Sizing Systems, Port Richey, FL, USA) operated at a wavelength of 632.8 nm. All samples were diluted with 5% glucose solution and measurements were conducted at 25°C in triplicate.
Preparation of the ammonium gradient for drug encapsulation
The ammonium gradients of the liposomes were established using a dialysis method monitored by ion pair high- performance liquid chromatography (IP-HPLC). Liposome suspensions (2 mL) consisting of HSPC/CH/mPEG2000-DSPE (3:1:1, w/w/w) were placed in dialysis tubing with a molecular weight cutoff of 10 kDa and dialyzed against 500 mL of 5% glucose solution. The glucose solution was changed at intervals of half an hour, and the dialysis was ceased when the IP-HPLC results stayed constant. The dialyzed liposomes were diluted with the glucose solution to a final lipid concentration of 20 mg/mL. Subsequently, DOX solution (2 mg/mL) was added to the liposomal suspension to achieve a drug/lipid ratio of 1:10 (w/w). The loading process was carried out at 60°C for 30 minutes, and then the mixture was cooled rapidly in an ice/water bath (0°C–2°C) for 5 minutes.
Determination of encapsulation efficiency
Non-entrapped drug was removed by using size exclusion chromatography to pass the DOX-loaded vesicle dispersions through a Sephadex G-50 mini-column (10×70 mm). The separation of 0.2 mL liposomes from free DOX was carried out by eluting with distilled water (0.4 mL, four times), and the concentration of DOX was determined by an ultraviolet spectrophotometer (UV-1801 UV/VIS Spectrophotometer, Beijing Rayleigh Analytical Instrument Co., Ltd, People’s Republic of China) at a wavelength of 480 nm after lysis of the liposomes with 90% (v/v) isopropyl alcohol containing 0.75 M hydrochloric acid. The encapsulation efficiency was calculated as the percentage of DOX remaining in the liposomes after elution, and DOX concentration was calculated using the lipid concentration.
Solubility of DOX in different mediums
The solubility of DOX was investigated in NH4EDTA (200 mM) and (NH4)2SO4 (200 mM) solutions. The pH values of these salt solutions were adjusted with aqueous ammonia solution or hydrochloric acid. In brief, 100 mg of lyophilized DOX powder was dissolved with diluted water to a concentration of 10 mg/mL, and 0.2 mL of this solution was added to 0.8 mL of 250 mM NH4EDTA or (NH4)2SO4 solutions to obtain final drug concentrations in each solution of 2 mg/mL. Each sample was vortexed for 2 minutes and incubated at 25°C for 48 hours in a homeothermic oscillator with a velocity of 100 rpm under low-light conditions. The drug precipitate was separated from the supernatant by centrifugation (10,000 rpm for 15 minutes) and the DOX concentration in the supernatant was measured photometrically at a wavelength of 480 nm.
Release experiments in vitro for DOX-loaded liposomes
Two types of release medium were used in the release assays: a conventional phosphate-buffered saline (PBS) and a special PBS buffer containing ammonium chloride (80 mM), histidine (10 mM), penicillin (100 μg/mL), and streptomycin (100 μg/mL).33 The pH values of both PBS buffers were adjusted to 7.4 with hydrochloric acid and osmotic pressure was adjusted to 280–320 mOsm with glucose. Briefly, 2 mL of HSPC/CH/mPEG2000-DSPE (3:1:1, w/w/w) liposomes containing DOX (drug/lipid ratio 1:10, w/w) were placed in dialysis tubing with a molecular weight cutoff of 10 kDa and dialyzed against 198 mL release buffer. The dialysis process was performed at 37°C with a velocity of 75 rpm under low-light conditions. At indicated time points, aliquots were withdrawn and analyzed using a fluorescence spectrophotometer (650-60; Hitachi Ltd., Tokyo, Japan) with excitation and emission wavelengths set at 472 nm and 591 nm, respectively.
Long-term stability experiments
The liposomal formulations were assessed for stability over a period. HSPC/CH/mPEG2000-DSPE (3:1:1, w/w/w) liposomal formulations (10 mg/mL) containing DOX (drug/lipid ratio 1:10, w/w) and driven by NH4EDTA (200 mM) or (NH4)2SO4 gradient (200 mM) (which is marked as NH4EDTA-L and (NH4)2SO4-L in the following texts, respectively) were stored at 4°C±2°C for a period of 3 months. Stability was evaluated in terms of appearance, particle size, and encapsulation efficiency at selected time intervals during the storage period by methods described above.
Antitumor activity tests
Antitumor activities of DOX preparations were investigated by a xenograft mouse model of liver cancer (H22). In brief, tumors were established in 6–8 weeks old Kunming male mice on Day 0 by injecting H22 tumor cells (provided by the Cancer Institute of Liaoning Province, Shenyang, People’s Republic of China) subcutaneously at the right axillary flank. Treatments consisted of multiple dosing on Days 6, 9, 12, and 15, and the animals were sacrificed on Day 16. Sixty tumor-bearing mice were randomly divided into six groups (n=10 per group). For the treatment groups, drug dosages are listed in Table 1, and the control group received injections of normal saline. During this period, mice had free access to food and water and their weights were recorded from the day of tumor injection to the day of execution. Necropsies were conducted to obtain the tumor, spleen, thymus gland, and heart. Tumor inhibition ratio, spleen index, and thymus gland index were calculated and served as indicators for the evaluation of antitumor activity and immunotoxicity.
Table 1.
The dosage of DOX for pharmacodynamic test
| Group | DOX solution | NH4EDTA-L
|
(NH4)2SO4-L | ||
|---|---|---|---|---|---|
| Low | Medium | High | |||
| Dosage (mg/kg) | 5 | 2.5 | 5 | 10 | 5 |
Abbreviations: DOX, doxorubicin; EDTA, ethylenediaminetetraacetic acid; L, doxorubicin-loaded liposomes.
Tissue distribution by repeated injection
Mice bearing H22 tumors were randomly divided into six groups (n=10 per group) for tissue distribution studies. The formulations administered were DOX solution, NH4EDTA-L, and (NH4)2SO4-L, and doses were the same as those used in the antitumor activity tests. Treatments consisted of multiple dosing on Days 6, 9, 12, and 15 after inoculation with H22 cells. Blood samples were collected at 24 hours after the last drug administration via the retroorbital collection method. Briefly, 0.1 mL blood samples were diluted to 5 mL with methanol/water (50:50, v/v) containing 0.3 M HCl and centrifuged immediately at 10,000 rpm for 10 minutes, and the supernatants were kept for analysis. The heart, liver, spleen, lungs, kidneys, and brain of each mouse were rapidly excised following blood collection and weighed. Tissue samples of 0.05 g were excised and all tissue samples were homogenized. The homogenates were diluted to 5 mL with methanol/water (50:50, v/v) containing 0.3 M HCl and centrifuged immediately at 10,000 rpm for 10 minutes, and the supernatants were kept for analysis. After DOX extraction from the serum and tissue-based samples, DOX levels were determined by fluorescence.
Histopathological studies of cardiotoxicity
Cardiac tissue from ventricles was excised from the mice used in the tissue distribution experiments, washed by normal saline, fixed in 10% formalin solution, cleaned by ethanol, and embedded in paraffin wax. The embedded samples were placed into the tissue slicer to make ultra-thin slices. After heating at 70°C, the slices were stained with hematoxylin and eosin and observed under a fluorescence microscope (IX71; Olympus Corporation, Tokyo, USA).
Statistical analysis
Statistical comparisons were performed using Student’s t-test for two groups and one-way analysis of variance for multiple groups. The significance threshold was set such that P-values less than 0.05 were considered to be statistically significant, and P-values less than 0.01 were considered to be extremely significant.
Results
Solubility of DOX in different mediums
As shown in Figure 1, the solubility of DOX in (NH4)2SO4 and NH4EDTA solutions at 25°C was dependent on pH. The solubility of DOX in 200 mM (NH4)2SO4 solution was almost invariable from pH 4 to pH 7 (approximately 0.23 mg/mL) and decreased gradually as pH was raised above pH 7. In 200 mM NH4EDTA solution, the solubility of DOX decreased sharply from 1.92 mg/mL (at pH 4) to 0.23 mg/mL (at pH 5), and the variation in solubility was comparable to that of (NH4)2SO4 solution above pH 5. The solubility of DOX in (NH4)2SO4 and NH4EDTA solutions at pH 4 was further examined by dilution with distilled water. As shown in Figure 2, DOX formed a precipitate in (NH4)2SO4 solution after centrifugation, while it was well dispersed in the NH4EDTA solution. However, when diluted with isochoric distilled water, the supernatant of the (NH4)2SO4 solution appeared to be homogeneous, while that of the NH4EDTA solution appeared layered.
Figure 1.
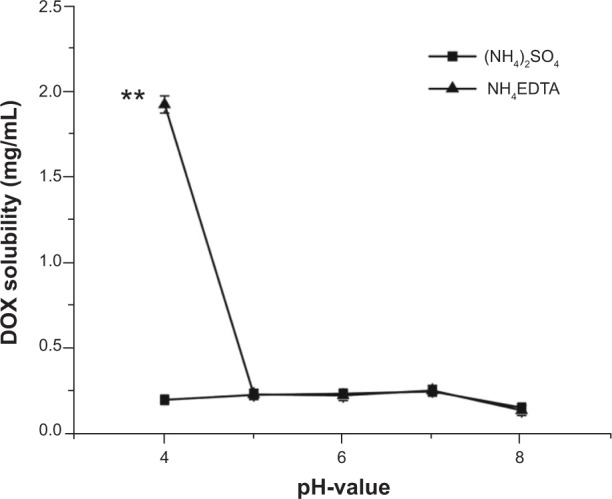
A comparison of the solubilities of DOX in different salt solutions as a function of pH. The solubilities of DOX were quantified after a 24-hour incubation of 2 mg DOX within 1 mL of 200 mM salt solutions of different pH at 25°C.
Note: **P<0.01 versus (NH4)2SO4 group.
Abbreviations: DOX, doxorubicin; EDTA, ethylenediaminetetraacetic acid.
Figure 2.

Photographs of mixtures of DOX with (1) (NH4)2SO4 and (2) NH4EDTA at 25°C and pH 4 (A) before centrifugation, (B) after centrifugation, and (C) diluted with isochoric distilled water.
Abbreviations: DOX, doxorubicin; EDTA, ethylenediaminetetraacetic acid.
Release experiments in vitro for DOX-loaded liposomes
Drug release experiments were performed for the quantitative comparison of the release kinetics of DOX from NH4EDTA-L and (NH4)2SO4-L, and the results are shown in Figure 3. When the release medium was conventional PBS (pH 7.4), the drug release rates from both liposomes over 24 hours were less than 5% with no significant difference between them (P>0.05). When the release medium was a special PBS buffer (pH 7.4) with ammonium chloride (80 mM), the release of encapsulated drug from NH4EDTA-L was much slower than (NH4)2SO4-L, and the total amounts of released DOX were 11.4% and 23.7%, respectively. Furthermore, it was noted that was a small difference between the release profiles of the two DOX liposomes. For the NH4EDTA-L preparation, release occurred at a constant slow and steady rate, while release from the (NH4)2SO4-L preparation followed a biphasic pattern, in which the first phase of drug release was a rapid response that lasted nearly 5 hours, which was followed by a slow and prolonged second phase.
Figure 3.
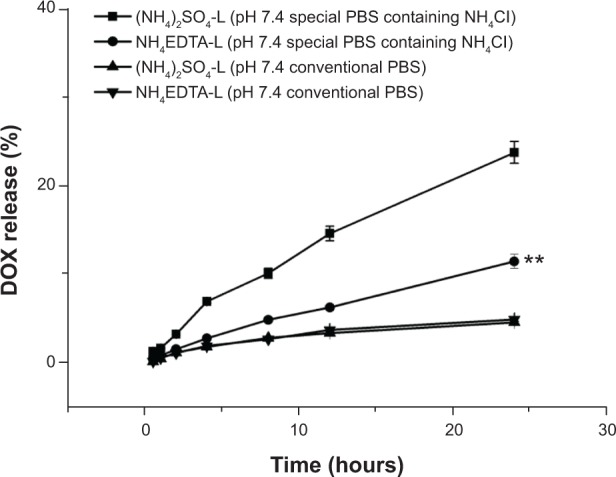
A comparison of the release of DOX encapsulated in liposomes in vitro via different ion gradients: 200 mM NH4EDTA solution and 200 mM (NH4)2SO4 solutions. Drug release profiles were analyzed in conventional PBS buffer (pH 7.4) and a special PBS buffer with ammonium chloride (80 mM), histidine (10 mM), penicillin (100 μg/mL), and streptomycin (100 μg/mL) (pH 7.4).
Note: **P<0.01 versus (NH4)2SO4-L in PBS buffer with ammonium chloride.
Abbreviations: DOX, doxorubicin; EDTA, ethylenediaminetetraacetic acid; L, doxorubicin-loaded liposomes; PBS, phosphate-buffered saline.
Long-term stability experiments
The long-term stability of the liposomal formulations was determined during 90 days of storage at 4°C±2°C, and changes in particle sizes and entrapment efficiencies are shown in Table 2. When stored at 4°C±2°C for 1 month, neither formulation showed obvious changes in entrapment efficiency (P>0.05), but both showed a slight increase in particle size (P<0.05). Precipitation was observed at Day 60 in the (NH4)2SO4-L preparation, but not in the NH4EDTA-L preparation. Over the 3-month study period, the NH4EDTA-L preparation showed constant entrapment efficiency and an increase in particle size from 79.4±1.87 nm to 106.2±3.55 nm.
Table 2.
Long-term stability of HSPC/CH/mPEG2000-DSPE (3/1/1, w/w/w) liposomes loaded with doxorubicin (drug/lipid ratio 1:10, w/w) via 200 mM NH4EDTA and (NH4)2SO4 gradient stored at 4°C±2°C
| Day | NH4EDTA-L
|
(NH4)2SO4-L
|
||||
|---|---|---|---|---|---|---|
| Appearance | Particle size ± SD (nm; n =3) | Entrapment efficiency ± SD (%; n=3) | Appearance | Particle size ± SD (nm; n=3) | Entrapment efficiency ± SD (%; n=3) | |
| 0 | + | 79.4±1.87 | 95.54±0.59 | + | 76.9±2.15 | 95.07±0.78 |
| 15 | + | 80.9±2.83 | 97.29±1.16 | + | 82.5±8.00 | 95.92±0.74 |
| 30 | + | 87.1±3.25 | 96.02±1.35 | + | 87.4±5.48 | 95.52±0.78 |
| 60 | + | 93.1±4.85 | 95.60±1.04 | ↓ | – | – |
| 90 | + | 106.2±3.55 | 95.32±1.05 | ↓ | – | – |
Notes: + represents cherry red liposomes, clear, without precipitation, phase separation, and bacterium; ↓ represents cherry red liposomes, precipitation, or bacterium; and – represents no data.
Abbreviations: CH, cholesterol; EDTA, ethylenediaminetetraacetic acid; HSPC, hydrogenated soy phosphatidylcholine; L, doxorubicin-loaded liposomes; mPEG2000-DSPE, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-(methoxy[polyethylene glycol]-2000); SD, standard deviation; w/w, weight/weight.
Antitumor activity tests
Antitumor activities of DOX solution, NH4EDTA-L DOX preparations (different concentrations), and the (NH4)2SO4-L preparation were evaluated in a rapidly growing H22 liver cancer xenograft model in mice. As shown in Figure 4A, all of the DOX preparations (solution and liposomes) produced extremely significant antitumor effects (P<0.01 versus saline). The antitumor effects observed in all liposome groups were greater than those of the free DOX solution group (P<0.05). There was no significant difference (P>0.05) among the groups that received different doses (low, medium, and high) of the NH4EDTA-L preparation. Tumor inhibition was comparable for equivalent doses of the NH4EDTA-L and (NH4)2SO4-L preparations, but the numbers of deaths in these groups were zero and two, respectively.
Figure 4.
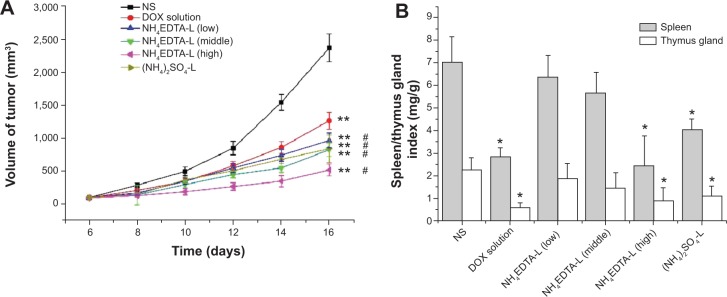
(A) Tumor inhibition effects of different DOX formulations. The drug doses were 5.0 mg/kg for DOX solution and (NH4)2SO4-L, and 2.5, 5.0, 10.0 mg/kg for the low, middle, and high concentration of NH4EDTA-L, respectively. (B) Damage of different formulations on immune organs of H22-bearing mice.
Notes: The data represent mean ± standard deviation. *P<0.05 versus NS-treated mice; **P<0.01 versus NS-treated mice; #P<0.05 versus DOX solution-treated mice.
Abbreviations: DOX, doxorubicin; EDTA, ethylenediaminetetraacetic acid; L, doxorubicin-loaded liposomes; NS, normal saline.
The body weights of tumor-bearing mice were also determined. For the saline-treated group, body weights increased from Day 0–12 as the tumor grew. After Day 12, the tumors in several mice progressed to a degree that resulted in loss of life quality and weight loss. For the DOX solution group, persistent weight loss was observed from the first treatment (6 days after inoculation), and four deaths occurred before the group was sacrificed. For the DOX-loaded liposome groups, low- and medium-dose NH4EDTA-L groups showed similar body weight changes; weight loss was observed from the second drug administration and there were no deaths during the observation period. Body weights at sacrifice were similar to those recorded on the first treatment day. For (NH4)2SO4-L and high-dose NH4EDTA-L groups, significant weight loss occurred after the second administration and two and three deaths were observed, respectively, after the third treatment.
Spleen index and thymus gland index were taken as indicators (weight of the spleen or thymus gland/body weight of the mouse, expressed in mg/g) to evaluate liposome-related damage to immune organs. As described in Figure 4B, compared with the saline-treated group, no significant weight loss was observed in the spleens or the thymus glands from low-dose and medium-dose NH4EDTA-L groups (P>0.05), but significant weight loss was observed in organs collected from the DOX solution, high-dose NH4EDTA-L, and (NH4)2SO4-L groups (P<0.05). Compared with an equivalent dose of the NH4EDTA-L preparation, spleen weight loss was significantly greater in mice treated with the (NH4)2SO4-L preparation (P<0.05).
Tissue distribution studies
In the in vivo distribution studies, the disposition of DOX after intravenous administration of each preparation was investigated. As seen in Figure 5A, the distribution of DOX in the tumor and all measured tissues was very low after intravenous administration of DOX solution, which was owing to its short half-life and the rapid elimination that occurred without the protection of vehicles. In the case of liposomal preparations, NH4EDTA-L (low, medium, and high doses) and (NH4)2SO4-L showed excellent targeting to the tumor, as indicated by a significant difference in disposition compared to tumor tissue from the free DOX solution-treated group (P<0.01). Compared with the equivalent dose of (NH4)2SO4-L, tissue accumulation after NH4EDTA-L administration was lower in the heart (by 45%), the liver (by 16%), and the spleen (by 72%), but higher in tumors (by 28%). Variation in dosage of each preparation also affected tissue distribution; as shown in Figure 5B, the drug concentrations measured in all tissues increased at various rates as the administered dosage increased. For example, when the dose was increased from 5 mg/kg to 10 mg/kg, measured DOX increased by 92% in tumors, 258% in the liver, 2,323% in the spleen, and 55% in the heart. In addition, the damage that was caused by drug administration to the liver and spleen increased sharply.
Figure 5.

Tissue distributions of DOX in mice after intravenous administration of (A) 5 mg/kg DOX solution, NH4EDTA-L, and (NH4)2SO4-L and (B) 5.0 mg/kg DOX solution and different doses of NH4EDTA-L (2.5, 5.0, and 10.0 mg/kg).
Notes: The data represent mean ± standard deviation (n=10). Differences between the groups versus DOX solution are statistically significant at the P<0.01 level.
Abbreviations: DOX, doxorubicin; EDTA, ethylenediaminetetraacetic acid; L, doxorubicin-loaded liposomes.
Histopathological studies of cardiotoxicity
Slice samples of ventricles were observed under a fluorescence microscope using a 40× objective. Vacuolization, edema, and inflammatory cell infiltration were not observed in any tested cardiac tissue samples. However, all of the experimental groups showed fragmentation of the myocardium to different degrees, which were markedly different from normal Henle’s fissures. Typical images are shown in Figure 6, and the myofibrillar damage sites are indicated by black circles. The ranking of cardiotoxicity produced in all of the groups, as indicated by the area and severity of the damage sites, was as follows: saline-treated < NH4EDTA-L (low-dose) < NH4EDTA-L (medium-dose) < (NH4)2SO4-L < NH4EDTA-L (high-dose) < free DOX-treated. All liposomal formulation groups showed less DOX-induced cardiotoxicity than the free DOX solution group. Compared with equivalent doses of the NH4EDTA-L preparations, the (NH4)2SO4-L-treated group showed significantly greater toxicity, which was similar to that observed in the high-dose NH4EDTA-L group.
Figure 6.

Typical images of pathological classification of myocardium injury of different DOX formulations: (A) NS; (B) DOX solution; (C) NH4EDTA-L (low-dose); (D) NH4EDTA-L (medium-dose); (E) NH4EDTA-L (high-dose); and (F) (NH4)2SO4-L.
Notes: Photographs were obtained under a fluorescence microscope (IX71; Olympus Corporation, Tokyo, Japan) using a 40× objective. The black circles point out the damaged sites of myocardium fragmentation.
Abbreviations: DOX, doxorubicin; EDTA, ethylenediaminetetraacetic acid; L, doxorubicin-loaded liposomes; NS, normal saline.
Discussion
Drug retention characteristics of liposomal formulations affect in vitro stability and in vivo dissolution rates, and indirectly affect the activity and toxicity of the loaded drugs.34–36 If the loaded drug forms an insoluble complex with the hydration medium in the liposomes, and the dissolution rate of the complex is slower than that of the drug departing through the bilayer, then increased drug retention will be observed.31 In addition to drug properties, retention is also dependent on the anionic varieties present and concentrations of the inner aqueous medium. When citric acid and ammonium sulfate gradients were employed to load DOX into liposomes, precipitation was observed as fiber bundles by cryo-electron microscopy with no observable membrane invagination.34 A similar result was obtained in liposomes loaded with idarubicin by an NH4EDTA gradient.31 In the current solubility studies, the biggest difference in DOX solubility between NH4EDTA and (NH4)2SO4 solutions appeared at pH 4. The solubility of DOX seemed ten times greater in the NH4EDTA solution than in the (NH4)2SO4 solution. In fact, DOX formed a gel-like complex that could not be precipitated by centrifugation. This difference in the solubility of DOX in different hydration mediums also resulted in differences in release rates in vitro.
Two types of media were utilized for in vitro release profile tests because conventional PBS could not differentiate the two formulations sufficiently within 24 hours. A special PBS solution with ammonium chloride, which led to the depletion of the transmembrane gradient and the release of encapsulated drugs, was employed to amplify the discrepancy between the DOX liposomes loaded by various gradients. Results showed that DOX release from the NH4EDTA-L preparation was much slower than release from the (NH4)2SO4-L preparation. For the NH4EDTA-L preparation, almost all of the DOX formed a gel-like compound, which resulted in a steady and nearly linear release rate. In contrast, DOX encapsulated in the liposomes in the (NH4)2SO4-L preparation was in the form of a saturated solution containing a DOX-(NH4)2SO4 precipitate, and the release followed a biphasic pattern. The first phase of release involved transmembrane release of the dissolved drug, and the second phase involved drug dissolving from the precipitate, which was followed by transmembrane release.
Additionally, the long-term stability experiments showed that the NH4EDTA-L preparations were highly stable when stored at 4°C±2°C for 3 months. In contrast, the (NH4)2SO4-L preparation showed a decreased loading ability for DOX after 2 months of storage, as indicated by a precipitate at the bottom of the bottle. The outstanding stability of NH4EDTA-L in the same storage conditions could be caused by the chelation of metal ions by intraliposomal EDTA, which inhibited the catalytic oxidation of phospholipids by metal ions.37 Furthermore, EDTA could inhibit bacterial contamination by chelating divalent metal ions that are essential for microbial growth.19 These advantages of EDTA are important for laboratory research, because they facilitate the production of stable lipid formulations for which long-term stability can be easily estimated.
In the clinic, repeated injections of antitumor formulations are generally used to treat cancers, so the antitumor activities and tissue distributions of the two DOX preparations after repeated administration were investigated. Compared with an equivalent dose of (NH4)2SO4-L, the accumulation of NH4EDTA-L was significantly greater in tumors, but lower in the liver, spleen, and heart. Although the tumor inhibition ratios showed no significant differences, toxicity caused by NH4EDTA-L was very low and no drug-related deaths were observed during the experiments.
The similar inhibition ratios observed for the NH4EDTA-L and (NH4)2SO4-L preparations, despite the high tumor accumulation produced by the former, might be associated with the controlled release of DOX. Because of this delayed DOX release, the available drug concentration produced by NH4EDTA-L administration was similar to that of (NH4)2SO4-L administration. Furthermore, the primary antitumor mechanism of DOX has been reported to be the formation of a complex with DNA by intercalation between base pairs or interaction with topoisomerase II. However, the generation of ROS, which results in DNA damage, lipid peroxidation, and apoptosis, might also have an effect on antitumor activity. Thus, when transition metal ions were chelated by EDTA, antitumor activity might be influenced by a reduction in ROS.23,38
From the spleen index and thymus gland index results, the superiority of the NH4EDTA-L preparation was quite apparent, because of lesser tissue accumulation in these organs and the chelating effect of EDTA. DOX cytotoxicity generally manifests as high levels of ROS and low levels of antioxidants and protective enzymes. Transition metal ions promote free radical reactions through enzymatic and nonenzymatic pathways; in the enzymatic pathway, the metal ions accept or donate single electrons to cellular oxidoreductases (eg, cytochrome P450 reductase, nicotinamide adenine dinucleotide dehydrogenase of complex I, xanthine oxidase), while in nonenzymatic pathways metal ions form compounds with DOX and thus generate superoxide anions, singlet oxygen, and hydrogen peroxide.23,25,39 Thus, it was presumed that the lower toxicity of NH4EDTA-L was owing to its chelation of metal ions, which inhibited and/or deregulated the immune system, and resulted in binding of receptor sites, suppression of proper enzyme functions, reduced antibody response, disordered hormone functions, undesirable upregulation of the immune system, and autoimmune diseases.40
Tissue distributions and histopathological studies of cardiotoxicity indicated that liposomal formulations show a lower risk of DOX-induced cardiotoxicity. Furthermore, NH4EDTA-L possessed a clear advantage over (NH4)2SO4-L at equivalent drug concentrations. A concentration dependent trend of toxicity among the three NH4EDTA-L groups was also observed. Due to the slow release rate of the liposomes, liposomal preparations could achieve high accumulation in targeted tissues because of the enhanced permeation and retention effect. Thus, the drug would reach relatively low levels in other tissues, and lessen the risk of drug-related toxicity. Within the two liposomal formulations in this study, NH4EDTA-L presented a notable advantage over (NH4)2SO4-L with respect to DOX-induced cardiotoxicity when an equivalent dose was administered. The primary reason for this result may have been that EDTA reduced the generation of ROS by chelating transition metal ions, which led to cardiomyocyte membrane damage and the occurrence and development of DOX-related cardiomyopathy. Additionally, as described above, excessive calcium in vivo can cause hypercalcinemia and promote the migration of cancer cells to the bones. Therefore, when EDTA is employed in a drug formulation extra calcium might be eliminated owing to its chelating effect, and suffering of associated off-target complications may be reduced.
The most valuable clinical implication of these studies on NH4EDTA-L is that it can reduce the cardiotoxicity induced by DOX without affecting the antitumor activity of the drug. However, the primary limitation of these novel liposomes is that drug accumulation in the spleen and liver remains high, and thus the risk of damage to these organs increases with the dose of the drug. To date, the authors have encapsulated several other amphiphilic weak bases, such as topotecan and huperzine A, via a transmembrane NH4EDTA gradient method. All tested drugs showed rapid accumulation in the vesicles, good long-term stability, considerable antitumor activity, and low cytotoxic activity, compared with drugs encapsulated by ammonium sulfate gradient or citric acid gradient. These findings led to the conclusion that this particular method could provide new opportunities for the clinical use of many chemotherapy drugs that have been limited by their severe side effects.
Conclusion
Use of the NH4EDTA gradient method to load doxorubicin into liposomes produced rapid and efficient drug encapsulation, and the liposomes showed slow drug release and good long-term stability. Furthermore, compared with liposomes prepared by (NH4)2SO4 gradient, doxorubicin loaded into liposomes via an NH4EDTA gradient produced significantly reduced toxicity and mortality without altered antitumor effects.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Domingo JL. Developmental toxicity of metal chelating agents. Reprod Toxicol. 1998;12(5):499–510. doi: 10.1016/s0890-6238(98)00036-7. [DOI] [PubMed] [Google Scholar]
- 2.Flora SJ, Pachauri V. Chelation in metal intoxication. Int J Environ Res Public Health. 2010;7(7):2745–2788. doi: 10.3390/ijerph7072745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gordon GF. EDTA and Chelation Therapy: History and Mechanisms of Action. Payson, AZ: Gordon Research Institute; 2000. [Google Scholar]
- 4.Riordan HD, Cheraskin E, Dirks M. Mineral excretion associated with EDTA chelation therapy. Journal of Advancement in Medicine. 1990;3(2):111–123. [Google Scholar]
- 5.Waters RS, Bryden NA, Patterson KY, Veillon C, Anderson RA. EDTA chelation effects on urinary losses of cadmium, calcium, chromium, cobalt, copper, lead, magnesium, and zinc. Biol Trace Elem Res. 2001;83(3):207–221. doi: 10.1385/BTER:83:3:207. [DOI] [PubMed] [Google Scholar]
- 6.Born T, Kontoghiorghe CN, Spyrou A, Kolnagou A, Kontoghiorghes GJ. EDTA chelation reappraisal following new clinical trials and regular use in millions of patients: review of preliminary findings and risk/benefit assessment. Toxicol Mech Methods. 2013;23(1):11–17. doi: 10.3109/15376516.2012.730562. [DOI] [PubMed] [Google Scholar]
- 7.Clarke NE. Atherosclerosis, occlusive vascular disease, and EDTA. Am J Cardiol. 1960;6(2):233–236. doi: 10.1016/0002-9149(60)90309-x. [DOI] [PubMed] [Google Scholar]
- 8.Villarruz MV, Dans A, Tan F. Chelation therapy for atherosclerotic cardiovascular disease [review] Cochrane Database Syst Rev. 2002;4:CD002785. doi: 10.1002/14651858.CD002785. [DOI] [PubMed] [Google Scholar]
- 9.Kitchell JR, Palmon F, Jr, Aytan N, Meltzer LE. The treatment of coronary artery disease with disodium EDTA: a reappraisal. Am J Cardiol. 1963;11(4):501–506. doi: 10.1016/0002-9149(63)90011-0. [DOI] [PubMed] [Google Scholar]
- 10.Blumer W, Cranton EM. Ninety percent reduction in cancer mortality after EDTA chelation therapy with EDTA. Journal of Advancement in Medicine. 1989;2(1–2):183–188. [Google Scholar]
- 11.Seely DM, Wu P, Mills EJ. EDTA chelation therapy for cardiovascular disease: a systematic review. BMC Cardiovasc Disord. 2005;5:32. doi: 10.1186/1471-2261-5-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buss JL, Torti FM, Torti SV. The role of iron chelation in cancer therapy. Curr Med Chem. 2003;10(12):1021–1034. doi: 10.2174/0929867033457638. [DOI] [PubMed] [Google Scholar]
- 13.Cranton EM, Frackelton JP. Free oxygen radical pathology and EDTA chelation therapy: mechanisms of action. Journal of Advancement in Medicine. 1998;11(4):277–310. [Google Scholar]
- 14.Hucho F, Kogler H, Pribilla I, Schiebler W, Tripier D, Inventors, Hoechst Aktiengesellschaft, assignee Use of ethylenediaminetetraacetic acid as inhibitor of calcium/calmoduline-dependent protein kinase II or protein kinase C. 1990. May 10, European patent CA2002637 A1.
- 15.Mundy GR, Guise TA. Hypercalcemia of malignancy. Am J Med. 1997;103(2):134–145. doi: 10.1016/s0002-9343(97)80047-2. [DOI] [PubMed] [Google Scholar]
- 16.Body JJ. Bone metastases and tumor-induced hypercalcemia. Curr Opin Oncol. 1992;4(4):624–631. doi: 10.1097/00001622-199208000-00005. [DOI] [PubMed] [Google Scholar]
- 17.Abdi S, Ali A. Role of ROS modified human DNA in the pathogenesis and etiology of cancer. Cancer Lett. 1999;142(1):1–9. doi: 10.1016/s0304-3835(99)00112-3. [DOI] [PubMed] [Google Scholar]
- 18.Martinez-Cayuela M. Oxygen free radicals and human disease. Biochimie. 1995;77(3):147–161. doi: 10.1016/0300-9084(96)88119-3. [DOI] [PubMed] [Google Scholar]
- 19.Thompson KA, Goodale DB. The recent development of propofol (Diprivan®) Intensive Care Med. 2000;26(Suppl 4):S400–S404. doi: 10.1007/pl00003783. [DOI] [PubMed] [Google Scholar]
- 20.Weiss RB. The anthracyclines: will we ever find a better doxorubicin? Semin Oncol. 1992;19(6):670–686. [PubMed] [Google Scholar]
- 21.Basser RL, Green MD. Strategies for prevention of anthracycline cardiotoxicity. Cancer Treat Rev. 1993;19(1):57–77. doi: 10.1016/0305-7372(93)90027-o. [DOI] [PubMed] [Google Scholar]
- 22.Dudka J, Burdan F, Korga A, et al. Intensification of doxorubicin-related oxidative stress in the heart by hypothyroidism is not related to the expression of cytochrome P450 NADPH-reductase and inducible nitric oxide synthase, as well as activity of xanthine oxidase. Oxid Med Cell Longev. 2012;2012:139327. doi: 10.1155/2012/139327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tokarska-Schlattner M, Zaugg M, Zuppinger C, Wallimann T, Schlattner U. New insights into doxorubicin-induced cardiotoxicity: the critical role of cellular energetics. J Mol Cell Cardiol. 2006;41(3):389–405. doi: 10.1016/j.yjmcc.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 24.Ortega R, Deves G, Bohic S, Simionovici A, Ménez B, Bonnin-Mosbah M. Iron distribution in cancer cells following doxorubicin exposure using proton and X-ray synchrotron radiation microprobes. Nucl Instrum Methods Phys Res B. 2001;181(1):480–484. [Google Scholar]
- 25.Octavia Y, Tocchetti CG, Gabrielson KL, Janssens S, Crijns HJ, Moens AL. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol. 2012;52(6):1213–1225. doi: 10.1016/j.yjmcc.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 26.Chen Y, Wan Y, Wang Y, Zhang H, Jiao Z. Anticancer efficacy enhancement and attenuation of side effects of doxorubicin with titanium dioxide nanoparticles. Int J Nanomedicine. 2011;6:2321–2326. doi: 10.2147/IJN.S25460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dudka J, Gieroba R, Korga A, et al. Different effects of resveratrol on dose-related doxorubicin-induced heart and liver toxicity. Evid Based Complement Alternat Med. 2012;2012:606183. doi: 10.1155/2012/606183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hruby M, Konak C, Ulbrich K. Polymeric micellar pH-sensitive drug delivery system for doxorubicin. J Control Release. 2005;103(1):137–148. doi: 10.1016/j.jconrel.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 29.Mitra S, Gaur U, Ghosh P, Maitra A. Tumour targeted delivery of encapsulated dextran–doxorubicin conjugate using chitosan nanoparticles as carrier. J Control Release. 2001;74(1–3):317–323. doi: 10.1016/s0168-3659(01)00342-x. [DOI] [PubMed] [Google Scholar]
- 30.Gabizon A, Martin F. Polyethylene glycol-coated (pegylated) liposomal doxorubicin. Rationale for use in solid tumors. Drugs. 1997;54(Suppl 4):15–21. doi: 10.2165/00003495-199700544-00005. [DOI] [PubMed] [Google Scholar]
- 31.Gubernator J, Chwastek G, Korycinska M, et al. The encapsulation of idarubicin within liposomes using the novel EDTA ion gradient method ensures improved drug retention in vitro and in vivo. J Control Release. 2010;146(1):68–75. doi: 10.1016/j.jconrel.2010.05.021. [DOI] [PubMed] [Google Scholar]
- 32.Yang Y, Ma Y, Wang S. A novel method to load topotecan into liposomes driven by a transmembrane NH4EDTA gradient. Eur J Pharm Biopharm. 2012;80(2):332–339. doi: 10.1016/j.ejpb.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 33.Cui J, Li C, Guo W, et al. Direct comparison of two pegylated liposomal doxorubicin formulations: is AUC predictive for toxicity and efficacy? J Control Release. 2007;118(2):204–215. doi: 10.1016/j.jconrel.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 34.Li X, Hirsh DJ, Cabral-Lilly D, et al. Doxorubicin physical state in solution and inside liposomes loaded via a pH gradient. Biochim Biophys Acta. 1998;1415(1):23–40. doi: 10.1016/s0005-2736(98)00175-8. [DOI] [PubMed] [Google Scholar]
- 35.Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta. 1993;1151(2):201–215. doi: 10.1016/0005-2736(93)90105-9. [DOI] [PubMed] [Google Scholar]
- 36.Fritze A, Hens F, Kimpfler A, Schubert R, Peschka-Süss R. Remote loading of doxorubicin into liposomes driven by a transmembrane phosphate gradient. Biochim Biophys Acta. 2006;1758(10):1633–1640. doi: 10.1016/j.bbamem.2006.05.028. [DOI] [PubMed] [Google Scholar]
- 37.Mei L, Decker EA, McClements DJ. Evidence of iron association with emulsion droplets and its impact on lipid oxidation. J Agric Food Chem. 1998;46(12):5072–5077. [Google Scholar]
- 38.Matalon ST, Ornoy A, Lishner M. Review of the potential effects of three commonly used antineoplastic and immunosuppressive drugs (cyclophosphamide, azathioprine, doxorubicin on the embryo and placenta) Reprod Toxicol. 2004;18(2):219–230. doi: 10.1016/j.reprotox.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 39.Minotti G, Recalcati S, Menna P, Salvatorelli E, Corna G, Cairo G. Doxorubicin cardiotoxicity and the control of iron metabolism: quinone-dependent and independent mechanisms. Methods Enzymol. 2004;378:340–361. doi: 10.1016/S0076-6879(04)78025-8. [DOI] [PubMed] [Google Scholar]
- 40.Dugi D, Takemoto A. The role of heavy metal detoxification in heart disease and cancers: a pilot study in detoxification of heavy metals; Paper presented at: WESCON Biomedicine and Bioengineering Conference; September 24, 2002; Anaheim, CA, USA. [Google Scholar]