

Abstract
Numerous tyrosine kinase inhibitors (TKIs) targeting c-Met are currently in clinical trials for several cancers. Their efficacy is limited due to the development of resistance. The present study aims to elucidate this mechanism of c-Met TKI resistance by investigating key mTOR and Wnt signaling proteins in melanoma cell lines resistant to SU11274, a c-Met TKI. Xenografts from RU melanoma cells treated with c-Met TKIs SU11274 and JNJ38877605 showed a 7- and 6-fold reduction in tumor size, respectively. Resistant cells displayed upregulation of phosphorylated c-Met, mTOR, p70S6Kinase, 4E-BP1, ERK, LRP6, and active β-catenin. In addition, GATA-6, a Wnt signaling regulator, was upregulated, and Axin, a negative regulator of the Wnt pathway, was downregulated in resistant cells. Modulation of these mTOR and Wnt pathway proteins was also prevented by combination treatment with SU11274, everolimus, an mTOR inhibitor, and XAV939, a Wnt inhibitor. Treatment with everolimus, resulted in 56% growth inhibition, and a triple combination of SU11274, everolimus and XAV939, resulted in 95% growth inhibition in RU cells. The V600E BRAF mutation was found to be positive only in MU cells. Combination treatment with a c-Met TKI and a BRAF inhibitor displayed a synergistic effect in reducing MU cell viability. These studies indicate activation of mTOR and Wnt signaling pathways in c-Met TKI resistant melanoma cells and suggest that concurrent targeting of c-Met, mTOR, and Wnt pathways and BRAF may improve efficacy over traditional TKI monotherapy in melanoma patients.
Keywords: c-Met, melanoma, resistance, mTOR, Wnt, SU11274, BRAF
Introduction
c-Met is a receptor tyrosine kinase (RTK) that plays a key role in the growth, metastasis, and angiogenesis of melanoma cells.1,2 Several signaling pathways are activated when c-Met binds to its ligand hepatocyte growth factor (HGF), a multifunctional growth factor that acts as a mitogen, motogen, and morphogen for multiple epithelial cell types.3,4 Studies have shown the combination of vemurafenib, a BRAF inhibitor, with siRNA targeting MET to be effective in inhibiting cell growth and reducing cell invasion and migration in melanoma cells with MET amplification.5 Vemurafenib targets the V600E BRAF mutation and is one of the most effective FDA approved molecularly targeted therapies for treatment of advanced melanoma.6,7 Interestingly, the V600E mutation is expressed in 82% of all benign nevi,8 suggesting this mutation alone is insufficient to promote tumorigenesis and may require additional pathways or mutational events to enable malignant melanoma transformation. Another important therapeutic target for melanoma is mutant NRAS, which may confer resistance to therapies such as vemurafenib, against which current therapies are ineffective.9 Studies have shown that NRAS mutants are sensitive to c-Met inhibition, resulting in downregulation of Akt phosphorylation, tumor cell proliferation, migration, and subsequently induction of apoptosis.10 The above studies show the importance of c-Met activation in melanoma progression and in several other cancers.11,12
It has been shown that overexpression and activation of c-Met plays a significant role in tumor development and metastasis in melanoma.1 We and other investigators have identified c-Met as a possible target in treating melanoma using TKIs such as SU11274 and JNJ38877605.1,13,14 Both are c-Met TKIs that function by inhibiting c-Met phosphorylation in vitro and in vivo in a variety of tumors.15-18 We have previously shown that SU11274 inhibits growth of melanoma cells in vitro by inducing apoptosis and differentiation.1 Although TKIs against c-Met are on the cutting-edge of cancer therapy, their individual efficacies are limited19 due to the development of resistance and subsequent potential for tumor recurrence.20
Currently, the pathway and mechanism of c-Met inhibitor resistance is largely unknown. A recent study suggests that amplification of MET and KRAS genes may mediate resistance to MET kinase inhibitors by subsequently inducing high activity of the mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-OH kinase (PI3K)/Akt pathways.16,21 In another study, a point mutation in the c-Met activation loop (Y1230H) was found to confer resistance due to decreased binding ability of c-Met inhibitors.19,21 Overall, further studies are necessary to elucidate the mechanism of c-Met inhibitor resistance in melanoma.
Several studies using c-Met inhibitors, alone or in combination therapy, for melanoma are also presently in clinical trials and may prove to be effective in improving efficacy and overcoming resistance (http://www.clinicaltrials.gov; NCT00940225, NCT01820364, and NCT01835184). In a phase II, multi-center study, a BRAF inhibitor, LGX818, is being studied in combination with c-Met inhibitor INC280 in adult patients with locally advanced or metastatic BRAF V600E melanoma (NCT01820364). Another study was conducted on dose escalation of tivantinib (ARQ197) in combination with sorafenib in adult patients with advanced solid tumors, including melanoma (NCT00827177). In phase II clinical trials, cabozantinib, an effective c-Met and VEGFR inhibitor,22 alone (NCT00940225) and in combination with vemurafenib (NCT01835184), is being tested in patients with advanced melanoma and in patients with metastatic melanoma,23 respectively. However, the outcomes have not yet been reported.
In the present study, the efficacy of c-Met inhibitors, SU11274 and JNJ38877605, an orally administered inhibitor, were studied in vivo and found to be comparable. To improve the efficacy of c-Met TKI combination therapies, we investigated the modes of c-Met TKI resistance in SU11274 resistant melanoma cell lines. The efficacy of c-Met, mTOR, and Wnt inhibitor combinations on resistant melanoma cells was tested in vitro, and a combination of all three inhibitors was found to be very effective. Key proteins inhibited in the Wnt canonical pathway after combination treatment include LRP6, a co-receptor for the Wnt/β-catenin signaling pathway24 and Axin, which is required for LRP6 phosphorylation and is a “rate limiting factor” for assembly of the β-catenin destruction complex.25-28 Additionally, GATA-6, which is phosphorylated by ERK,29 is upregulated in several cancers,30 and its induced expression leads to the trans-activation of the Wnt7b promoter, ultimately resulting in increased activation of the Wnt canonical pathway.31,32 Therefore, we propose a c-Met inhibitor resistance mechanism in melanoma, and suggest that simultaneous targeting of c-Met, mTOR and Wnt pathways with inhibitors could overcome this resistance. The present combination therapy may be the basis for future therapies to enhance the efficacy of c-Met TKIs, prevent resistance, and improve melanoma patient outcomes.
Results
SU11274 inhibits in vivo growth of HGF producing RU-Parental (RU-P) cells
In this study, four cell lines were used (EP-P, MU-P, RU-P, and WK-P). These cell lines demonstrate abundant expression of c-Met,1 indicating that c-Met inhibitors may be effective in these cell lines. HGF-Met signaling has been demonstrated to have an important role in the induction, invasiveness and promotion of melanoma, both in vitro and in vivo.1,13,33 Therefore, we evaluated the HGF levels in all five cell lines and found that RU-P (285 pg/mL) and WK-P (69 pg/mL) cells produced high levels of HGF (Fig. 1A). No detectable levels of HGF were found in EP-P and MU-P cell lines (data not shown). Since RU cells express considerably higher levels of HGF, and are an established tumor model,34 the in vivo efficacy of SU11274 was evaluated in a xenograft model using the RU-P melanoma cell line. SU11274 treatment reduced tumor volume by 7-fold as compared with control (Fig. 1B). Formalin-fixed paraffin-embedded (FFPE) tissues from mice treated with SU11274 or diluent control for p-c-Met (Y1234/1235) expression by immunohistochemistry (IHC) were further analyzed. Tumor sections from mice treated with SU11274 were found to show downregulation of expression of p-c-Met as compared with the diluent control (Fig. 1C). CD31 staining revealed that treatment with SU11274 reduced the number of blood vessels by 79.8% ± 1.5% (P < 0.001) suggesting that inhibition of vessel formation may be a mechanism whereby SU11274 inhibits tumor growth (Fig. 1D). Furthermore, SU11274 treatment decreased VEGF expression and increased TSP-1 expression, as seen by IHC (Fig. 1E). These results imply that inhibition of c-Met phosphorylation has a significant effect on tumor proliferation and maintenance.

Figure 1. Intratumoral TKI treatment reduces tumor size in vivo. (A) Production of HGF by melanoma cell lines. RU-P cells produced 4-fold higher amounts of HGF compared with WK-P cells in conditioned medium as determined by HGF ELISA kit. (B) Five million RU-P melanoma cells were injected subcutaneously into the hind flanks of Rag1−/− mice. Tumors were allowed to develop for a week after which daily intratumoral doses of SU11274 or vehicle were given for 4 wk. SU11274 treated RU-P tumor xenografts showed a 7-fold reduction in tumor size in comparison to control mice. Seven mice xenografts in each group were evaluated for this study. (C) Melanoma tumor sections from mice treated with SU11274 showed downregulation of p-c-Met compared with control mice (D) Immunostaining of CD31 in RU-P tumor xenografts in control and SU11274 treated mice. There was a 79.8% (± 1.5%) (P < 0.001) decrease in the number of blood vessels when counted in 10 microscopic fields. (E) A decrease in VEGF and an increase of TSP1 were found after treatment with SU11274, suggesting decreased angiogenesis.
RU-P melanoma cells are inhibited by JNJ38877605 in vivo
To study the therapeutic efficacy of JNJ38877605, an orally bioavailable c-Met TKI, in vivo studies were performed. Mice bearing RU-P melanoma cell tumor xenografts were treated orally with 20 mg/kg JNJ38877605 or vehicle for three weeks. Similar to SU11274, it was determined that JNJ38877605 significantly reduced tumor size by 6-fold (124 ± 57 mm2 and 17 ± 11 mm2, P < 0.03), as compared with control (vehicle) (Fig. 2A). Tumors treated with JNJ38877605 showed a significant reduction in expression of p-c-Met (Y1234/1235), as seen by IHC in small residual tumor nodules (Fig. 2B). These results indicate that the reduction in p-c-Met after administration of JNJ38877605 has a significant effect on tumor proliferation. Treatment with JNJ38877605 also resulted in 80% ± 2% (P < 0.001) reduction in blood vessels, as seen by CD31 staining, suggesting that inhibition of vessel formation may be one of the mechanisms by which JNJ38877605 inhibits tumor growth (Fig. 2C). Similar to SU11274 treatment, JNJ38877605 decreased VEGF expression and increased TSP-1 expression, as seen by IHC (Fig. 2D). These data indicate that JNJ38877605 could be a promising orally administered therapeutic option for treating HGF-producing melanoma.
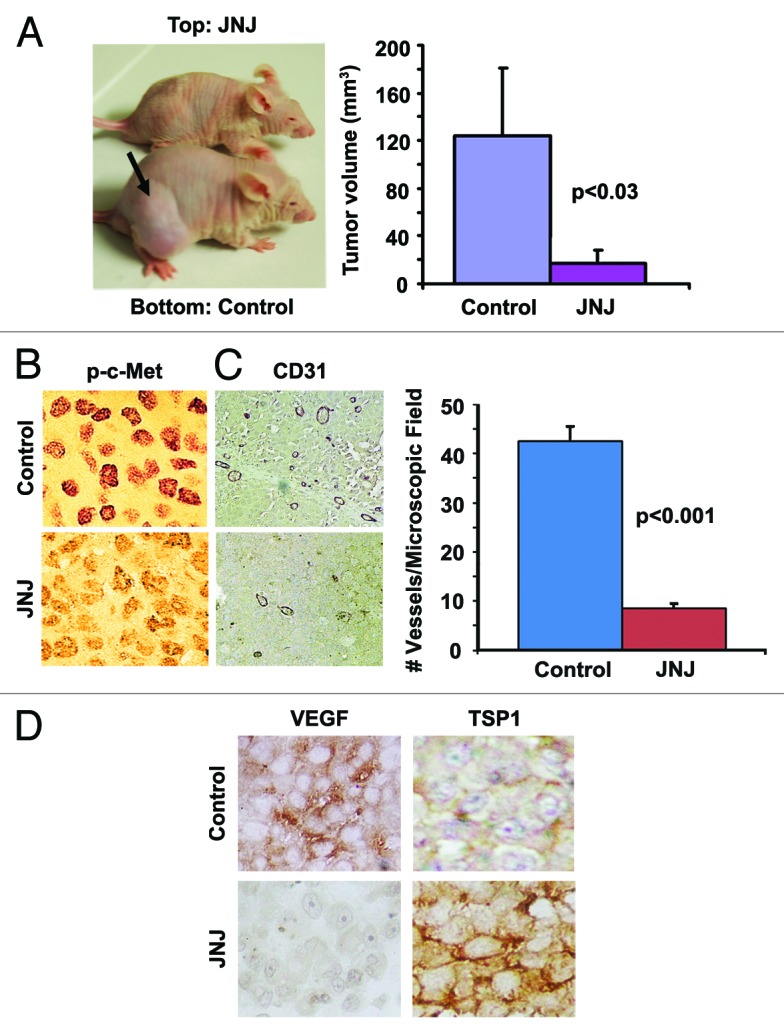
Figure 2. Oral TKI treatment reduces tumor size in vivo. Five million RU-P melanoma cells were injected subcutaneously into the hind flanks of nu/nu mice. Tumors were allowed to develop for a week after which daily oral doses of JNJ38877605 or vehicle were given for 3 wk. (A)Treatment with JNJ38877605 reduced tumor size by 6-fold when compared with control mice. (B) Immunostaining of control and JNJ38877605-treated RU-P tumor xenografts with p-c-Met antibody showed decrease in p-c-Met after treatment with JNJ38877605. (C) Immunostaining of control and JNJ38877605 treated RU-P tumor xenografts with CD31 antibody indicate treatment with JNJ38877605 decreased the number of blood vessels in melanoma. There was an 80% (± 2%) decrease in the number of blood vessels when counted in 10 microscopic fields after treatment with JNJ38877605. (D) Immunostaining of control and JNJ38877605-treated RU-P tumor xenografts with VEGF and TSP1 antibody showed a decrease in VEGF and an increase of TSP1 with JNJ38877605 treatment suggesting decreased angiogenesis.
Resistance to SU11274 in MU and RU melanoma cells is not mediated by mutations in the c-Met tyrosine kinase domain (TKD)
To study the mechanism of c-Met TKI resistance, MU and RU cells were made resistant to SU11274 (MU-R, RU-R), as described in the materials and methods section. IC50 values for SU11274 were found to be 1.5 μM and 1.0 μM for MU-P and RU-P cell lines, respectively, and 10 μM in both resistant cell lines (Table S1). Gene sequencing of exons 15–21 of the c-Met TKD revealed no relevant mutations associated with SU11274 resistance in either parental or resistant MU and RU cell lines, and demonstrated 100% homology with the reference NCBI gene sequence (NM_000245).
MU-R cells are cross-resistant to tivantinib; BRAF and c-Met inhibitors work effectively to reduce growth in both MU-P and MU-R cells with the V600E BRAF mutation
To determine BRAF mutation status in MU and RU cells, sequencing of exon 15 of the BRAF gene was performed and showed that MU cells possess a V600E mutation that sensitizes them to BRAF inhibitors35 while RU cells possess wt-BRAF.5 Therefore, the cross-resistance of SU11274 resistant MU cells to tivantinib was studied in vitro. Tivantinib, a clinically effective inhibitor, predominantly inhibits c-Met in vitro at a submicromolar range.36 Before determining the efficacy of tivantinib and vemurafenib combination therapy, dose response studies for tivantinib (0.05–0.5 µM) and vemurafenib (0.01–0.5 µM) were conducted (Figs. S1 and S2). To determine cross resistance to tivantinib, MU-P and MU-R cells were treated with tivantinib for 96 h, after which an MTT cell viability assay was performed, as described earlier.37 Two-tenths micromolar tivantinib inhibited cell growth of MU-P and MU-R by 29.60% (± 1.32%) and 5.65% (± 1.5%), respectively (n = 6, P < 0.001). Although tivantinib inhibited MU-P cells significantly, it had minimal effect on MU-R cells. Furthermore, MU-R cells displayed a 5-fold decrease in sensitivity to tivantinib compared with MU-P cells (Fig. 3A).
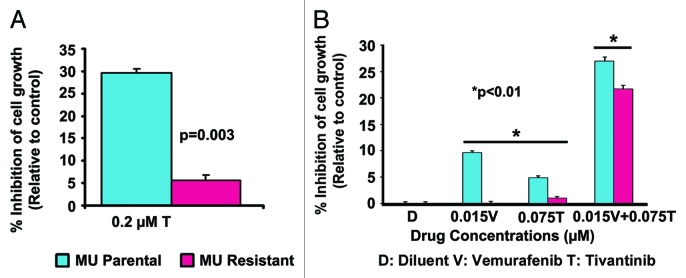
Figure 3. Effect of a combination of vemurafenib and tivantinib on cell growth in MU cells. (A) MU-R cells showed a 5-fold decrease in sensitivity to the anti-proliferative effect of tivantinib compared with parental cells (P = 0.003) indicating cell lines are resistant to tivantinib. (B) Two thousand MU-P/R cells were plated and treated after 24 h with vemurafenib (0.015 μM) and tivantinib (0.075 μM) individually and in combination (0.015 μM vemurafenib + 0.075 μM tivantinib) after which an MTT viability assay was performed after 96 h. MU-P/R cells were inhibited significantly more by vemurafenib and tivantinib in combination (27% and 22%), (P < 0.01) in comparison to single drug treated cells (< 10%).
To further study the effects of BRAF and c-Met inhibitors in MU cells, vemurafenib, a BRAF inhibitor and tivantinib, a c-Met inhibitor in clinical trials,38,39 were used. Cells were treated with tivantinib at concentrations ranging from 0.05 to 0.5 μM or vemurafenib ranging from 0.01 to 0.5 μM for 96 h, after which an MTT viability assay was performed. Tivantinib inhibited growth of parental cells by 4–48%, whereas resistant cells were inhibited by 2–34% (Fig. S1). Vemurafenib inhibited growth of parental cells by 18–69%, whereas resistant cell growth was inhibited by 13–77% (Fig. S2). MU-P and MU-R cells were then treated with 0.015 µM vemurafenib and 0.075 µM tivantinib, individually and in combination. Minimal inhibition (<10%) was seen in response to vemurafenib and tivantinib as single therapeutic agents at these low concentrations (MU-P 9.6%/4.8% and MU-R 0%/1% vemurafenib/tivantinib) (Fig. 3B). Interestingly, these concentrations in combination therapy inhibited the MU-P (27%) and MU-R (22%) cells more significantly (P < 0.01), as analyzed by ANOVA (Fig. 3B). These results indicate that tivantinib and vemurafenib have a synergistic effect, as CI values were found to be < 1 using CalcuSyn software (Biosoft), and suggest that vemurafenib in combination with c-Met inhibitors may benefit melanoma patients having the V600E mutation and expressing c-Met.
Effect of HGF on c-Met and β-catenin phosphorylation in MU-P and MU-R cells
To further investigate the mechanism of c-Met TKI resistance in MU-R cells, key signaling proteins that have significant roles in melanoma and other cancers were studied. Earlier studies indicate that Wnt/EGFR and EGFR/c-Met crosstalk promotes tumorigenesis.37,40 These studies led us to investigate the role of the Wnt pathway in mediating c-Met inhibitor resistance. MU-R cells exhibited 1.5- and 2.4-fold upregulation of p-c-Met (Y1234/1235) and p-c-Met (Y1003), respectively, in the absence of HGF compared with MU-P cells (Fig. 4A). Additionally, time course experiments for p-c-Met and active β-catenin, a transcription factor in the Wnt pathway, were conducted. A 3- to 5-fold increase in p-c-Met (Y1234/1235) expression in MU-R cells was observed following HGF stimulation relative to control. Basal levels of active β-catenin were found to be 3-fold higher in the absence of HGF and remained elevated (2.5-fold) for 7.5 min after HGF treatment in MU-R cells, compared with those in MU-P cells. Following HGF treatment, levels of p-c-Met and active β-catenin both remained elevated for at least 60 min in MU-R cells, compared with 30 min in MU-P cells (Fig. 4B). These results indicate increased stabilization of p-c-Met suggesting crosstalk with the Wnt pathway, and that upregulation of the Wnt pathway may play a role in mediating SU11274 resistance in MU-R cells.
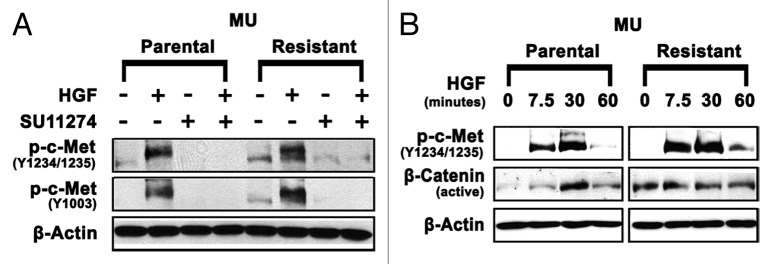
Figure 4. Upregulation of p-c-Met and active β-catenin in MU-R cells. (A) Cells were starved overnight in media supplemented with 0.5% BSA, and then treated with or without 10 µM SU11274 for 24 h. Cells were stimulated with 40 ng/mL of HGF for 7.5 min, after which immunoblotting analysis was performed. Upregulation of p-c-Met Y1003 (2.4-fold) and p-c-Met Y1234/1235 (1.5-fold) in MU-R cells in absence of HGF was observed, and a 3–5-fold increase in p-c-Met Y1234/1235 was seen after HGF treatment. (B) In MU-R cells, HGF induced p-c-Met and active β-catenin signaling was prolonged by 30 min compared with MU-P cells. Cells were starved for 24 h and then stimulated with 40 ng/ml HGF. Immunoblotting indicated that in MU-R cells, HGF activated p-c-Met (Y1234/1235) and basal levels of active β-catenin were also 3-fold higher in the absence of HGF and remained high (2.5-fold) for 7.5 min after HGF treatment in MU-R cells compared with those in MU-P cells at 0 min incubation.
HGF induces activation of mTOR, p70S6Kinase, 4E-BP1, Akt, and ERK in MU-R and RU-R cells
Upregulation of the Akt/mTOR pathway is often observed in multiple types of cancer, including melanoma,41,42 leading to cancer cell survival, growth and drug resistance. Therefore, potential proteins mediating c-Met TKI resistance in MU and RU cell lines were further investigated including mTOR, a key regulator of cancer cells.43 RU-R cells displayed 8- and 3-fold upregulation of p-mTOR (S2448) ± HGF, respectively, relative to RU-P cells. MU-R cells displayed 2- and 3-fold upregulation of p-mTOR (S2448) ± HGF, respectively, relative to MU-P cells (Fig. 5A). Interestingly, a 2-fold upregulation of p-p70S6kinase (T389), in the presence of HGF and a 4- and 2-fold upregulation of p-4E-BP1 (T37/46) ± SU11274, respectively, was seen in MU-R cells (Fig. 5B). Additionally, a 2- and 10-fold upregulation of p-Akt (S473) was found in MU-R cells treated with SU11274 ± HGF, respectively. Finally, p-ERK (T202/Y204), a downstream target of c-Met, was upregulated 2-fold in the presence of HGF (Fig. 5B). These results suggest that mTOR, or other upstream receptors, may upregulate p-p70S6kinase and p-4E-BP1 to mediate tumor resistance to c-Met inhibitors.
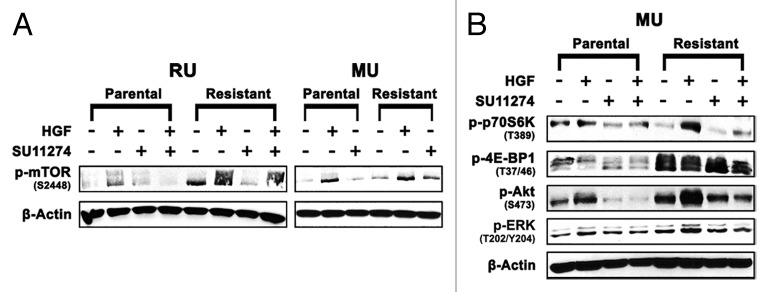
Figure 5. Upregulation of mTOR pathway proteins. (A) p-mTOR (S2448) is upregulated (2–8-fold ± HGF) in resistant (MU/RU) cell lines. (B) Upregulation of p-p70S6Kinase (T389) (2-fold) and p-4E-BP1 (T37/46) (2–4-fold ± HGF), which are downstream signaling proteins of mTOR, are upregulated in MU-R cell line. Additionally, downstream targets of c-Met, p-ERK (T202/Y204) were upregulated 2-fold in the presence of HGF and p-Akt (S473) was upregulated 2- and 10-fold in the absence and presence of HGF respectively.
Upregulation of mTOR and Wnt pathway proteins in c-Met inhibitor resistant MU cells is inhibited by combination therapy with mTOR, Wnt, and c-Met inhibitors
To determine if treatment with a combination of c-Met, mTOR and Wnt inhibitors could affect modulation of c-Met and key proteins in mTOR and Wnt pathways, immunoblotting was performed as described above. HGF-mediated upregulation of p-c-Met (3.0-fold) in MU-R cells was completely inhibited with combination therapy, which was comparable to that of MU-P cells. The mTOR pathway proteins, p-Akt (1.5-fold) and p-p70S6 kinase (1.5-fold), were upregulated in MU-R cells in the presence of HGF, as compared with MU-P cells (Fig. S3A). Furthermore, a 4.5-fold upregulation of p-Akt, in the absence of HGF, was observed in MU-R cells. In addition, a 3.0- and 2.5-fold upregulation of p-Akt ± HGF, respectively, was inhibited after combination treatment in MU-R cells, as compared with untreated MU-R cells (Fig. S3A). p-LRP6, a key regulator of the Wnt pathway, was upregulated by 1.5-fold ± HGF in MU-R cells, which was comparable to that of MU-P cells. Axin, a negative regulator of the Wnt pathway,44 was downregulated by 3.0- and 1.5-fold ± HGF, respectively, in MU-R cells, as compared with MU-P cells. Furthermore, GATA-6 (4.0-fold) and p-ERK (3.5-fold) were upregulated in MU-R cells, as compared with MU-P cells, in the absence of HGF. Upregulation of p-LRP6, GATA-6 and p-ERK was also inhibited with combination treatment in MU-R cells, as compared with untreated MU-R cells (Fig. S3B).
MU-R and RU-R cells are inhibited by everolimus and XAV939
The mTOR and Wnt signaling pathways have been shown to play a major role in conferring resistance to chemotherapy, and targeting key proteins in these pathways has become an important therapeutic approach in cancer.45,46 Therefore, the drugs everolimus and XAV939, which inhibit mTOR and Wnt pathway proteins, respectively,47,48 were selected for this combination inhibitor study. RU and MU cells were treated with everolimus or XAV939, alone and in combination with SU11274. Combination drug concentrations were determined from drug titration experiments (data not shown) and previous studies by us and other investigators.49,50 Treatment with 1 µM everolimus inhibited both RU-P and RU-R cell growth by 56% (Fig. 6A). Administration of 1 µM everolimus in combination with 5 µM SU11274 and 15 µM XAV939, a Wnt inhibitor, inhibited RU-P and RU-R cell growth by 95% (Fig. 6A). Treatment with 1 µM everolimus inhibited MU-P and MU-R cell growth 50% and 30%, respectively (Fig. 6B). Administration of 1 µM everolimus in combination with 5 µM SU11274 and 15 µM XAV939 inhibited MU-P and MU-R cell growth 78% and 65%, respectively (Fig. 6B). To verify the results of the MTT assay, cell numbers were assessed using trypan blue exclusion to examine viable cells after inhibitor treatment, and similar results were obtained (Fig. S4). Concentrations higher than the IC50 of SU11274 were used for in vitro combination experiments since the resistant cell lines were grown to develop resistance in higher concentrations of SU11274. These results suggest that mTOR and Wnt pathways may play an essential role in c-Met TKI resistance, and that simultaneous targeting of mTOR, Wnt pathways, and c-Met may prove to be an effective approach for melanoma therapy (Fig. 7). Further studies are warranted in this direction.

Figure 6. Combination therapy with mTOR, Wnt, and c-Met inhibitors is effective in c-Met resistant cell lines. 2000 MU-P/R and RU-P/R cells were plated and treated with everolimus (1 μM), SU11274 (5 μM), and XAV939 (15 μM) individually and in combinations after 24 h and an MTT viability assay was performed after 96 h. (A) Growth of RU-R cells is inhibited by 95% when using everolimus (1 μM) in the presence of SU11274 (5 μM) and XAV939 (15 μM). Inhibition was comparable to that seen in parental cells. (B) In MU-R cells, 65% inhibition of growth occurred when everolimus was used with both SU11274 and XAV939. Significant differences (RU-R or MU-R, P < 0.001) were observed between treatment with single drug (S, E, and X), double drug combinations (S+E, S+X, and E+X) and triple drug combination (S+E+X) as seen by repeated measures ANOVA.

Figure 7. Proposed role of mTOR and Wnt pathways in c-Met inhibitor resistance. Increased resistance to c-Met inhibitors could be due to overexpression of c-Met and consequent activation of c-Met-dependent PI3K/Akt/mTOR, RAS/RAF/MEK/ERK (MAPK) or alternative signaling pathways, such as the Wnt pathway. Upregulation of the Akt/mTOR pathway in c-Met TKI resistant cells leads to increased cell survival and proliferation. Furthermore, we postulate that in c-Met resistant cells, phosphorylation of ERK by the RAS/RAF pathway may lead to phosphorylation of GATA-6.29 Upregulated GATA-689 (Fig. S3) also stimulates the Wnt pathway,31 which may result in accumulation of β-catenin in the nucleus,78 leading to melanoma tumorigenicity.45
Discussion
Molecularly targeted TKIs are integral in the treatment of melanoma.7 However, the development of resistance to TKIs has brought about a new challenge for researchers and clinicians. Therefore, we studied the mechanism of c-Met TKI resistance by investigating key signaling pathways potentially involved in this resistance in two melanoma cell lines, one of which is positive for the V600E BRAF mutation. We demonstrate that combination of a c-Met TKI and a BRAF inhibitor improved therapeutic efficacy in V600E mutant cell lines. mTOR and Wnt signaling pathways were also found to play a key role in c-Met inhibitor resistance, and combinatorial therapy with c-Met, mTOR, BRAF, and Wnt inhibitors was effective in overcoming c-Met TKI resistance. In the present study, we suggest a mechanism of c-Met inhibitor resistance in melanoma and a novel combination therapy to overcome this resistance.
To confirm the validity of using c-Met TKIs as a possible monotherapy in melanoma treatment, we compared the in vivo efficacy of two c-Met TKIs: SU11274, delivered intratumorally, and JNJ38877605, delivered orally. Results indicate that SU11274 and JNJ38877605 significantly reduce RU tumor size in mice, suggesting c-Met inhibition as a promising therapeutic option for HGF producing, c-Met TKI sensitive tumors in melanoma patients. These studies also show that inhibition of vessel formation by decreased VEGF expression and increased TSP-1 expression may be a result of c-Met inhibition, by which both SU11274 and JNJ38877605 decrease tumor growth. These results are comparable to the results obtained after treating lung cancer tumors in vivo with PHA665752.51
Studies have indicated that development of resistance may greatly reduce the efficacy of c-Met TKI therapy.19,52 Therefore, effective treatment strategies may require combination of multiple inhibitors to improve patient outcomes.53 Previous in vitro studies were focused on mutations, including the Y1230 TKD phosphorylation site,19 upregulation of KRAS,16 and expression of a novel SND-1 BRAF fusion protein.54 The present study is concentrated on identifying alternative signaling pathways involved in conferring resistance to c-Met TKI, while ruling out c-Met TKD mutations. To facilitate our investigation, we studied two melanoma model cell lines (MU-P and RU-P) that are known to express high levels of p-c-Met,1 which we made resistant to c-Met TKIs by exposure to sequentially increasing doses of the c-Met inhibitor SU11274. All cell lines used in the present study had an abundant expression of c-Met as seen in earlier studies,1 indicating that c-Met inhibitors might be effective in these cell lines.
Similar to c-Met, mTOR has previously been studied in many human tumor entities55-57 and its activation is associated with malignant melanomas (73%), to a lesser extent with benign nevi (4%),58 and poor patient outcomes.59 mTOR activation by Akt has been shown through both the direct inhibition of tuberous sclerosis 2 (TSC2), a negative regulator of mTOR,60 and by inhibition of AMPK-mediated phosphorylation of TSC2.61 Interestingly, phosphorylation of mTOR, p70S6kinase, and 4E-BP1 is upregulated in resistant MU and RU cells. These data substantiate involvement of the mTOR pathway in resistant cells, as p70S6kinase and 4E-BP1 are downstream targets of mTOR and are used to assess mTOR activity.43 Modulation of the mTOR pathway in resistant cells implies that this alternative cellular signaling pathway may play a role in the mechanism of acquired resistance to c-Met TKIs.
Additionally, upregulation of active β-catenin suggests involvement of the Wnt pathway in resistant MU and RU cells. Further studies determining the nuclear translocation of β-catenin are being performed. Simultaneous activation of Wnt and mTOR pathways suggests crosstalk, which is consistent with previous studies demonstrating that overactive Wnt signaling modulates mTOR and plays a role in tumorigenicity.62 In addition, increased transcriptional activity of β-catenin-TCF/LEF-1 by Akt has been demonstrated through inhibition of GSK-3 and/or by direct phosphorylation of β-catenin at Ser552, which enhances β-catenin nuclear accumulation.63 Wnt pathway involvement is also supported by prevalence of the Wnt5a ligand in invasive and metastatic melanoma64 and a 5-fold increase in its expression in 50% of primary malignant melanomas.65 Strong Wnt5a ligand expression is associated with melanoma progression and poor prognosis as well.66 While results indicate that p-c-Met is upregulated and remains stable longer in MU-R cells as compared with MU-P cells, treatment with SU11274 still inhibits p-c-Met in resistant cells. This suggests that resistant cells do not exclusively utilize c-Met as a means to survive, but rather employ multiple signaling pathways to bypass a c-Met blockade. This is substantiated by the results in MU melanoma cells, indicating that triple drug combination treatment is more effective than single drug treatment. This may be due to upregulation of the Akt pathway, suggesting that other alternative pathways should be inhibited in MU cells to achieve more complete growth inhibition. Furthermore, sequencing of the c-Met gene demonstrated that acquired resistance to SU11274 was not associated with mutations altering protein structure and activity. This is consistent with observations that c-Met TKI-resistant cell lines continue to show downregulated p-c-Met following inhibitor treatment.
As refractory melanoma demonstrates the ability to proliferate despite SU11274 and JNJ38877605 mediated c-Met blockade, targeting alternative pathways such as Wnt and mTOR in combination with c-Met may be required to inhibit cell growth. To further validate the role of mTOR and Wnt pathways in c-Met TKI resistance, MU-P, MU-R, RU-P, and RU-R cell lines were treated with their respective inhibitors, everolimus and XAV939, as single agents or in combination with SU11274. Inhibition of mTOR and Wnt alone did not significantly inhibit the growth of MU-R cells, which are V600E positive, suggesting the role of c-Met and BRAF as the primary drivers of proliferation. However, a combination of everolimus, XAV939, and SU11274 was able to overcome resistance to c-Met TKI in MU and RU cell lines.
Various c-Met inhibitors are currently in early melanoma clinical trials, as c-Met signaling has been shown to have an important role in melanoma biology. Thus, the efficacy and resistance mechanisms of c-Met inhibitors should be studied in melanomas positive for the BRAF mutation.67 The synergistic effect observed with c-Met and BRAF inhibitors in BRAF mutated MU-P and MU-R cell lines suggests that this combinatorial therapy may be an effective treatment option for patients sensitive to c-Met TKIs, as well as patients with primary or acquired c-Met TKI resistance. Additionally, this combination might be most effective if used in patients whose tumors express both a V600E BRAF mutation and elevated c-Met levels. Interestingly, a preclinical study on BRAF inhibitor resistance in BRAF mutant melanoma patients also demonstrated that MAPK and PI3K/Akt signaling pathways are activated due to overexpression of c-Met, resulting in resistance to BRAF inhibitors.68 Therefore, clinical evidence of V600E and elevated p-c-Met may be a strong indication for combination therapy as a first line treatment; however, more research in this area is needed. Additionally, in V600E negative patients, c-Met monotherapy or c-Met, Wnt and mTOR inhibitor combinations could be effective.
In a recent study, we demonstrated that the c-Met TKI resistance mechanism may act through mTOR and Wnt pathways in non-small cell lung cancer cells (NSCLC).69 These results support our earlier studies involving melanoma, which indicate that the c-Met inhibitor, SU11274, can modulate the Akt/mTOR pathway.1 Additionally, the activation of Wnt signaling by HGF has been demonstrated through accumulation and nuclear translocation of β-catenin, both in vitro and in vivo.70,71 Recent studies also indicate cross talk between HGF/c-Met and Wnt/β-catenin pathways to be a key factor in the progression of HGF-induced tumors.72 In the present study, we observed that combination therapy using c-Met, mTOR, and Wnt inhibitors proved to be more effective in inhibiting cell proliferation than single or double inhibitor treatment. We also observed upregulation of downstream signaling proteins of the mTOR pathway, p-p70S6K and p-4E-BP1,73 and upregulation of β-catenin, a key downstream effector in the Wnt/β-catenin signaling pathway,74 in MU-R cells. After triple combination treatment, we show inhibition of upregulation of keys proteins, such as p-c-Met, p-p70S6Kinase, p-Akt, GATA-6, p-ERK, and p-LRP6, in MU-R cells. In addition, Axin, a key negative regulator of Wnt/β-catenin signaling,75 was downregulated 28,44,76 in MU-R cells, compared with MU-P cells (Fig. S3).
Currently, the mechanism of c-Met inhibitor resistance which is being observed in patients77 is not known. We propose a possible resistance mechanism by which phosphorylation of ERK by the RAS/RAF (MAPK) pathway may lead to either the inhibition of GSK-3 and/or the phosphorylation of GATA-6.29 As a result of GSK-3 inhibition, β-catenin is phosphorylated and accumulates in the nucleus and induces Wnt gene expression,78 thereby activating the Wnt pathway, which leads to melanoma cell growth and proliferation. Activated GATA-6 also stimulates the Wnt pathway79, possibly resulting in melanoma tumorigenicity. In the present study on c-Met-resistant cells, we observed overexpression of c-Met and upregulation of key proteins of the Akt/mTOR and Wnt/β-catenin pathways, suggesting their possible role in resistance to c-Met inhibitors as depicted in Figure 7. These studies, for the first time, shed light on a possible role of these pathways in c-Met inhibitor resistance in melanoma.
In conclusion, this study demonstrates that resistance to c-Met TKIs in melanoma may be driven, in part, by the upregulation of alternative signaling pathways and may not necessarily be associated with TKD mutations. Additionally, c-Met TKI resistance may be overcome with a more detailed understanding of individual melanoma tumor biology and the use of proper therapeutic combinations of c-Met, Wnt, and mTOR inhibitors. Finally, assessment of p-c-Met expression, the V600E BRAF mutation, and the activity of key proteins such as mTOR and Wnt in individual melanoma tumors could enable clinicians to better design targeted combination therapies to improve patient outcomes.
Materials and Methods
Reagents
Molecular inhibitors and growth factor: SU11274 (Sigma-Aldrich, CAS 658084-23-2), XAV939 (Sigma-Aldrich, CAS 284028-89-3) JNJ38877605 (Selleck, CAS 943540-75-8) Vemurafenib (PLX4032) (Chemietek, CAS 1029872-54-5), tivantinib (ARQ197) (Chemietek, CAS 905854-02-6) and Everolimus (LC Laboratories, CAS 159351-69-6). All inhibitors were suspended in DMSO and stored in 0.1 mL aliquots at −20 °C. HGF (Peprotech, 100-39).
Antibodies
Phosphospecific antibodies for Akt (S473, 4051), mTOR (S2448, 5536), p70S6Kinase (T389, 9205), 4E-BP1 (T37/46, 7854), c-Met (Y1234/1235, 3077), ERK1/2 (T202/Y204, 9101), and LRP6 (S1490, 2568), as well as Axin1 (2087) were obtained from Cell Signaling Technology. Following antibodies were obtained from Santa Cruz Biotechnology: VEGF (sc-152), TSP-1 (sc-12312), CD31 (sc-1506), and GATA-6 (sc-9055). Anti-active β-catenin (05-665, Millipore), anti-β-actin antibody (Sigma-Aldrich, A5441), phosphorylated c-Met antibody (Y1003) (Invitrogen, 44-882G) were used. All antibodies were used according to manufacturer’s instructions.
Cell lines and cell culture
Parental MU, RU, EP and WK (MU-P, RU-P, EP-P and WK-P) cell lines were obtained by explant culture.80,81 Cells were incubated at 37 °C and 7% CO2 and maintained in MEM medium (Thermo Fisher Scientific, MT-10-010-CM) and supplemented with 10% (v/v) fetal bovine serum (Atlanta Biologicals, S11150) and 1% (v/v) antibiotic/antimycotic (Invitrogen, 15240). The cells were trypsinized on a weekly basis with 0.05% trypsin at 1:4 split ratio. MU and RU melanoma cell lines were obtained as described previously.81-83 These cell lines have been well characterized and used in several studies, as described above. The purity and origin of melanoma cells were ensured with antibodies specific to melanoma, S-100 and HMB-45, as described earlier.80,82
Measurement of HGF production
EP-P, MU-P, RU-P, and WK-P cell lines were screened for HGF production by the human HGF ELISA kit (Ray Biotech, ELH-HGF-001) according to manufacturer’s instructions. Standards of human HGF were prepared at concentrations of 3–2000 pg/mL in duplicate for construction of the standard curve. One hundred microliters of HGF standards and undiluted supernatant from cells were loaded into a 96-well plate coated with an immobilized anti-human HGF antibody and allowed to incubate for 2.5 h at room temperature. HGF bound to the immobilized antibody was detected with biotinylated anti-human HGF antibody. HRP-conjugated streptavidin was then applied, developed, and detected spectrophotometrically at 450 nm.
Animals
Five-week-old male nu/nu (Taconic) and Rag1−/− on Balb/c background (Jackson Laboratories) mice used in the study were housed in the pathogen-free animal facility at the University of Illinois, College of Medicine at Rockford.
Ethical handling of animals for in vivo tumor analysis
Animals were monitored once daily. Animals were euthanized if tumor size exceeded 2 cm or if they developed erosions, displayed reduced motor activity, lost ability to drink or feed, or displayed weight loss greater than 20% compared with untreated controls. Animals did not suffer discomfort, distress, or pain in addition to that described above during the experiment. Animals were euthanized by CO2 inhalation, thus minimizing any potential discomfort, pain, or distress and were further subjected to cervical dislocation to ensure death. This is consistent with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association. Veterinary care was provided by experienced veterinary personnel at the University of Illinois, College of Medicine at Rockford. This facility is AAALAC accredited. All animal protocols were approved by the university regulated biological resource committee.
Antitumor activity of SU11274 and JNJ38877605 in vivo
Antitumor activity of SU11274 was assessed in vivo in RU-P xenografts. Five million RU-P cells suspended in HBSS were injected subcutaneously into the hind flanks of mice to establish xenograft models. Tumors were allowed to grow for one week and then treated daily with 50 μg SU11274 or diluent (100 µL 2% DMSO) daily by intratumoral (IT) injection for 4 wk. Following treatment, tumor volume was measured weekly by a blinded observer with digital calipers (Fisher Scientific, 06-664-16). Antitumor activity of JNJ38877605 was assessed in vivo in RU-P xenografts as described above. Palpable tumors were treated orally with 20 mg/kg JNJ38877605 in 20% Captisol, 0.25% PVP in 0.1 N HCl (vehicle) or vehicle alone as control for three weeks. Following treatment, tumor volume was measured weekly by a blinded observer with digital calipers (Fisher Scientific). Tumor volume was calculated according to the formula: volume = (length × width2) / 2.
Immunohistochemistry (IHC)
RU-P cell xenografts were resected from the mouse flank following treatment with TKIs or vehicle as described above. Tumors were formalin fixed, paraffin-embedded (FFPE) and sectioned at the Rockford Memorial Hospital pathology lab. Immunostaining procedures were performed as described earlier.1,84 Appropriate negative controls for immunostaining were prepared by omitting the primary antibody step and substituting it with non-immune rabbit serum. Antibodies against p-c-Met, CD31, VEGF, and TSP-1 were used according to manufacturer instructions. Horseradish peroxidase (HRP) was conjugated to secondary antibody using the Vector ABC kit, developed with DAB, and counterstained with Mayer’s hematoxylin. Blood vessel density in tumor sections was determined by averaging the number of vessels in 10 microscopic fields at 20× magnification.
Establishment of SU11274 drug resistant cell lines and evaluation of resistance
SU11274 drug resistant cells were established by exposure to increasing concentrations of SU11274 (0.5–12 μM). MU and RU cells were initially cultured in DMEM containing SU11274 at a concentration of 0.5 μM and then the cells were sub-cultured weekly in DMEM with 0.5–1 μM increases in concentrations of SU11274. Finally, the resultant cell lines that grew exponentially in the presence of high concentrations were designated as drug resistant cell lines, and named as MU-R and RU-R cell lines respectively. Five individual clones from each cell line were isolated, expanded, and assessed for stable resistance after each serial passage.85 The clone for each cell line that was most stable and least susceptible to the drug was selected for further studies. IC50 values for SU11274 were calculated for each cell line (Table S1) using Sigma Plot V12.0 (SYSTAT Software). SU11274 resistant MU and RU cells (MU-R, RU-R) were cultured in the absence of SU11274 for 12 passages, passed on a weekly basis, and found to retain resistance. Resistant clones were cultured in concentrations of SU11274, 7- to 10-fold higher compared with parental cells. For evaluation of resistance in cell lines, MU-P, MU-R, RU-P, and RU-R cells were seeded in 6-well plates, starved in 0.5% BSA for 24 h, then treated with SU11274 (0.5–10 µM) or tivantinib (0.2 µM) for 24–96 h.
Preparation of lysates and immunoblotting
Cells were lysed in buffer containing 20 mM Tris (pH 8.0), 150 mM NaCl, 10% glycerol, 1% NP40, 0.42% NaF, 1 mM PMSF, 1 mM sodium ortho-vanadate, and protease inhibitor cocktail (Roche Applied Sciences, 11 697 498 001) as described previously.1,86 Lysates were prepared for SDS-PAGE by adding 50 μg of protein to 4× Laemelli’s loading buffer (250 mM TRIS-HCl pH 6.8, 8% SDS, 40% glycerol, 8% β-mercaptoethanol, and 0.02% bromophenol blue) (Boston Bioproducts, BP-110NR). Samples were denatured at 100 °C for 8 min and centrifuged for 30 s at 14 000 rpm, prior to electrophoresis. Protein samples and dual colored protein standards (Bio-rad, 161-0374) were loaded onto a 10% polyacrylamide gel and electrophoresed for 1.5 h at 90 V in a Mini-Protean Tetra Cell (Bio-Rad, 165-8004) using the PowerPac Basic Power Supply (Bio-Rad, 164-5052) and Running buffer (25 mM Tris-HCL pH 8.3, 0.19 M glycine, and 0.1% SDS) (Boston Bioproducts, BP-150). Proteins were transferred onto a nitrocellulose membrane (BioRad, 162-0112) using the Trans-Blot SD Semi-Dry Electrophoretic Transfer Cell (BioRad, 170-3940) in transfer buffer (0.025 M Tris-HCL pH 8.4, glycine 0.19 M, and 20% methanol) (Boston Bioproducts, BP-190) at 18 V for 45 min. Membranes were then immunoblotted for proteins of interest. β-actin served as a loading control. Immunoblots were visualized using enhanced chemiluminescence (Thermo Fisher Scientific, 32106). Densitometry was performed using ImageJ software.
Cell viability assays
Cell viability was measured by MTT colorimetric dye reduction assay (Sigma, M5655) according to manufacturer instructions. Two thousand cells per well of MU-P, MU-R, RU-P, and RU-R were plated in a 96-well plate in replicates of six and treated after 24 h with inhibitors. After 96 h of treatment, MTT reagent was added, allowed to incubate for 3 h, and solubilized. Absorbance was measured at a wavelength of 570 nm (corrected for background at 690 nm). Cell viability was determined relative to the control. Dose response curves of Tivantinib (0.05–0.5 μM) and Vemurafenib (0.01–0.5 μM) were conducted and results are shown in Figures S1 and S2, respectively. Dose response curves of SU11274 were conducted by us previously as well. MTT data was confirmed by measuring cell proliferation and assessing the number of viable cells using the trypan blue exclusion method, as seen in Figure S4. Cells were plated and after 24 h, treated with diluent (drug-free media) or everolimus (1 μM), SU11274 (5 μM), and XAV939 (15 μM), individually and in combinations for 96 h. The numbers of viable cells were counted, subsequent to trypsinization, as described previously.37,87
DNA sequencing of BRAF and MET genes
Five million MU-P, MU-R, RU-P and RU-R cells were plated on 150 mm diameter petri dishes. DNA was extracted using the Qiagen DNeasy® Blood and Tissue kit (69504) according to manufacturer instructions. PCR using AmpliTaq Gold® PCR Master Mix (Applied Biosystems, 4398876) was then performed to amplify exons 15–21 of the MET gene containing the TKD as reported earlier84 and exon 15 of the BRAF gene to identify V600E mutation status as previously described.88 PCR products were purified using the GeneJETTM PCR Purification Kit (Fermentas, K0701) and were sequenced at the University of Illinois DNA Services Facility. Forward and reverse sequences were then aligned against the MET gene sequence (NM_000245) using NCBI blast or discontinuous-megablast algorithms to determine homology.
Statistical analysis
Statistical analyses were performed using SPSS 17.0 software. Repeated measures ANOVA with multiple pairwise comparisons as well as custom contrasts with Bonferroni adjustments were performed. Statistical significance was determined with α at 0.05. To confirm the differences between treatments, a paired two-tailed Student t test was also used. For all analysis performed, a P value of less than 0.05 was considered to be statistically significant. Synergism of tivantinib and vemurafenib was verified using the BIOSOFT CalcuSyn software, where the isobologram graphs showed combinatorial index (CI) values below 1.0, based on the medial effect model.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This research was funded by the University of Illinois at Chicago Cancer Center 2009 Pilot Grant Program. The authors would like to acknowledge the Research Open Access Publishing (ROAAP) Fund of the University of Illinois at Chicago for financial support toward the open access publishing fee for this article.
References
- 1.Puri N, Ahmed S, Janamanchi V, Tretiakova M, Zumba O, Krausz T, Jagadeeswaran R, Salgia R. c-Met is a potentially new therapeutic target for treatment of human melanoma. Clin Cancer Res. 2007;13:2246–53. doi: 10.1158/1078-0432.CCR-06-0776. [DOI] [PubMed] [Google Scholar]
- 2.Lee YJ, Kim DH, Lee SH, Kim DW, Nam HS, Cho MK. Expression of the c-Met Proteins in Malignant Skin Cancers. Ann Dermatol. 2011;23:33–8. doi: 10.5021/ad.2011.23.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee JH, Han SU, Cho H, Jennings B, Gerrard B, Dean M, Schmidt L, Zbar B, Vande Woude GF. A novel germ line juxtamembrane Met mutation in human gastric cancer. Oncogene. 2000;19:4947–53. doi: 10.1038/sj.onc.1203874. [DOI] [PubMed] [Google Scholar]
- 4.Nakamura T, Sakai K, Nakamura T, Matsumoto K. Hepatocyte growth factor twenty years on: Much more than a growth factor. J Gastroenterol Hepatol. 2011;26(Suppl 1):188–202. doi: 10.1111/j.1440-1746.2010.06549.x. [DOI] [PubMed] [Google Scholar]
- 5.Vergani E, Vallacchi V, Frigerio S, Deho P, Mondellini P, Perego P, Cassinelli G, Lanzi C, Testi MA, Rivoltini L, et al. Identification of MET and SRC activation in melanoma cell lines showing primary resistance to PLX4032. Neoplasia. 2011;13:1132–42. doi: 10.1593/neo.111102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharma A, Shah SR, Illum H, Dowell J. Vemurafenib: targeted inhibition of mutated BRAF for treatment of advanced melanoma and its potential in other malignancies. Drugs. 2012;72:2207–22. doi: 10.2165/11640870-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 7.Dummer R, Flaherty KT. Resistance patterns with tyrosine kinase inhibitors in melanoma: new insights. Curr Opin Oncol. 2012;24:150–4. doi: 10.1097/CCO.0b013e32834fca92. [DOI] [PubMed] [Google Scholar]
- 8.Pollock PM, Harper UL, Hansen KS, Yudt LM, Stark M, Robbins CM, Moses TY, Hostetter G, Wagner U, Kakareka J, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- 9.Kelleher FC, McArthur GA. Targeting NRAS in melanoma. Cancer J. 2012;18:132–6. doi: 10.1097/PPO.0b013e31824ba4df. [DOI] [PubMed] [Google Scholar]
- 10.Chattopadhyay C, Ellerhorst JA, Ekmekcioglu S, Greene VR, Davies MA, Grimm EA. Association of activated c-Met with NRAS-mutated human melanomas. Int J Cancer. 2012;131:E56–65. doi: 10.1002/ijc.26487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sattler M, Salgia R. c-Met and hepatocyte growth factor: potential as novel targets in cancer therapy. Curr Oncol Rep. 2007;9:102–8. doi: 10.1007/s11912-007-0005-4. [DOI] [PubMed] [Google Scholar]
- 12.Kim ES, Salgia R. MET pathway as a therapeutic target. J Thorac Oncol. 2009;4:444–7. doi: 10.1097/JTO.0b013e31819d6f91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Otsuka T, Takayama H, Sharp R, Celli G, LaRochelle WJ, Bottaro DP, Ellmore N, Vieira W, Owens JW, Anver M, et al. c-Met autocrine activation induces development of malignant melanoma and acquisition of the metastatic phenotype. Cancer Res. 1998;58:5157–67. [PubMed] [Google Scholar]
- 14.Cruz J, Reis-Filho JS, Silva P, Lopes JM. Expression of c-met tyrosine kinase receptor is biologically and prognostically relevant for primary cutaneous malignant melanomas. Oncology. 2003;65:72–82. doi: 10.1159/000071207. [DOI] [PubMed] [Google Scholar]
- 15.Sattler M, Pride YB, Ma P, Gramlich JL, Chu SC, Quinnan LA, Shirazian S, Liang C, Podar K, Christensen JG, et al. A novel small molecule met inhibitor induces apoptosis in cells transformed by the oncogenic TPR-MET tyrosine kinase. Cancer Res. 2003;63:5462–9. [PubMed] [Google Scholar]
- 16.Cepero V, Sierra JR, Corso S, Ghiso E, Casorzo L, Perera T, Comoglio PM, Giordano S. MET and KRAS gene amplification mediates acquired resistance to MET tyrosine kinase inhibitors. Cancer Res. 2010;70:7580–90. doi: 10.1158/0008-5472.CAN-10-0436. [DOI] [PubMed] [Google Scholar]
- 17.King P, Jansens B, Verhulst T, Argent R, Kumari R, Watson S, Andries L, Mevellec L, Page M, Perera T. JNJ-38877605: A selective oral Met inhibitor for the treatment of cancer. Cancer Res [Internet] 2010; 70(8 Suppl):Abstract 3628. Available from: http://cancerres.aacrjournals.org/cgi/content/meeting_abstract/70/8_MeetingAbstracts/3628
- 18.Perera T, Lavrijssen T, Janssens B, Geerts T, King P, Mevellec L, Cummings M, Lu T, Johnson D, Page M. JNJ-38877605: a selective Met kinase inhibitor inducing regression of Met-driven tumor models. Presented at the 99th AACR Annual Meeting; 2008 Apr 12–16; San Diego (CA): Abstract nr 4837. [Google Scholar]
- 19.Qi J, McTigue MA, Rogers A, Lifshits E, Christensen JG, Jänne PA, Engelman JA. Multiple mutations and bypass mechanisms can contribute to development of acquired resistance to MET inhibitors. Cancer Res. 2011;71:1081–91. doi: 10.1158/0008-5472.CAN-10-1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sierra JR, Cepero V, Giordano S. Molecular mechanisms of acquired resistance to tyrosine kinase targeted therapy. Mol Cancer. 2010;9:75. doi: 10.1186/1476-4598-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sierra JR, Tsao MS. c-MET as a potential therapeutic target and biomarker in cancer. Ther Adv Med Oncol. 2011;3(Suppl):S21–35. doi: 10.1177/1758834011422557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, Qian F, Chu F, Bentzien F, Cancilla B, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther. 2011;10:2298–308. doi: 10.1158/1535-7163.MCT-11-0264. [DOI] [PubMed] [Google Scholar]
- 23.Gordon SM, Kluger HM, Shapiro G, Kurzrock R, Edelman G, Samuel TA, Moussa AH, Ramies DA, Laird AD, Schimmoller F, et al. Activity of cabozantinib (XL184) in metastatic melanoma: Results from a phase II randomized discontinuation trial (RDT). J Clin Oncol 2012; 30(suppl): abstr 8531. Available from: http://meetinglibrary.asco.org/content/94473-114
- 24.He X, Semenov M, Tamai K, Zeng X. LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: arrows point the way. Development. 2004;131:1663–77. doi: 10.1242/dev.01117. [DOI] [PubMed] [Google Scholar]
- 25.Zeng X, Huang H, Tamai K, Zhang X, Harada Y, Yokota C, Almeida K, Wang J, Doble B, Woodgett J, et al. Initiation of Wnt signaling: control of Wnt coreceptor Lrp6 phosphorylation/activation via frizzled, dishevelled and axin functions. Development. 2008;135:367–75. doi: 10.1242/dev.013540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee E, Salic A, Krüger R, Heinrich R, Kirschner MW. The roles of APC and Axin derived from experimental and theoretical analysis of the Wnt pathway. PLoS Biol. 2003;1:E10. doi: 10.1371/journal.pbio.0000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salic A, Lee E, Mayer L, Kirschner MW. Control of beta-catenin stability: reconstitution of the cytoplasmic steps of the wnt pathway in Xenopus egg extracts. Mol Cell. 2000;5:523–32. doi: 10.1016/S1097-2765(00)80446-3. [DOI] [PubMed] [Google Scholar]
- 28.Kim MJ, Chia IV, Costantini F. SUMOylation target sites at the C terminus protect Axin from ubiquitination and confer protein stability. FASEB J. 2008;22:3785–94. doi: 10.1096/fj.08-113910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adachi Y, Shibai Y, Mitsushita J, Shang WH, Hirose K, Kamata T. Oncogenic Ras upregulates NADPH oxidase 1 gene expression through MEK-ERK-dependent phosphorylation of GATA-6. Oncogene. 2008;27:4921–32. doi: 10.1038/onc.2008.133. [DOI] [PubMed] [Google Scholar]
- 30.Bhattacharya S, Mathew G, Jayne DG, Pelengaris S, Khan M. 15-lipoxygenase-1 in colorectal cancer: a review. Tumour Biol. 2009;30:185–99. doi: 10.1159/000236864. [DOI] [PubMed] [Google Scholar]
- 31.Weidenfeld J, Shu W, Zhang L, Millar SE, Morrisey EE. The WNT7b promoter is regulated by TTF-1, GATA6, and Foxa2 in lung epithelium. J Biol Chem. 2002;277:21061–70. doi: 10.1074/jbc.M111702200. [DOI] [PubMed] [Google Scholar]
- 32.Zhong Y, Wang Z, Fu B, Pan F, Yachida S, Dhara M, Albesiano E, Li L, Naito Y, Vilardell F, et al. GATA6 activates Wnt signaling in pancreatic cancer by negatively regulating the Wnt antagonist Dickkopf-1. PLoS One. 2011;6:e22129. doi: 10.1371/journal.pone.0022129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li G, Schaider H, Satyamoorthy K, Hanakawa Y, Hashimoto K, Herlyn M. Downregulation of E-cadherin and Desmoglein 1 by autocrine hepatocyte growth factor during melanoma development. Oncogene. 2001;20:8125–35. doi: 10.1038/sj.onc.1205034. [DOI] [PubMed] [Google Scholar]
- 34.Stender IM, Etoh T, Tsuchida T, Nakagawa H, Byers HR, Imokawa G, Ishibashi Y. Metastatic behavior and tumorigenicity of a human melanoma cell line (MM-RU) after injection into nude mice. J Dermatol. 1993;20:611–7. doi: 10.1111/j.1346-8138.1993.tb01349.x. [DOI] [PubMed] [Google Scholar]
- 35.Vultur A, Villanueva J, Herlyn M. Targeting BRAF in advanced melanoma: a first step toward manageable disease. Clin Cancer Res. 2011;17:1658–63. doi: 10.1158/1078-0432.CCR-10-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munshi N, Jeay S, Li Y, Chen CR, France DS, Ashwell MA, Hill J, Moussa MM, Leggett DS, Li CJ. ARQ 197, a novel and selective inhibitor of the human c-Met receptor tyrosine kinase with antitumor activity. Mol Cancer Ther. 2010;9:1544–53. doi: 10.1158/1535-7163.MCT-09-1173. [DOI] [PubMed] [Google Scholar]
- 37.Puri N, Salgia R. Synergism of EGFR and c-Met pathways, cross-talk and inhibition, in non-small cell lung cancer. J Carcinog. 2008;7:9. doi: 10.4103/1477-3163.44372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yap TA, Olmos D, Brunetto AT, Tunariu N, Barriuso J, Riisnaes R, Pope L, Clark J, Futreal A, Germuska M, et al. Phase I trial of a selective c-MET inhibitor ARQ 197 incorporating proof of mechanism pharmacodynamic studies. J Clin Oncol. 2011;29:1271–9. doi: 10.1200/JCO.2010.31.0367. [DOI] [PubMed] [Google Scholar]
- 39.Shaw HM, Nathan PD. Vemurafenib in melanoma. Expert Rev Anticancer Ther. 2013;13:513–22. doi: 10.1586/era.13.24. [DOI] [PubMed] [Google Scholar]
- 40.Hu T, Li C. Convergence between Wnt-β-catenin and EGFR signaling in cancer. Mol Cancer. 2010;9:236. doi: 10.1186/1476-4598-9-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Efeyan A, Sabatini DM. mTOR and cancer: many loops in one pathway. Curr Opin Cell Biol. 2010;22:169–76. doi: 10.1016/j.ceb.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jazirehi AR, Wenn PB, Damavand M. Therapeutic implications of targeting the PI3Kinase/AKT/mTOR signaling module in melanoma therapy. Am J Cancer Res. 2012;2:178–91. [PMC free article] [PubMed] [Google Scholar]
- 43.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 44.Yamamoto H, Kishida S, Kishida M, Ikeda S, Takada S, Kikuchi A. Phosphorylation of axin, a Wnt signal negative regulator, by glycogen synthase kinase-3beta regulates its stability. J Biol Chem. 1999;274:10681–4. doi: 10.1074/jbc.274.16.10681. [DOI] [PubMed] [Google Scholar]
- 45.Sinnberg T, Menzel M, Ewerth D, Sauer B, Schwarz M, Schaller M, Garbe C, Schittek B. β-Catenin signaling increases during melanoma progression and promotes tumor cell survival and chemoresistance. PLoS One. 2011;6:e23429. doi: 10.1371/journal.pone.0023429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mita MM, Mita A, Rowinsky EK. The molecular target of rapamycin (mTOR) as a therapeutic target against cancer. Cancer Biol Ther. 2003;2(Suppl 1):S169–77. [PubMed] [Google Scholar]
- 47.Pópulo H, Lopes JM, Soares P. The mTOR Signalling Pathway in Human Cancer. Int J Mol Sci. 2012;13:1886–918. doi: 10.3390/ijms13021886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614–20. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- 49.O’Reilly T, McSheehy PM. Biomarker Development for the Clinical Activity of the mTOR Inhibitor Everolimus (RAD001): Processes, Limitations, and Further Proposals. Transl Oncol. 2010;3:65–79. doi: 10.1593/tlo.09277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Busch AM, Johnson KC, Stan RV, Sanglikar A, Ahmed Y, Dmitrovsky E, Freemantle SJ. Evidence for tankyrases as antineoplastic targets in lung cancer. BMC Cancer. 2013;13:211. doi: 10.1186/1471-2407-13-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Puri N, Khramtsov A, Ahmed S, Nallasura V, Hetzel JT, Jagadeeswaran R, Karczmar G, Salgia R. A selective small molecule inhibitor of c-Met, PHA665752, inhibits tumorigenicity and angiogenesis in mouse lung cancer xenografts. Cancer Res. 2007;67:3529–34. doi: 10.1158/0008-5472.CAN-06-4416. [DOI] [PubMed] [Google Scholar]
- 52.Laux I, Goldman J, Just R, Brady K, Li J, Schwartz B, Savage R, Garmey E, Rosen L. Phase I dose escalation trial (ARQ 197–111) evaluating combination of selective c-Met inhibitor ARQ 197 and erlotinib. J Clin Oncol 2009; 27(15S):abstract 3549. Available from: http://meetinglibrary.asco.org/content/31504-65
- 53.Nakachi I, Naoki K, Soejima K, Kawada I, Watanabe H, Yasuda H, Nakayama S, Yoda S, Satomi R, Ikemura S, et al. The combination of multiple receptor tyrosine kinase inhibitor and mammalian target of rapamycin inhibitor overcomes erlotinib resistance in lung cancer cell lines through c-Met inhibition. Mol Cancer Res. 2010;8:1142–51. doi: 10.1158/1541-7786.MCR-09-0388. [DOI] [PubMed] [Google Scholar]
- 54.Dillon R, Nilsson CL, Shi SD, Lee NV, Krastins B, Greig MJ. Discovery of a novel B-Raf fusion protein related to c-Met drug resistance. J Proteome Res. 2011;10:5084–94. doi: 10.1021/pr200498v. [DOI] [PubMed] [Google Scholar]
- 55.Schmid K, Bago-Horvath Z, Berger W, Haitel A, Cejka D, Werzowa J, Filipits M, Herberger B, Hayden H, Sieghart W. Dual inhibition of EGFR and mTOR pathways in small cell lung cancer. Br J Cancer. 2010;103:622–8. doi: 10.1038/sj.bjc.6605761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dazert E, Hall MN. mTOR signaling in disease. Curr Opin Cell Biol. 2011;23:744–55. doi: 10.1016/j.ceb.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 58.Karbowniczek M, Spittle CS, Morrison T, Wu H, Henske EP. mTOR is activated in the majority of malignant melanomas. J Invest Dermatol. 2008;128:980–7. doi: 10.1038/sj.jid.5701074. [DOI] [PubMed] [Google Scholar]
- 59.Pópulo H, Soares P, Faustino A, Rocha AS, Silva P, Azevedo F, Lopes JM. mTOR pathway activation in cutaneous melanoma is associated with poorer prognosis characteristics. Pigment Cell Melanoma Res. 2011;24:254–7. doi: 10.1111/j.1755-148X.2010.00796.x. [DOI] [PubMed] [Google Scholar]
- 60.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–57. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 61.Hahn-Windgassen A, Nogueira V, Chen CC, Skeen JE, Sonenberg N, Hay N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J Biol Chem. 2005;280:32081–9. doi: 10.1074/jbc.M502876200. [DOI] [PubMed] [Google Scholar]
- 62.Choo AY, Roux PP, Blenis J. Mind the GAP: Wnt steps onto the mTORC1 train. Cell. 2006;126:834–6. doi: 10.1016/j.cell.2006.08.025. [DOI] [PubMed] [Google Scholar]
- 63.Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, Nika H, Mills GB, Kobayashi R, Hunter T, Lu Z. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem. 2007;282:11221–9. doi: 10.1074/jbc.M611871200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weeraratna AT, Jiang Y, Hostetter G, Rosenblatt K, Duray P, Bittner M, Trent JM. Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell. 2002;1:279–88. doi: 10.1016/S1535-6108(02)00045-4. [DOI] [PubMed] [Google Scholar]
- 65.Pham K, Milovanovic T, Barr RJ, Truong T, Holcombe RF. Wnt ligand expression in malignant melanoma: pilot study indicating correlation with histopathological features. Mol Pathol. 2003;56:280–5. doi: 10.1136/mp.56.5.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Da Forno PD, Pringle JH, Hutchinson P, Osborn J, Huang Q, Potter L, Hancox RA, Fletcher A, Saldanha GS. WNT5A expression increases during melanoma progression and correlates with outcome. Clin Cancer Res. 2008;14:5825–32. doi: 10.1158/1078-0432.CCR-07-5104. [DOI] [PubMed] [Google Scholar]
- 67.Flaherty KT. Chemotherapy and targeted therapy combinations in advanced melanoma. Clin Cancer Res. 2006;12:2366s–70s. doi: 10.1158/1078-0432.CCR-05-2505. [DOI] [PubMed] [Google Scholar]
- 68.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–4. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fong JT, Jacobs RJ, Moravec DN, Uppada SB, Botting GM, Nlend M, Puri N. Alternative signaling pathways as potential therapeutic targets for overcoming EGFR and c-Met inhibitor resistance in non-small cell lung cancer. PLoS One. 2013;8:e78398. doi: 10.1371/journal.pone.0078398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Apte U, Zeng G, Muller P, Tan X, Micsenyi A, Cieply B, Dai C, Liu Y, Kaestner KH, Monga SP. Activation of Wnt/beta-catenin pathway during hepatocyte growth factor-induced hepatomegaly in mice. Hepatology. 2006;44:992–1002. doi: 10.1002/hep.21317. [DOI] [PubMed] [Google Scholar]
- 71.Liou GI, Matragoon S, Samuel S, Behzadian MA, Tsai NT, Gu X, Roon P, Hunt DM, Hunt RC, Caldwell RB, et al. MAP kinase and beta-catenin signaling in HGF induced RPE migration. Mol Vis. 2002;8:483–93. [PubMed] [Google Scholar]
- 72.Huang FI, Chen YL, Chang CN, Yuan RH, Jeng YM. Hepatocyte growth factor activates Wnt pathway by transcriptional activation of LEF1 to facilitate tumor invasion. Carcinogenesis. 2012;33:1142–8. doi: 10.1093/carcin/bgs131. [DOI] [PubMed] [Google Scholar]
- 73.Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer. 2004;4:335–48. doi: 10.1038/nrc1362. [DOI] [PubMed] [Google Scholar]
- 74.Barker N. The canonical Wnt/beta-catenin signalling pathway. Methods Mol Biol. 2008;468:5–15. doi: 10.1007/978-1-59745-249-6_1. [DOI] [PubMed] [Google Scholar]
- 75.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cadigan KM, Liu YI. Wnt signaling: complexity at the surface. J Cell Sci. 2006;119:395–402. doi: 10.1242/jcs.02826. [DOI] [PubMed] [Google Scholar]
- 77.Kawada I, Hasina R, Arif Q, Mueller J, Smithberger E, Husain AN, Vokes EE, Salgia R. Dramatic antitumor effects of the dual MET/RON small-molecule inhibitor LY2801653 in non-small cell lung cancer. Cancer Res. 2014;74:884–95. doi: 10.1158/0008-5472.CAN-12-3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu D, Pan W. GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem Sci. 2010;35:161–8. doi: 10.1016/j.tibs.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yun MS, Kim SE, Jeon SH, Lee JS, Choi KY. Both ERK and Wnt/beta-catenin pathways are involved in Wnt3a-induced proliferation. J Cell Sci. 2005;118:313–22. doi: 10.1242/jcs.01601. [DOI] [PubMed] [Google Scholar]
- 80.Byers HR, Etoh T, Doherty JR, Sober AJ, Mihm MC., Jr. Cell migration and actin organization in cultured human primary, recurrent cutaneous and metastatic melanoma. Time-lapse and image analysis. Am J Pathol. 1991;139:423–35. [PMC free article] [PubMed] [Google Scholar]
- 81.Puri N, Eller MS, Byers HR, Dykstra S, Kubera J, Gilchrest BA. Telomere-based DNA damage responses: a new approach to melanoma. FASEB J. 2004;18:1373–81. doi: 10.1096/fj.04-1774com. [DOI] [PubMed] [Google Scholar]
- 82.Ramirez-Montagut T, Andrews DM, Ihara A, Pervaiz S, Pandolfi F, Van Den Elsen PJ, Waitkus R, Boyle LA, Hishii M, Kurnick JT. Melanoma antigen recognition by tumour-infiltrating T lymphocytes (TIL): effect of differential expression of melan-A/MART-1. Clin Exp Immunol. 2000;119:11–8. doi: 10.1046/j.1365-2249.2000.01089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pandolfi F, Trentin L, Boyle LA, Stamenkovic I, Byers HR, Colvin RB, Kurnick JT. Expression of cell adhesion molecules in human melanoma cell lines and their role in cytotoxicity mediated by tumor-infiltrating lymphocytes. Cancer. 1992;69:1165–73. doi: 10.1002/cncr.2820690517. [DOI] [PubMed] [Google Scholar]
- 84.Ma PC, Jagadeeswaran R, Jagadeesh S, Tretiakova MS, Nallasura V, Fox EA, Hansen M, Schaefer E, Naoki K, Lader A, et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. 2005;65:1479–88. doi: 10.1158/0008-5472.CAN-04-2650. [DOI] [PubMed] [Google Scholar]
- 85.Coley HM. Development of drug-resistant models. Methods Mol Med. 2004;88:267–73. doi: 10.1385/1-59259-406-9:267. [DOI] [PubMed] [Google Scholar]
- 86.Maulik G, Kijima T, Ma PC, Ghosh SK, Lin J, Shapiro GI, Schaefer E, Tibaldi E, Johnson BE, Salgia R. Modulation of the c-Met/hepatocyte growth factor pathway in small cell lung cancer. Clin Cancer Res. 2002;8:620–7. [PubMed] [Google Scholar]
- 87.Puri N, Pitman RT, Mulnix RE, Erickson T, Iness AN, Vitali C, Zhao Y, Salgia R. Non-small cell lung cancer is susceptible to induction of DNA damage responses and inhibition of angiogenesis by telomere overhang oligonucleotides. Cancer Lett. 2014;343:14–23. doi: 10.1016/j.canlet.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Turner DJ, Zirvi MA, Barany F, Elenitsas R, Seykora J. Detection of the BRAF V600E mutation in melanocytic lesions using the ligase detection reaction. J Cutan Pathol. 2005;32:334–9. doi: 10.1111/j.0303-6987.2005.00338.x. [DOI] [PubMed] [Google Scholar]
- 89.Belaguli NS, Aftab M, Rigi M, Zhang M, Albo D, Berger DH. GATA6 promotes colon cancer cell invasion by regulating urokinase plasminogen activator gene expression. Neoplasia. 2010;12:856–65. doi: 10.1593/neo.10224. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.