

Abstract
A common view is that an accidental increase in ploidy contributes to the evolution of neoplastic cells primarily by decreasing the fidelity of mitosis with extra chromosomes and centrosomes. This view implies that how neoplastic cells become polyploid is irrelevant, as it has been widely assumed. If this assumption is correct, then the oncogenic contribution of the pathways to polyploidy and thus their potential as targets for cancer prevention is determined by their incidence in the body. A lesson from plant evolution, in which an accidental increase in ploidy has a prevalent role, suggests that this assumption needs to be reconsidered.
Keywords: cancer; differentiation; allopolyploidy; autopolyploidy; cell hybrids; cell fusion; evolution; plants; polyploidy,reprogramming; tetraploidy
Cancers are commonly viewed as a result of Darwinian evolution that normal cells undergo by acquiring genomic aberrations.1-8 A common cause of these aberrations, according to the actively debated tetraploidy model of carcinogenesis,9-16 is an accidental increase in ploidy. An accidental increase in ploidy is also a common and extensively studied cause of evolution in plants,17,18 a potential homology to explore for clues to cancer development. This commentary suggests that one of these clues is the fact that the properties and the evolutionary potential of plants are defined by how they become tetraploid. (Box 1)
Box 1: Terminology
Euploid cells have an exact multiple of the haploid chromosome set. Diploid cells have two sets, tetraploid four sets, etc.
Aneuploid cells are those that are not euploid.
Polyploid cells are those that have more than two sets of chromosomes. Autopolyploid cells or organisms have more than two homologous sets of chromosomes, autotetraploid would have four.
Allopolyploid cells or organisms have more than two homologous sets of chromosomes derived from two or more species.
Hybrid is a cell derived from the fusion of two or more different cells, or an organism derived from crossing two or more species.
Saltational evolution produces new species rapidly, within one or few generations, while gradual evolution occurs by accumulation of minor traits.
Tetraploidy is thought to cause cancer primarily by increasing the incidence of chromosomal aberrations that result from overloading the mitotic machinery with extra chromosomes and from enabling multipolar mitoses with extra centrosomes,11,14,15,19-25 although other consequences of having extra chromosomes, such as increased DNA damage, are also considered.11,15,19,26 If doubling the number of chromosomes and centrosomes is indeed the primary oncogenic consequence of tetraploidy, then how cells become tetraploid, which can happen by endoreduplication, cytokinesis failure or cell fusion is irrelevant, as it has been tacitly assumed.15,19-28 If this assumption is correct, then the contribution of these three mechanisms to cancer and thus their potential as targets for cancer prevention is directly proportional to their incidence in the body. The role of polyploidy in plant evolution suggests that these assumptions may need to be reconsidered.
Two types of polyploidy are distinguished in plants and other organisms (Fig. 1): autopolyploidy results from duplicating the genome of one species, while allopolyploidy is an outcome of combining genomes of different species or sub-species.17,29-31 In other words, allopolyploid plants are polyploid hybrids. Although the ploidy of auto- and allopolyploid plants can be identical, their properties are not. While phenotypic manifestations of autopolyploidy may not always be obvious,17,29 allopolyploid plants have high phenotypic plasticity, are prevalent, adapt better to challenging environments and are prone to saltational evolution.17,18,29,32,33 “One example of rapid and superior adaptation is provided by the widespread dispersal of the invasive, recently formed allopolyploid, Spartina anglica, which contrasts with the relatively noninvasive nature of the parental species, which are presumed to be autopolyploids”.17 The properties of allopolyploid plants and the “footprints” of allopolyploidy in plant genomes led to the “diverge, merge, and diverge” model, according to which a majority of the flowering species have emerged from repeated cycles of allopolyploidy and divergence.34 Hence, the properties and the evolutionary potential of plants depend on how they become polyploid.
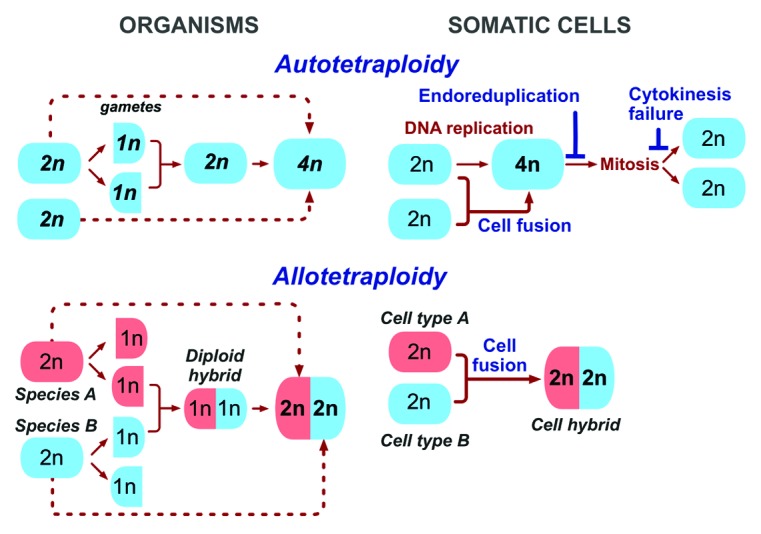
Figure 1. The pathways to tetraploidy in the evolution of species and somatic cells. Autotetraploid organisms result from whole genome duplication occurring either by doubling the genome of the zygote, or by the fusion of non-reduced gametes (dotted lines). Allotetraploid organisms are created by the fusion of gametes from different species with the subsequent duplication of the genome, or by fusion of non-reduced gametes (dotted lines). In somatic cells, tetraploidy is caused by either whole genome duplication (top panel), which can result from endoreduplication, cytokinesis failure, and fusion of identical cells, or by fusion that combines different cells (bottom panel). Note (dotted lines) that allopolyploids can be made by fusing somatic plant cells (protoplasts) in vitro and letting the resulting hybrids to develop into plants. 1n, 2n and 4n indicate haploid, diploid and tetraploid number of chromosomes.
The phenotypic diversity of allopolyploid plants reflects their genomic and epigenetic plasticity,35 whose scale and variety is reminiscent of that observed in cancer.
Genomic changes characteristic of allopolyploids are familiar to a cancer researcher, although in plants these changes are not called aberrations, as all plants are considered normal, even those that invade others. The list includes chromosome translocations, inversions, deletions, duplications, loss, amplification or reduction of repetitive sequences, extensive chromosome repatterning, horizontal transfer of genomic segments between the parental genomes, nonreciprocal homeologous recombination, repetitive sequences, transposon activation, and aneuploidy.18,34,36-38 At least some of the genomic rearrangements are not random and involve elimination of specific non-coding DNA sequences and intergenomic suppression of disease-resistant genes.18 Like genomic aberrations in cancer, genomic rearrangements in allopolyploids can range from simple to highly complex, persist through generations, or re-emerge after being in remission for generations.18,34-36,39
However substantial the genomic restructuring is, “by far, the most striking change to regulation of gene expression in allopolyploids…is that of epigenetic modification”.40 Some of these modifications are also similar to those found in cancers and include genome-wide alterations in cytosine methylation, gene silencing or activation, alteration of gene imprinting, gene non-functionalization, sub-functionalization, neo-functionalization, changes in mRNA splicing and in gene expression.30,35,41 Of particular interest are emergent, or so-called non-additive patterns of gene expression, as they can result in new, potentially advantageous phenotypes.29,40-42
To begin explaining how allopolyploidy causes genomic and epigenetic changes, Barbara McClintock suggested that uniting two distinct genomes in one nucleus is a type of stress, which she named “genomic shock”, that triggers restructuring and integration of the parental genomes,36 but what this stress is, how it is sensed, and how this information is translated into genome restructuring is still unknown.40,43
By analogy with the genomic shock, the cause of the gene expression changes has been termed the transcriptome shock,40 to indicate that uniting two different expression patterns is also a type of stress. The search for what the transcriptome shock is and how the merged regulatory gene expression networks reach a consensus is still in its infancy41-48 (see Box 2).
Box 2. Parental squabbles, state conflicts, and the Waddington landscape.
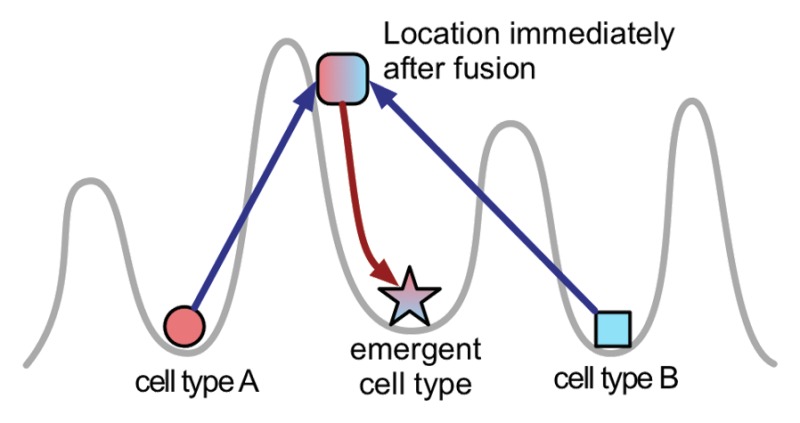
The relationship of the parental gene regulatory networks that are merged in allopolyploids has been compared with a conflict zone43 and parental squabbles.48 According to the colliding networks model,44 these analogies may be even deeper than they seem. The model is based on the notion that gene expression patterns correspond to points in the Waddington epigenetic landscape (sketched with the gray line), with the stable patterns (cell types) corresponding to the “valleys” known as attractors.49,50 Merging different cells nearly instantly creates a new pattern, which is initially an average of the two, on a slope of a “hill” in the landscape. This act of creation corresponds to the transcriptome shock.40 Being on a slope, the network “slides” into an attractor corresponding to a cell type distinct from either of the parental types, hence the emergent properties of the hybrids. This transition is manifested by changes in gene expression that appear chaotic and the instability that continues until the attractor is reached. Simulating this process computationally reveals that the transition is inherently uneven, with spontaneous bursts of rearrangements occurring after periods of gradual change.44 The cell can die in the process if an intermediate or the final gene expression pattern is incompatible with cell viability. The simulation also suggests that a majority of attractors correspond to abnormal cell types and that even minor variations in parental cells can change the properties of the progeny. The model posits that the laws that govern the fate of the merged networks in cell hybrids can be applicable to other complex systems derived from mergers, including families, businesses and countries.44
Symphiliosis, the Process of Intracellular Reconciliation
The characteristic features of allopolyploid plants—genomic restructuring, epigenetic plasticity and emergent gene expression patterns—are also the hallmarks of somatic cell hybrids in plants, animals and humans.44,51-59 Such commonality suggests that these features are manifestations of a basic, non-random (Box 2) and not yet fully understood intracellular process that is caused by merging two or more different cells into one and has a remarkable, perhaps unmatched capacity of producing cells and organisms with new properties (Fig. 2). Several aspects of this process have been named the genomic36 or transcriptome40 shock, a conflict zone,43 parental squabbles,48 nuclear reprogramming,60 and the problem of colliding networks.44 To acknowledge the existence of this process and to have a term to refer to it as a whole, I suggest calling it symphiliosis, from a Greek word for reconciliation, “συμφιλίωση”, and, accordingly, referring to all interrelated instabilities associated with symphiliosis, of which we probably know only some, as symphilial instability, or SIN. Hence, the defining features of allopolyploids are a result of symphiliosis rather than of the increase in ploidy per se.
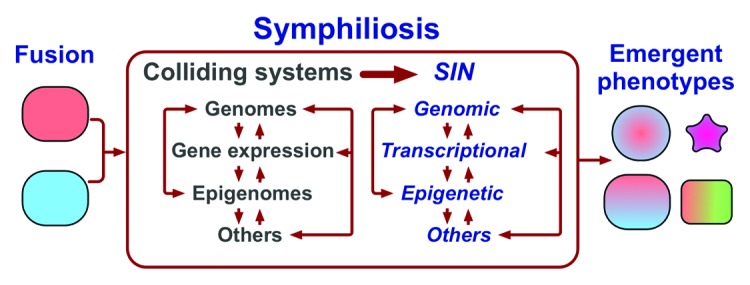
Figure 2. Fusion of different cells causes the collision of merged cellular systems, resulting in death or leading to symphiliosis, or reconciliation. Symphiliosis is manifested by interrelated instabilities summarily named symphilial instabilities (SIN) that are a result of integrating distinct sets of parental networks into one. Symphiliosis can produce emergent phenotypes, cause death, senescence, or continue permanently. “Other” systems can include mitochondria, which have their own genome, and interactions with other cells to give just two examples.
To emphasize the difference between the consequences of a ploidy increase and symphiliosis, let us consider the fact that each proliferating cell transiently becomes tetraploid during each cell cycle, from the moment it completes DNA replication and until it divides at the end of mitosis. Some cell types, such as hepatocytes,61 are commonly tetraploid or have a higher ploidy for their lifetime. Hence, tetraploidy per se is a normal and common condition in the life of a cell that does not necessarily affect its normal functioning. In contrast, cell fusion is normally restricted to a handful of processes, such as fertilization or formation of muscles, that involve predetermined cell types.62 The accidental fusion of different somatic cells, as it happens during viral infections,63 is akin to the marriage of randomly and blindly selected strangers, with similarly unexpected consequences.44
Do Allopolyploid Plants and Cancers Share Evolutionary Mechanisms?
The genomic and epigenetic plasticity shared by allopolyploid plants and cancers, and the common ability to nearly instantly, albeit on different time scales, produce new phenotypes beg the question of whether these similarities are superficial or reflect common underlying mechanisms or even causes. Barbara McClintock has already suggested the latter possibility by noting that “hybrids produced by crosses of distantly related Nicotiana species are known to give rise to tumors, some of which resemble teratomas”36 with similar effects observed in hybrids of some other species of plants and animals.64
A direct mechanistic link between allopolyploidy and cancer is implied by the model that cancers can result from the accidental fusion of different cells (reviewed in refs. 63 and 65–68). According to this model, premalignant cells can acquire the hallmarks of cancer—the abilities to invade and metastasize—by fusing to a normal cell, such as a macrophage or a lymphocyte, whose function is to travel throughout the body freely. This “marriage of convenience” can produce progeny that proliferate infinitely as one parent and invade multiple organs as the other. Fusion between neoplastic cells and between neoplastic and normal cells of various kind is thought to affect cancer progression by combining their properties, and by triggering symphilial instabilities, not unlike it is envisioned by the “diverge, merge, and diverge” model of plant evolution.34,69,70 Finally, the fusion of different cell types often results in the loss of gene expression patterns specific to either of the parental types, a phenomenon known as extinction44,71 would not be expected to include, a feature of aggressive cancers. Extinction also has the potential to revert the cell to an atavistic state.3 Overall, the cell fusion model implies that genomic and epigenetic changes found in allopolyploid plants and cancers are similar because some cancers are also allopolyploid, or in essence, that some cancers are homologous to invasive allopolyploid weeds.
Since allopolyploidy results from merging cells of different species, the question is whether cells in the body are sufficiently different to trigger symphiliosis if they are fused. Several facts support this possibility. First, as it is becoming apparent, the genomes of normal somatic cells in the same individual are not identical,73 although whether these differences are sufficient to cause symphiliosis is unknown. That they might be is suggested by the fact that a difference as small as that between variants of the same species is sufficient to trigger the effects of allopolyploidy in plants.72 Second, the cells of a premalignant lesion are often aneuploid, although not as extensively as cancer cells, and heterogeneous in their karyotypes,74 indicating gross genomic variation among neoplastic and between neoplastic and normal cells. Third, proliferating cells of identical ploidy can differ grossly in their genomes if they are at different phases of cell cycle, a difference that can cause premature chromosome condensation if such cells fuse.75 Finally, fusion of different cell types results in genomic aberrations and in emergent gene expression patterns, manifestations of symphiliosis, even if the fusing cells are euploid.44,51
The idea that cancer cells are new species that arise by becoming aneuploid,6,64,76 the notion that new species of plants can arise through karyotypic changes and the fact that allopolyploid plants can be created by fusing somatic cells77 together make the relationship between allopolyploids and neoplastic cell hybrids even more direct. An open question is whether symphiliosis can be triggered by horizontal transfer of information other that cell fusion, such as transfer of chromosomes and genomic fragments by apoptotic bodies and other vesicles78,79 or even by events that do not involve cell-cell interaction.
A Model: The Properties of Neoplastic Cells, as Properties of Plants, Depend On How These Cells Become Polyploid
Since symphiliosis results from fusing different cells rather than from an increase in ploidy per se, it is logical to expect that the contribution of cell fusion to neoplastic progression would be different from that of other pathways to polyploidy (Fig. 3). To borrow the terminology from plant biology, cytokinesis failure, endoreduplication, and fusion of identical cells would produce autotetraploid cells that contribute to carcinogenesis and tumor progression through the effects of increased ploidy and cell size, such as changes in gene dosage, and through chromosomal aberrations arising in mitosis. These changes would result in gradual evolution, with occasional radical phenotypic changes caused by a chromosome translocation or another aberration that affects a particularly critical gene. In contrast, fusion of different cells would result in allopolyploid clones, which, in addition to be affected by the consequences of tetraploidy, would enable saltational evolution by acquiring emergent properties through symphiliosis. Some of these properties, such as increased drug resistance, the ability to become dormant, or be invasive may be clinically relevant.65-67 Of particular interest are the possibilities that fusion of neoplastic cells to normal stem cells.66,80,81 or extinction resulting from fusion of different cell types is a source of cancer stem cells. If plant evolution is any guide, even if cell fusion is by far more rare in the body than other pathways to polyploidy, and the majority of the resulting allopolyploids vanish, having emergent properties can make the survivors more consequential for the emergence and progression of the disease.
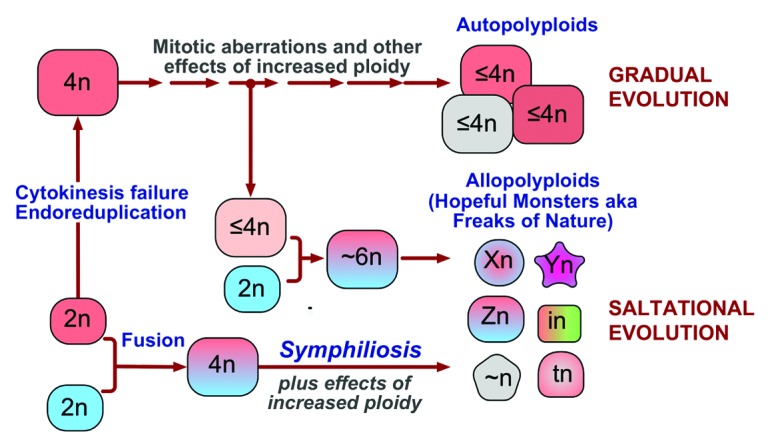
Figure 3. A model: The pathways to polyploidy have different roles in the evolution of neoplastic cells. To use plant terminology, cytokinesis failure and endoreduplication produce autopolyploid cells that are affected by mitotic aberrations and other consequences of increased ploidy, resulting in gradual evolution of the cells. Fusion of different cells results in allopolyploids, thus suddenly, within one generation, producing clones with emergent phenotypes and enabling saltational evolution. Cells of any ploidy can fuse, thus increasing the diversity of allopolyploids.
What Could This Model Help To Explain?
First, the increasingly intimate look into cancer genomes and the ability to trace clonal lineages in tumors82-84 has led to two interrelated observations: (1) that genomic aberrations in cancers include massive, characteristic, and previously unrecognized changes such as chromothripsis,85,86 chromoplexy,87 chromoanagenis,88 kataegis (thunderstorm),89 and firestorms;90 and (2) that cancer development can proceed not gradually, but through a “punctuated evolution”,87 which creates clones with new properties through single “catastrophic events”,85,88,91 meaning catastrophic to the patient, not to the cell.
What are these catastrophic events? Our discussion suggests cell fusion as a candidate, a possibility that has been considered only in passing and only in relation to premature chromosome condensation, a result of the fusion between cells that are at different phases of cell cycle.88,91 Fusion to lymphocytes could also explain the “footprints” of genome-modifying enzymes, such as AID,89 whose expression is normally restricted to these cells. The consequences of allo- and autopolyploidy in plants have been dissected, in part, by analyzing plants created by fusing somatic cells, protoplasts, in vitro.77 Similarly, identifying genomic and epigenetic “footprints” of endoreduplication, cytokinesis failure and cell fusion and comparing them to the aberrations observed in cancer may help to determine the relative contribution of these 3 pathways for cancer development and whether any of them can be responsible for gradual or saltational evolution in neoplasia.
Second, comparing cancer development to plant evolution may help to resolve the ongoing debate on whether tetraploidy and the consequent aneuploidy promotes cancer development or impedes it.27,92 The answer may depend on how tetraploidy is induced. An early view that polyploidy has contributed little to plant evolution has been retired by discovering how prevalent allopolyploids are.17,31,48 To quote, “like interspecific hybrids, newly formed allopolyploids display a range of novel phenotypes that are both favorable and unfavorable, but which are overall of questionable fitness. Although it might seem unlikely that these “freaks of nature” could contribute to the evolutionary race…new allopolyploid species were fit enough to beget the present multitude of seed-plant species”48. These “freaks of nature” or “hopeful monsters”,93,94 with either term applicable to cancer cells, prevail despite the multiple disadvantages of allopolyploids that prevent their propagation.17,29 Similarly, senescence95 and perhaps other tumor suppression mechanisms96 can prevent the proliferation of somatic hybrids. However, as in plants,17 the need to overcome roadblocks may be well compensated by the evolutionary benefits of creating novel phenotypes through symphiliosis.
Third, the model implies that massive epigenetic and gene expression changes can be associated with a ploidy increase without being caused by it, as it is the case in allopolyploids. Hence, general conclusions about the effects of tetraploidy in cancer could be premature if based on studies that use only one way to increase ploidy. For example, the effects of cytokinesis failure, which is a convenient and commonly used way to induce tetraploidy in vitro and in vivo, would not include such defining consequences of allopolyploidy as emergent gene expression patterns. Considering the possibility that not all tetraploid cells are created equal may help to reconcile experimental findings with the observations made in neoplastic lesions and to improve cancer diagnostics that relies on ploidy assessment.13 This would require developing approaches that can distinguish autopolyploid and allopolyploid cells in human tissues.
Fourth, some cancers of the same organ, with prostate cancer being an example,97 can remain tetraploid and stable over time, while others become aneuploid and progress quickly. The model we have discussed suggests that this difference could be explained by different causes of tetraploidy. Again, using plant evolution as a guide, autotetraploid cancers would be less aggressive than allotetraploid, in which symphilial chromosomal and other instabilities would facilitate the emergence of clones with new, potentially harmful properties. Whether a particular cancer is auto- or allotetraploid could depend on the presence of agents capable of fusing cells in premalignant and cancerous lesions, such as endogenous or infectious viruses, endogenous fusogenic proteins,63,98 or others.
Finally, the current view of tetraploidy in neoplasia implies that reverting a cell to diploidy through a reductive division99,100 would return its genome to a stable state, as the number of chromosomes and centrosomes returns to normal. Since symphilial instabilities are induced by merging different cells, rather than by a change in chromosome number, they are expected to persist even after the reversal of tetraploidy.52 This possibility implies that some diploid or pseudodiploid cancers can have allopolyploid origin.
To summarize, if the mechanisms that drive the evolution of polyploid plants also underlie the evolution of cancerous cells, as this commentary proposes, then learning what these mechanisms are would help us to prevent cancer, provide new tools for creating new plants, and teach us once again that studying plants can reveal the most interesting secrets of nature.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The author thanks Francine Carland, Leonid Chernomordik, Brian Coon, Grigori Enikolopov, Egor Lazebnik, Moshe Feldman, George Parris, Thomas Ried, and Howard Sharpe for comments, discussions, and encouragements, and Irinna Papangeli for suggesting the word symphiliosis.
References
- 1.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–8. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 2.Moreno E. Cancer: Darwinian tumour suppression. Nature. 2014;509:435–6. doi: 10.1038/nature13337. [DOI] [PubMed] [Google Scholar]
- 3.Vincent MD. Cancer: beyond speciation. Adv Cancer Res. 2011;112:283–350. doi: 10.1016/B978-0-12-387688-1.00010-7. [DOI] [PubMed] [Google Scholar]
- 4.Bignold LP. Variation, “evolution”, immortality and genetic instabilities in tumour cells. Cancer Lett. 2007;253:155–69. doi: 10.1016/j.canlet.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 5.Sprouffske K, Merlo LM, Gerrish PJ, Maley CC, Sniegowski PD. Cancer in light of experimental evolution. Curr Biol. 2012;22:R762–71. doi: 10.1016/j.cub.2012.06.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duesberg P, Rasnick D. Aneuploidy, the somatic mutation that makes cancer a species of its own. Cell Motil Cytoskeleton. 2000;47:81–107. doi: 10.1002/1097-0169(200010)47:2<81::AID-CM1>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 7.Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer. 2012;12:487–93. doi: 10.1038/nrc3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heng HH, Bremer SW, Stevens JB, Horne SD, Liu G, Abdallah BY, Ye KJ, Ye CJ. Chromosomal instability (CIN): what it is and why it is crucial to cancer evolution. Cancer Metastasis Rev. 2013;32:325–40. doi: 10.1007/s10555-013-9427-7. [DOI] [PubMed] [Google Scholar]
- 9.Boveri T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J Cell Sci. 2008;121(Suppl 1):1–84. doi: 10.1242/jcs.025742. [DOI] [PubMed] [Google Scholar]
- 10.Shackney SE, Smith CA, Miller BW, Burholt DR, Murtha K, Giles HR, Ketterer DM, Pollice AA. Model for the genetic evolution of human solid tumors. Cancer Res. 1989;49:3344–54. [PubMed] [Google Scholar]
- 11.Ricke RM, van Deursen JM. Aneuploidy in health, disease, and aging. J Cell Biol. 2013;201:11–21. doi: 10.1083/jcb.201301061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005;437:1043–7. doi: 10.1038/nature04217. [DOI] [PubMed] [Google Scholar]
- 13.Merlo LM, Wang LS, Pepper JW, Rabinovitch PS, Maley CC. Polyploidy, aneuploidy and the evolution of cancer. Adv Exp Med Biol. 2010;676:1–13. doi: 10.1007/978-1-4419-6199-0_1. [DOI] [PubMed] [Google Scholar]
- 14.Dewhurst SM, McGranahan N, Burrell RA, Rowan AJ, Grönroos E, Endesfelder D, Joshi T, Mouradov D, Gibbs P, Ward RL, et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov. 2014;4:175–85. doi: 10.1158/2159-8290.CD-13-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ganem NJ, Storchova Z, Pellman D. Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev. 2007;17:157–62. doi: 10.1016/j.gde.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 16.Padilla-Nash HM, Hathcock K, McNeil NE, Mack D, Hoeppner D, Ravin R, Knutsen T, Yonescu R, Wangsa D, Dorritie K, et al. Spontaneous transformation of murine epithelial cells requires the early acquisition of specific chromosomal aneuploidies and genomic imbalances. Genes Chromosomes Cancer. 2012;51:353–74. doi: 10.1002/gcc.21921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Comai L. The advantages and disadvantages of being polyploid. Nat Rev Genet. 2005;6:836–46. doi: 10.1038/nrg1711. [DOI] [PubMed] [Google Scholar]
- 18.Feldman M, Levy AA. Genome evolution due to allopolyploidization in wheat. Genetics. 2012;192:763–74. doi: 10.1534/genetics.112.146316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hayashi MT, Karlseder J. DNA damage associated with mitosis and cytokinesis failure. Oncogene. 2013;32:4593–601. doi: 10.1038/onc.2012.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krajcovic M, Overholtzer M. Mechanisms of ploidy increase in human cancers: a new role for cell cannibalism. Cancer Res. 2012;72:1596–601. doi: 10.1158/0008-5472.CAN-11-3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weaver BA, Cleveland DW. Decoding the links between mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell death. Cancer Cell. 2005;8:7–12. doi: 10.1016/j.ccr.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 22.Weaver BA, Cleveland DW. Does aneuploidy cause cancer? Curr Opin Cell Biol. 2006;18:658–67. doi: 10.1016/j.ceb.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 23.Ganem NJ, Pellman D. Linking abnormal mitosis to the acquisition of DNA damage. J Cell Biol. 2012;199:871–81. doi: 10.1083/jcb.201210040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.King RW. When 2+2=5: the origins and fates of aneuploid and tetraploid cells. Biochim Biophys Acta. 2008;1786:4–14. doi: 10.1016/j.bbcan.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sagona AP, Stenmark H. Cytokinesis and cancer. FEBS Lett. 2010;584:2652–61. doi: 10.1016/j.febslet.2010.03.044. [DOI] [PubMed] [Google Scholar]
- 26.Coward J, Harding A. Size Does Matter: Why Polyploid Tumor Cells are Critical Drug Targets in the War on Cancer. Front Oncol. 2014;4:123. doi: 10.3389/fonc.2014.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ganem NJ, Pellman D. Limiting the proliferation of polyploid cells. Cell. 2007;131:437–40. doi: 10.1016/j.cell.2007.10.024. [DOI] [PubMed] [Google Scholar]
- 28.Fox DT, Duronio RJ. Endoreplication and polyploidy: insights into development and disease. Development. 2013;140:3–12. doi: 10.1242/dev.080531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen ZJ. Molecular mechanisms of polyploidy and hybrid vigor. Trends Plant Sci. 2010;15:57–71. doi: 10.1016/j.tplants.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Otto SP. The evolutionary consequences of polyploidy. Cell. 2007;131:452–62. doi: 10.1016/j.cell.2007.10.022. [DOI] [PubMed] [Google Scholar]
- 31.Otto SP, Whitton J. Polyploid incidence and evolution. Annu Rev Genet. 2000;34:401–37. doi: 10.1146/annurev.genet.34.1.401. [DOI] [PubMed] [Google Scholar]
- 32.Brochmann C, Brysting AK, Alsos IG, Borgen L, Grundt HH, Scheen AC, Elven R. Polyploidy in arctic plants. Biol J Linn Soc Lond. 2004;82:521–36. doi: 10.1111/j.1095-8312.2004.00337.x. [DOI] [Google Scholar]
- 33.Leitch AR, Leitch IJ. Genomic plasticity and the diversity of polyploid plants. Science. 2008;320:481–3. doi: 10.1126/science.1153585. [DOI] [PubMed] [Google Scholar]
- 34.Tayalé A, Parisod C. Natural pathways to polyploidy in plants and consequences for genome reorganization. Cytogenet Genome Res. 2013;140:79–96. doi: 10.1159/000351318. [DOI] [PubMed] [Google Scholar]
- 35.Ma XF, Gustafson JP. Genome evolution of allopolyploids: a process of cytological and genetic diploidization. Cytogenet Genome Res. 2005;109:236–49. doi: 10.1159/000082406. [DOI] [PubMed] [Google Scholar]
- 36.McClintock B. The significance of responses of the genome to challenge. Science. 1984;226:792–801. doi: 10.1126/science.15739260. [DOI] [PubMed] [Google Scholar]
- 37.Xiong Z, Gaeta RT, Pires JC. Homoeologous shuffling and chromosome compensation maintain genome balance in resynthesized allopolyploid Brassica napus. Proc Natl Acad Sci U S A. 2011;108:7908–13. doi: 10.1073/pnas.1014138108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Comai L, Tyagi AP, Winter K, Holmes-Davis R, Reynolds SH, Stevens Y, Byers B. Phenotypic instability and rapid gene silencing in newly formed arabidopsis allotetraploids. Plant Cell. 2000;12:1551–68. doi: 10.1105/tpc.12.9.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feldman M, Levy AA, Fahima T, Korol A. Genomic asymmetry in allopolyploid plants: wheat as a model. J Exp Bot. 2012;63:5045–59. doi: 10.1093/jxb/ers192. [DOI] [PubMed] [Google Scholar]
- 40.Hegarty M, Coate J, Sherman-Broyles S, Abbott R, Hiscock S, Doyle J. Lessons from natural and artificial polyploids in higher plants. Cytogenet Genome Res. 2013;140:204–25. doi: 10.1159/000353361. [DOI] [PubMed] [Google Scholar]
- 41.Jackson S, Chen ZJ. Genomic and expression plasticity of polyploidy. Curr Opin Plant Biol. 2010;13:153–9. doi: 10.1016/j.pbi.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Osborn TC, Pires JC, Birchler JA, Auger DL, Chen ZJ, Lee HS, Comai L, Madlung A, Doerge RW, Colot V, et al. Understanding mechanisms of novel gene expression in polyploids. Trends Genet. 2003;19:141–7. doi: 10.1016/S0168-9525(03)00015-5. [DOI] [PubMed] [Google Scholar]
- 43.Jones RN, Hegarty M. Order out of chaos in the hybrid plant nucleus. Cytogenet Genome Res. 2009;126:376–89. doi: 10.1159/000266171. [DOI] [PubMed] [Google Scholar]
- 44.Koulakov AA, Lazebnik Y. The problem of colliding networks and its relation to cell fusion and cancer. Biophys J. 2012;103:2011–20. doi: 10.1016/j.bpj.2012.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Smet R, Van de Peer Y. Redundancy and rewiring of genetic networks following genome-wide duplication events. Curr Opin Plant Biol. 2012;15:168–76. doi: 10.1016/j.pbi.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 46.Flagel LE, Wendel JF, Udall JA. Duplicate gene evolution, homoeologous recombination, and transcriptome characterization in allopolyploid cotton. BMC Genomics. 2012;13:302. doi: 10.1186/1471-2164-13-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flagel L, Udall J, Nettleton D, Wendel J. Duplicate gene expression in allopolyploid Gossypium reveals two temporally distinct phases of expression evolution. BMC Biol. 2008;6:16. doi: 10.1186/1741-7007-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pignatta D, Comai L. Parental squabbles and genome expression: lessons from the polyploids. J Biol. 2009;8:43. doi: 10.1186/jbiol140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Macarthur BD, Ma’ayan A, Lemischka IR. Systems biology of stem cell fate and cellular reprogramming. Nat Rev Mol Cell Biol. 2009;10:672–81. doi: 10.1038/nrm2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Waddington CH. The strategy of the genes: a discussion of some aspects of theoretical biology. London: Allen & Unwin, 1957. [Google Scholar]
- 51.Ringertz NR, Savage RE. Cell hybrids. New York: Academic Press, 1976. [Google Scholar]
- 52.Rappa G, Mercapide J, Lorico A. Spontaneous formation of tumorigenic hybrids between breast cancer and multipotent stromal cells is a source of tumor heterogeneity. Am J Pathol. 2012;180:2504–15. doi: 10.1016/j.ajpath.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Quintana-Bustamante O, Grueso E, Garcia-Escudero R, Arza E, Alvarez-Barrientos A, Fabregat I, Garcia-Bravo M, Meza NW, Segovia JC. Cell fusion reprogramming leads to a specific hepatic expression pattern during mouse bone marrow derived hepatocyte formation in vivo. PLoS One. 2012;7:e33945. doi: 10.1371/journal.pone.0033945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Powell AE, Anderson EC, Davies PS, Silk AD, Pelz C, Impey S, Wong MH. Fusion between Intestinal epithelial cells and macrophages in a cancer context results in nuclear reprogramming. Cancer Res. 2011;71:1497–505. doi: 10.1158/0008-5472.CAN-10-3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Palermo A, Doyonnas R, Bhutani N, Pomerantz J, Alkan O, Blau HM. Nuclear reprogramming in heterokaryons is rapid, extensive, and bidirectional. FASEB J. 2009;23:1431–40. doi: 10.1096/fj.08-122903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Massa S, Junker S, Matthias P. Molecular mechanisms of extinction: old findings and new ideas. Int J Biochem Cell Biol. 2000;32:23–40. doi: 10.1016/S1357-2725(99)00102-8. [DOI] [PubMed] [Google Scholar]
- 57.Mallet J. Hybrid speciation. Nature. 2007;446:279–83. doi: 10.1038/nature05706. [DOI] [PubMed] [Google Scholar]
- 58.Curril IM, Koide M, Yang CH, Segal A, Wellman GC, Spees JL. Incomplete reprogramming after fusion of human multipotent stromal cells and bronchial epithelial cells. FASEB J. 2010;24:4856–64. doi: 10.1096/fj.09-152991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cowan CA, Atienza J, Melton DA, Eggan K. Nuclear reprogramming of somatic cells after fusion with human embryonic stem cells. Science. 2005;309:1369–73. doi: 10.1126/science.1116447. [DOI] [PubMed] [Google Scholar]
- 60.Yamanaka S, Blau HM. Nuclear reprogramming to a pluripotent state by three approaches. Nature. 2010;465:704–12. doi: 10.1038/nature09229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Celton-Morizur S, Merlen G, Couton D, Desdouets C. Polyploidy and liver proliferation: central role of insulin signaling. Cell Cycle. 2010;9:460–6. doi: 10.4161/cc.9.3.10542. [DOI] [PubMed] [Google Scholar]
- 62.Ogle BM, Cascalho M, Platt JL. Biological implications of cell fusion. Nat Rev Mol Cell Biol. 2005;6:567–75. doi: 10.1038/nrm1678. [DOI] [PubMed] [Google Scholar]
- 63.Duelli D, Lazebnik Y. Cell-to-cell fusion as a link between viruses and cancer. Nat Rev Cancer. 2007;7:968–76. doi: 10.1038/nrc2272. [DOI] [PubMed] [Google Scholar]
- 64.Huxley J. Cancer Biology - Comparative and Genetic. Biol Rev Camb Philos Soc. 1956;31:474–514. doi: 10.1111/j.1469-185X.1956.tb01558.x. [DOI] [Google Scholar]
- 65.Parris GE. Historical perspective of cell-cell fusion in cancer initiation and progression. Crit Rev Oncog. 2013;18:1–18. doi: 10.1615/CritRevOncog.v18.i1-2.20. [DOI] [PubMed] [Google Scholar]
- 66.Duelli D, Lazebnik Y. Cell fusion: a hidden enemy? Cancer Cell. 2003;3:445–8. doi: 10.1016/S1535-6108(03)00114-4. [DOI] [PubMed] [Google Scholar]
- 67.Pawelek JM, Chakraborty AK. Fusion of tumour cells with bone marrow-derived cells: a unifying explanation for metastasis. Nat Rev Cancer. 2008;8:377–86. doi: 10.1038/nrc2371. [DOI] [PubMed] [Google Scholar]
- 68.Clawson GA. Cancer. Fusion for moving. Science. 2013;342:699–700. doi: 10.1126/science.1244270. [DOI] [PubMed] [Google Scholar]
- 69.Parris GE. The cell clone ecology hypothesis and the cell fusion model of cancer progression and metastasis: history and experimental support. Med Hypotheses. 2006;66:76–83. doi: 10.1016/j.mehy.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 70.Parris GE. Clinically significant cancer evolves from transient mutated and/or aneuploid neoplasia by cell fusion to form unstable syncytia that give rise to ecologically viable parasite species. Med Hypotheses. 2005;65:846–50. doi: 10.1016/j.mehy.2005.05.036. [DOI] [PubMed] [Google Scholar]
- 71.Bulla GA, Luong Q, Shrestha S, Reeb S, Hickman S. Genome-wide analysis of hepatic gene silencing in mammalian cell hybrids. Genomics. 2010;96:323–32. doi: 10.1016/j.ygeno.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 72.Groszmann M, Greaves IK, Albertyn ZI, Scofield GN, Peacock WJ, Dennis ES. Changes in 24-nt siRNA levels in Arabidopsis hybrids suggest an epigenetic contribution to hybrid vigor. Proc Natl Acad Sci U S A. 2011;108:2617–22. doi: 10.1073/pnas.1019217108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McConnell MJ, Lindberg MR, Brennand KJ, Piper JC, Voet T, Cowing-Zitron C, Shumilina S, Lasken RS, Vermeesch JR, Hall IM, et al. Mosaic copy number variation in human neurons. Science. 2013;342:632–7. doi: 10.1126/science.1243472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ried T, Hu Y, Difilippantonio MJ, Ghadimi BM, Grade M, Camps J. The consequences of chromosomal aneuploidy on the transcriptome of cancer cells. Biochim Biophys Acta. 2012;1819:784–93. doi: 10.1016/j.bbagrm.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Johnson RT, Rao PN. Mammalian cell fusion: induction of premature chromosome condensation in interphase nuclei. Nature. 1970;226:717–22. doi: 10.1038/226717a0. [DOI] [PubMed] [Google Scholar]
- 76.Vincent MD. The animal within: carcinogenesis and the clonal evolution of cancer cells are speciation events sensu stricto. Evolution. 2010;64:1173–83. doi: 10.1111/j.1558-5646.2009.00942.x. [DOI] [PubMed] [Google Scholar]
- 77.Bassene JB, Froelicher Y, Dubois C, Ferrer RM, Navarro L, Ollitrault P, Ancillo G. Non-additive gene regulation in a citrus allotetraploid somatic hybrid between C. reticulata Blanco and C. limon (L.) Burm. Heredity (Edinb) 2010;105:299–308. doi: 10.1038/hdy.2009.162. [DOI] [PubMed] [Google Scholar]
- 78.García-Olmo DC, Picazo MG, García-Olmo D. Transformation of non-tumor host cells during tumor progression: theories and evidence. Expert Opin Biol Ther. 2012;12(Suppl 1):S199–207. doi: 10.1517/14712598.2012.681370. [DOI] [PubMed] [Google Scholar]
- 79.Holmgren L. Horizontal gene transfer: you are what you eat. Biochem Biophys Res Commun. 2010;396:147–51. doi: 10.1016/j.bbrc.2010.04.026. [DOI] [PubMed] [Google Scholar]
- 80.Bjerkvig R, Tysnes BB, Aboody KS, Najbauer J, Terzis AJ. Opinion: the origin of the cancer stem cell: current controversies and new insights. Nat Rev Cancer. 2005;5:899–904. doi: 10.1038/nrc1740. [DOI] [PubMed] [Google Scholar]
- 81.He X, Tsang TC, Pipes BL, Ablin RJ, Harris DT. A stem cell fusion model of carcinogenesis. J Exp Ther Oncol. 2005;5:101–9. [PubMed] [Google Scholar]
- 82.Navin NE. Tumor evolution in response to chemotherapy: phenotype versus genotype. Cell Rep. 2014;6:417–9. doi: 10.1016/j.celrep.2014.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Almendro V, Cheng YK, Randles A, Itzkovitz S, Marusyk A, Ametller E, Gonzalez-Farre X, Muñoz M, Russnes HG, Helland A, et al. Inference of tumor evolution during chemotherapy by computational modeling and in situ analysis of genetic and phenotypic cellular diversity. Cell Rep. 2014;6:514–27. doi: 10.1016/j.celrep.2013.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–4. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Forment JV, Kaidi A, Jackson SP. Chromothripsis and cancer: causes and consequences of chromosome shattering. Nat Rev Cancer. 2012;12:663–70. doi: 10.1038/nrc3352. [DOI] [PubMed] [Google Scholar]
- 87.Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, Ghandi M, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013;153:666–77. doi: 10.1016/j.cell.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Holland AJ, Cleveland DW. Chromoanagenesis and cancer: mechanisms and consequences of localized, complex chromosomal rearrangements. Nat Med. 2012;18:1630–8. doi: 10.1038/nm.2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, et al. Breast Cancer Working Group of the International Cancer Genome Consortium Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–93. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hicks J, Krasnitz A, Lakshmi B, Navin NE, Riggs M, Leibu E, Esposito D, Alexander J, Troge J, Grubor V, et al. Novel patterns of genome rearrangement and their association with survival in breast cancer. Genome Res. 2006;16:1465–79. doi: 10.1101/gr.5460106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rajapakse I, Scalzo D, Groudine M. Losing control: cancer’s catastrophic transition. Nucleus. 2011;2:249–52. doi: 10.4161/nucl.2.4.16506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tang YC, Amon A. Gene copy-number alterations: a cost-benefit analysis. Cell. 2013;152:394–405. doi: 10.1016/j.cell.2012.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Theissen G. Saltational evolution: hopeful monsters are here to stay. Theory in biosciences = Theorie in den Biowissenschaften. 2009;128:43–51. doi: 10.1007/s12064-009-0058-z. [DOI] [PubMed] [Google Scholar]
- 94.Parris GE. The Hopeful Monster Finds a Mate and Founds a New Species. 2011 [Google Scholar]
- 95.Chuprin A, Gal H, Biron-Shental T, Biran A, Amiel A, Rozenblatt S, Krizhanovsky V. Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev. 2013;27:2356–66. doi: 10.1101/gad.227512.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Duelli DM, Hearn S, Myers MP, Lazebnik Y. A primate virus generates transformed human cells by fusion. J Cell Biol. 2005;171:493–503. doi: 10.1083/jcb.200507069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Forsslund G, Esposti PL, Nilsson B, Zetterberg A. The prognostic significance of nuclear DNA content in prostatic carcinoma. Cancer. 1992;69:1432–9. doi: 10.1002/1097-0142(19920315)69:6<1432::AID-CNCR2820690621>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 98.Avinoam O, Podbilewicz B. Eukaryotic cell-cell fusion families. Current topics in membranes 2011; 68:209-34. [DOI] [PubMed] [Google Scholar]
- 99.Duncan AW, Taylor MH, Hickey RD, Hanlon Newell AE, Lenzi ML, Olson SB, Finegold MJ, Grompe M. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature. 2010;467:707–10. doi: 10.1038/nature09414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Teplitz RL, Gustafson PE, Pellett OL. Chromosomal distribution in interspecific invitro hybrid cells. Exp Cell Res. 1968;52:379–91. doi: 10.1016/0014-4827(68)90479-5. [DOI] [PubMed] [Google Scholar]