

Abstract
Chemokine CXCL12 promotes CXCR4-dependent chemotaxis of cancer cells to characteristic organs and tissues, leading to metastatic disease. This study was designed to investigate how cells expressing CXCR7 regulate chemotaxis of a separate population of CXCR4 cells under physiologic conditions in which cells are exposed to gradients of CXCL12. We recapitulated a cancer-stroma microenvironment by patterning CXCR4-expressing cancer cells in microchannels at spatially defined positions relative to CXCL12-producing cells and CXCR7-expressing cells. CXCR7 scavenges and degrades CXCL12, which has been proposed to facilitate CXCR4-dependent chemotaxis through a source-sink model. Using the microchannel device, we demonstrated that chemotaxis of CXCR4 cells depended critically on the presence and location of CXCR7 cells (sink) relative to chemokine secreting cells (source). Furthermore, inhibiting CXCR4 on migrating cells or CXCR7 on sink cells blocked CXCR4-dependent chemotaxis toward CXCL12, showing that the device can identify new therapeutic agents that block migration by targeting chemoattractant scavenging receptors. Our system enables efficient chemotaxis under much shallower yet more physiological chemoattractant gradients by generating an in vitro microenvironment where combinations of cellular products may be secreted along with formation of a chemoattractant gradient. In addition to elucidating mechanisms of CXCL-12 mediated chemotaxis, this simple and robust method can be broadly useful for engineering multiple microenvironments to investigate intercellular communication.
Introduction
Chemotaxis, which is the directional migration of cells guided by gradients of signaling molecules, is essential for processes including embryogenesis, immune cell trafficking, atherosclerosis, and cancer metastasis.1–4 While gradients of chemokines or other signaling molecules are required for chemotaxis, the physiological mechanisms for generating these gradients and the steepness of a gradient required for chemotaxis remain poorly defined. To effectively target chemotaxis to prevent cancer progression as well as treat other diseases, there is a need for new, experimentally tractable technologies to investigate chemotactic gradients and cell migration under more physiological microenvironments.
Microfluidic systems have been used previously to stably define complex gradients of chemoattractants and clarify relationships between gradient shapes and chemotaxis.5–10 However, these systems do not recapitulate critical physiologic interactions that surrounding stromal cells have on generating and reshaping the gradient profile to which migrating cells respond.11,12 These systems require relatively steep gradients of chemoattract molecules for efficient migration13,14 at least in part because of a lack of autocrine/paracrine factors that play an important role in cell migration.8,10 These autocrine/paracrine factors are washed out in systems that require flow to establish and maintain gradients. Other systems have used source cells to generate gradients, but those devices do not facilitate precise arrangements of multiple cell types required for the systematic analysis of more complex cell-gradient interactions.15,16 Gradients in the in vivo microenvironment are affected by multiple cell types that can produce, scavenge, and/or respond to a specific chemokine. This complex interplay has a direct effect on how cells in the local area function. Therefore, models that better recapitulate intercellular interactions would serve as a more accurate chemotaxis assay and also a predictor of effects drugs have on migrating cells. To overcome limitations of existing systems, we developed a new microchannel device to recapitulate cellular interactions in microenvironments such as those present in primary breast cancer. This microfluidic platform provides two key advantages: (i) hydrodynamic positioning of multiple cell types at precise positions with microchannels;17 and (ii) establishing microenvironments with small effective culture volumes (ECV)18 that enable source and sink cells to rapidly condition culture media to establish and maintain concentration gradients. We hypothesized that recreation of a more physiologic in vitro microenvironment, where combinations of cellular products may be secreted along with the chemo-attractant, would induce efficient chemotaxis with much shallower, yet more physiologic, chemoattractant gradients.
To establish the capabilities of this microfluidic device for studies of chemotaxis, we focused on functions of chemokine CXCL12,2 a molecule proposed to promote metastasis in breast cancer and more than 20 other human malignancies. Studies show that fibroblasts in primary human breast tumor secrete CXCL12,19 which bind to receptor CXCR4 on breast cancer cells to promote chemotaxis. CXCR7,20 a recently identified second receptor for CXCL12, also is expressed by stromal cells in the tumor microenvironment and subsets of breast cancer cells,21 but functions of CXCR7 in CXCL12-dependent chemotaxis remain poorly defined.22,23 A recent study in zebrafish suggests that gradients of CXCL12 form by a source-sink model with CXCR7 on somatic cells sequestering this chemokine, which was required for CXCR4-dependent migration of germ cells.24 However, the complex environment in vivo precludes facile, systematic evaluation of the importance of relative cell positions, combinations of receptors, or effects of potential inhibitors of chemotaxis. Using our microfluidic system, we show that chemotaxis of CXCR4-expressing cells toward CXCL12-producing cells (source) is critically-dependent on the presence and location of CXCR7-expressing cells (sink). Furthermore, we demonstrate that inhibition of CXCR7 reduces migration of CXCR4 cells despite an overall increase in the concentration of the chemoattractant CXCL12.
Results and discussion
The microfluidic device consists of two PDMS layers of microchannels separated by a semi-permeable membrane (Fig. 1A). The top layer has a dead-end channel to facilitate capturing of cells. The bottom layer has three channels whose locations dictate patterning of captured cells in the top layer. To pattern multiple cell types as shown in Fig. 1B, each cell type is introduced sequentially into each inlet of the top channel, and culture media is introduced into the other two inlets to focus cells on each bottom channel using multiple laminar flows. Cells are patterned over a specific bottom channel by keeping its outlet open and other outlets closed. CXCR4-expressing cells (X4) labeled with Hoechst (blue) were patterned between CXCL12-producing cells (L12) labeled with CellTracker red and CXCR7-expressing cells (X7) labeled with CellTracker green with 200 mm gaps between cells. Time lapse images show progressive migration of X4 cells toward L12 cells (Fig. 1C).
Fig. 1.
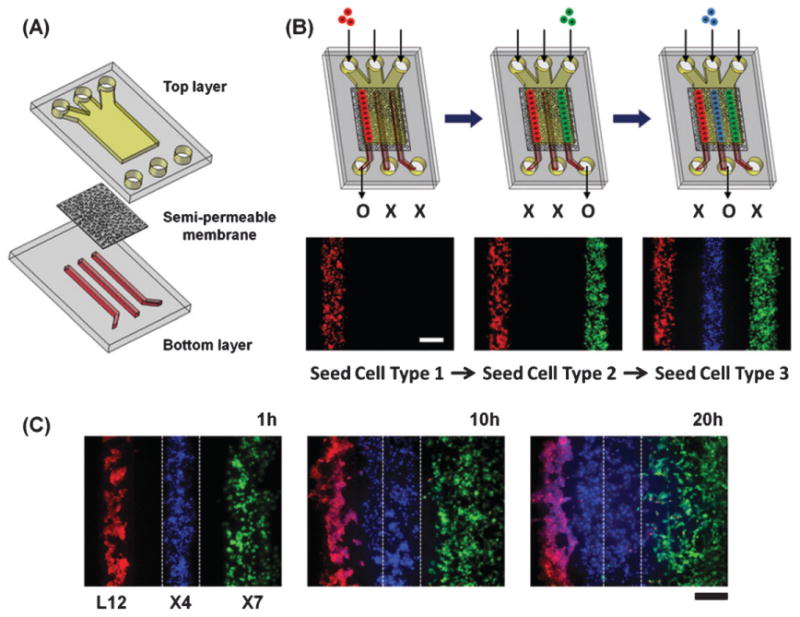
Microfluidic system for patterning source, sink, and migrating cells in defined positions. (A) Schematic illustration of the device. Two layers of PDMS channels are separated by a semi-permeable membrane. The top layer is a straight channel with a dead end where cells are patterned. (B) The cellular seeding process; three types of cells are sequentially introduced into each inlet of the top channel. Cells are patterned over select bottom channels by keeping the outlet for that respective channel open and the other two bottom channel outlets closed. (C) Time lapse images of X4 cell migration. X4 cells (blue) were patterned between L12 cells (red) and X7 cells (green) with 200 mm gaps and co-cultured in a device. Fluorescent images were taken 1 h, 10 h, and 20 h after seeding the cells. Scale bar: 200 μm.
The source-sink model suggests that X7 cells are needed to form chemotactic gradients of CXCL12 and that relative positions and spacing of L12, X4, and X7 cells are key determinants for CXCL12-dependent migration of X4 cells. Flexibility of cell seeding within the device allowed us to systematically test effects of X7 cells by patterning X4 cells: (i) with L12 cells but without X7 cells (Fig. 2A); (ii) with L12 cells and X7 cells but with the positions of X4 and X7 cells switched (Fig. 2B); (iii) between L12 cells and X7 cells but with wider gaps (400 mm) between cell types (Fig. 2C); or (iv) with L12 cells and GFP cells that lack CXCR7 (Fig. 2D). Fig. 2E shows quantified data for migration of X4 cells based on movement of the collective center of X4 cells from the center of the middle bottom channel. The X4 cells did not show directional migration without X7 cells (i), with X7 cells positioned between L12 and X4 cells (ii), or with GFP cells lacking CXCR7 (iv). Chemotaxis of X4 cells was decreased by 70% with wider gaps between cell populations (iii). As seen in Fig. 2, X4 cellular migration depends critically on the presence and location of X7 cells.
Fig. 2.
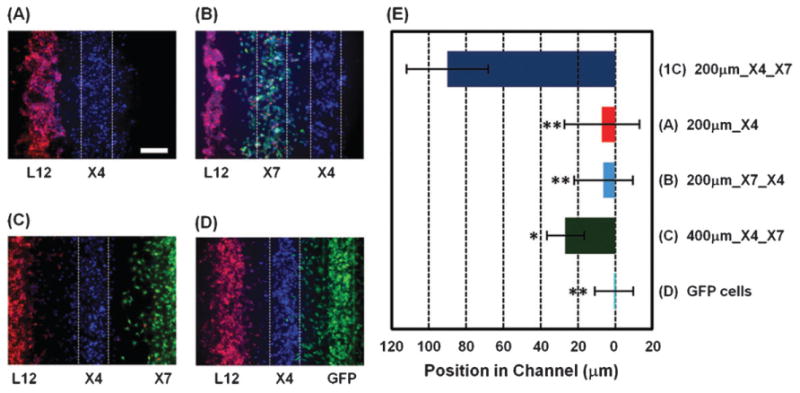
Migration of CXCR4 expressing cells is modulated by CXCR7 expressing cells. X4 cells were co-cultured with L12 cells but without X7 cells (A), with both L12 cells and X7 cells but with the positions of the X4 and X7 cells inverted (B), with wider gaps (400 mm) between the X4 cells and both the L12 cells and X7 cells (C), or with X7 cells replaced with GFP cells that do not express CXCR7 (D). Fluorescent images were taken 20 h after seeding cells. (E) Quantification of X4 cell migration under each condition (n = 9–16 independent experiments for each condition). (1C) shows the result of Fig. 1C at 20 h. The X-axis shows distance from the center of the middle bottom channel where X4 cells are initially positioned. Scale bars: 200 μm. * p < 0.001, ** p < 0.0001.
We further evaluated effects of cellular positioning on chemotaxis by either mixing X4 and X7 cells or patterning them juxtaposed (Fig. 3A and B). Fig. 3F shows quantified data for migration of X4 cells based on movement of the collective center of X4 cells. X4 cells showed directional migration towards L12 cells only when they were patterned side-by-side with X7 cells. We also evaluated migration of cells expressing both CXCR4 and CXCR7 (X4/X7) (Fig. 3C). X4/X7 cells did not show any directional migration, suggesting that each receptor has to be expressed on different cells for efficient chemotaxis at least in some types of cells. Furthermore, X4 cells did not show any directional migration when they were cultured without L12 cells or with GFP cells lacking CXCR7 (Fig. 3D and E). These results further indicate that the location of the sink cells is crucial for chemotaxis.
Fig. 3.
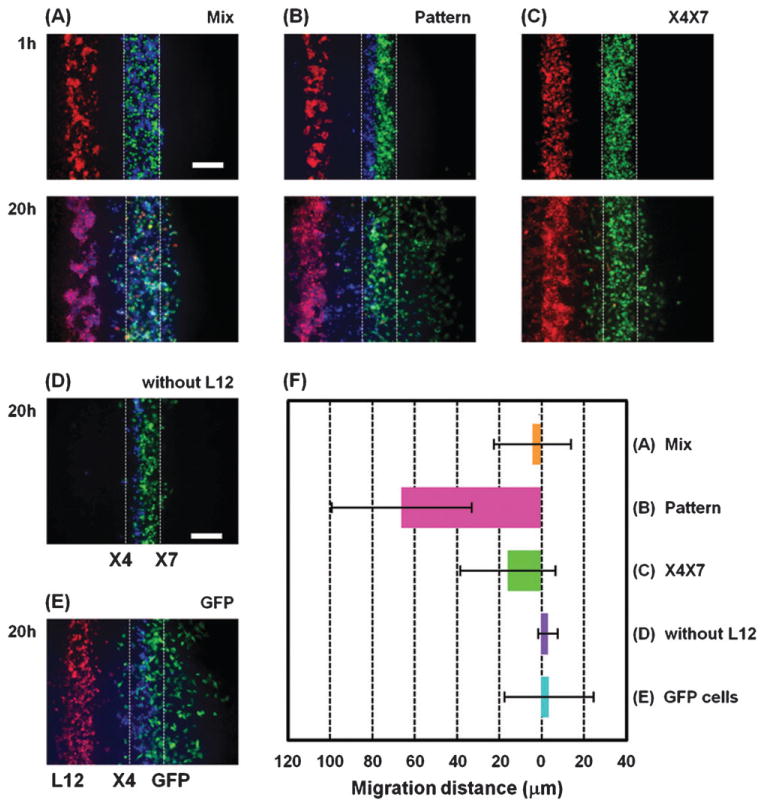
Effect of closely positioning CXCR4 and CXCR7 expressing cells together. (A, B) X4 cells (blue) and X7 cells (green) were co-patterned over the middle channel of the lower layer by either randomly mixing together (A) or by patterning side-by-side (B). In (C) MDA-MB-231 cells stably expressing both CXCR4 and CXCR7 (green) were patterned over the middle channel of the lower layer. For comparison, X4 cells (blue) were patterned side-by-side to X7 cells (green) without L12 cells (red) in (D) and X4 cells (blue) and GFP cells that do not express CXCR7 (green) were co-patterned over the middle channel of the lower layer in a side-by-side manner in (E). Fluorescent images were taken 1 h and 20 h after seeding the cells. (F) Quantification of X4 cell migration under each condition (n = 5–10 independent experiments for each condition). The X-axis shows distance from the center of the initial position of X4 cells. Scale bars: 200 μm.
To understand positional effects of source and sink cells on gradients of CXCL12 in the device, we developed a finite element model to simulate the produced gradient (Fig. 4A). It has been shown that cells migrate based on the magnitude of the concentration gradient across the cell. The specific gradient is the change in concentration over a cell distance divided by the average concentration over that distance. The specific gradient (Fig. 4B) show that a significant gradient is established only when X4 cells are patterned between the L12 and X7 cells. In addition, the gradient is slightly changed (∼ 1.21 ×) by changing spacing between source and sink cells from 200 μm to 400 μm, which significantly altered chemotactic response. This level of relatively small change in concentration gradient has been shown to induce significantly different chemotactic responses in the same cell type previously,8 although more complex effects may be at play in our system that includes multiple cell types. The model does suggests that X4 cells can migrate towards L12 cells at significantly smaller specific gradients (8.5 × 10–4/μm) of L12 compared to previous reports (1.5 × 10–2 /μm; ∼18× difference)8. This is likely due to presence of synergistic cell-produced factors preserved in the fluidically static microfabricated chemotaxis chamber that recreates a chemically complex but more physiological cancer microenvironment.8,10 It should be noted that our estimated specific gradient value is similar to predicted in vivo specific gradient value (∼7.5 × 10–4/μm) of IL-8 for neutrophil migration as determined by a mathematical model.13 Therefore, physiological steepness of chemotactic gradients is much shallower than that required for efficient migration in most in vitro systems.13,14,25,26, An additional interesting observation from our study is that co-expression of CXCR7 may interfere with CXCR4-dependent chemotaxis under some conditions since X4/X7 cells did not show any directional migration even though the slope of the gradient is predicted to be the steepest. Co-expression of CXCR4 and CXCR7 in the same cell has been reported to diminish ligand-dependent activation of G protein signaling pathways by CXCR4, which could account for reduced chemotaxis.27
Fig. 4.
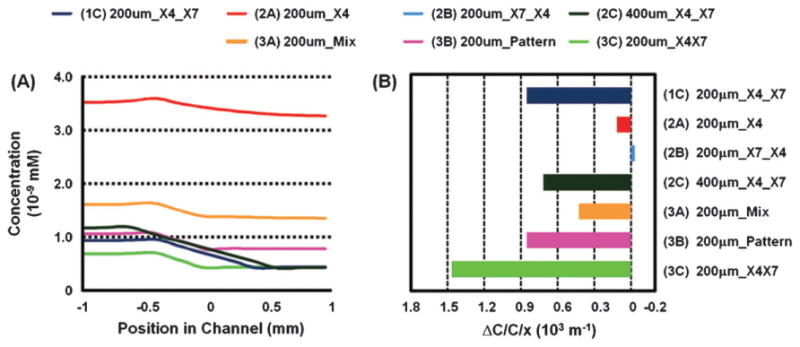
CXCL12 gradients generated by microfluidically patterned source and sink cells. (A) Simulated gradient profiles of CXCL12 in channels after 20 h in culture for the various cellular patterns generated. The X axis shows distance from the center of the middle bottom channel where X4 cells are typically seeded. (B) Estimated specific gradients over X4 cells for the different cellular patterns. The magnitude of the specific gradient matches well the observed trends in degree of migration except for condition 3C.
To determine to what extent CXCL12-dependent chemo-taxis could be interrupted by targeting either migrating or sink cells, we tested specific inhibitors of CXCR4 or CXCR7. As expected, X4 cell migration was reduced significantly by a CXCR4 inhibitor (1 mM AMD3100) (Fig. 5A). Importantly, directed cell migration of the X4 cells was also inhibited by a CXCR7 inhibitor (100 nM CCX733), a receptor present only on the sink cells (Fig. 5B) (p < 0.001). These results indicate that the CXCR4 mediated directed migration of X4 cells towards L12 cells requires CXCL12 scavenging by CXCR7-expressing cells under physiological ECVs. The results also demonstrate that the microfluidic system can be used to test and identify compounds that block cell migration either through direct effects on migrating cells or indirectly by targeting sink cells.
Fig. 5.
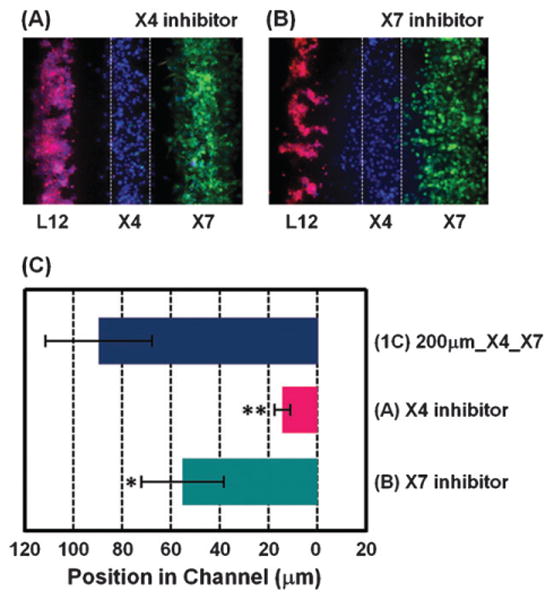
Evaluation of inhibitors on CXCL-12-dependent chemotaxis. CXCR4 inhibitor (1 mM AMD3100) (A) or CXCR7 inhibitor (100 nM CCX733) (B) was added in the device right after seeding the cells. Fluorescent images were taken 20 h after seeding cells. (C) Quantification of X4 cell migration (n = 9–16 independent experiments for each condition). (1C) shows the result of Fig. 1C at 20 h. X-axis shows distance from the center of the middle bottom channel. Scale bars: 200 mm. * p o 0.001, ** p o 0.0001.
Conclusion
Although a large number of chemotaxis devices have been reported,5,6 there are currently no methods to investigate interdependent effects of source and sink cells to establish effective chemotactic microenvironments. Recently, in vivo studies have proposed the essential role of sink cells for cell migration,24,28 yet there have been no appropriate in vitro models to rigorously test the source-sink model of chemotaxis. Studies of the source-sink model and other proposed mechanisms for gradient formation require microchannel devices rather than standard cell culture because chemokines from source cells can diffuse quickly, whereas cellular uptake and removal of chemokines are only felt locally.18 The role of sink cells to enhance chemotaxis is underappreciated and previously undocumented for cancer cell migration. Our microfluidic device shows that: (i) CXCR7 cells are necessary for migration of CXCR4-expressing cells toward CXCL12 and (ii) spatial positions of cells are also critical factors. Moreover, we determined that CXCL12-CXCR4 chemotaxis can be inhibited by targeting receptors not only on the migrating cells (CXCR4) but also on the sink cells (CXCR7), suggesting that receptors on sink cells may be novel therapeutic targets to prevent or treat cancer. Reduction in directed migration through inhibition of sink cells is somewhat counter intuitive since the chemoattractant concentration increases, but this is consistent with previous publications showing that chemotaxis is directed by the fractional concentration difference across the cell8,9 and not simply towards a higher concentration of a chemoattractant. Compared to other in vitro chemotaxis devices, cancer cells in our system exhibit efficient chemotaxis under much shallower yet more physiological chemoattractant gradients, most likely due to presence of cell secreted sensitization factors other than CXCL128,10 that are retained within the microdevice. In addition to cancer biology, the effects of sink cells on cell migration may be essential for a wide range of normal processes in development and immune cell trafficking. The methods and tools described should enable a broad range of biological studies.
Experimental
Cell culture
Breast cancer cells (MDA-MB-231 cell line; ATCC) and human embryonic kidney cells (HEK293 cell line; ATCC) were cultured in Dulbecco's Modified Eagle's Medium (DMEM; 11965; Invitrogen) containing 10% v/v fetal bovine serum (FBS; 10082; Gibco), 100 U/mL penicillin, and 100 U/mL streptomycin. MDA-MB-231 cells were stably transduced either with EGFP (GFP), CXCR4 (X4), CXCR7 (X7), CXCR4 and CXCR7 (X4/X7).29 HEK293T cells were stably transduced with CXCL12-cherry (L12).30 L12 cells, X7 cells, and X4 cells were stained with CellTracker red CMTPX (1.5 mM), CellTracker green CMFDA (10 mM), and Hoechst 33342 (1.6 mM) for 1 h before seeding the cells, respectively. To establish that X7 cells accumulate CXCL12-cherry, we collected CXCL12-cherry from supernatants of L12 cells.29 Supernatants were diluted in phenol red free DMEM (Invitrogen) at a 1 : 5 ratio and incubated with X7 cells for 30 min. Fluorescence images were obtained with a 40X objective (Olympus) (Supplementary Fig. 1). We also quantified CXCR7-dependent uptake of CXCL12 using a bioluminescent chemokine comprised of CXCR7-dependent uptake of CXCL12 using a bioluminescent chemokine comprised of CXCR7 fused to Gaussia luciferase as described previously29 (Supplementary Fig. 1).
Fabrication of microfluidic devices and cell seeding
The devices consist of two layers of microchannels separated by a semi-permeable membrane (Fig. 1A). The fabrication method of the devices was reported previously.31 The upper layer comprised of a straight channel having three inlet channels with a height of 200 μm is designed with a dead-end to facilitate cell capture. The lower layer consists of three channels with a width of 200 μm and a height of 100 μm. The semi-porous membrane is made of polycarbonate (TMP04700; Fisher), with 5 μm diameter pores and 10 μm thick. The microchannels were fabricated from poly(dimethylsiloxane) (PDMS) formed from prepolymer (Sylgard 184, Dow Corning) at a ratio of 1 : 10 base to curing agent using a soft lithographic method. The membrane was bound to the PDMS channels using a thin layer of liquid PDMS prepolymer as mortar. To allow introduction of solutions into the channels, the outlets of the bottom layer were connected with tubing. Prior to the experiment, 30 μg/mL of Collagen type IV (Sigma) was introduced into the channels after plasma oxidization and incubated for 2 h to promote cellular adhesion.
Cells were introduced into the upper channel using gravity-driven flow. As shown in Fig. 1B, different types of cell suspensions (typically, 104 cells) were sequentially introduced into each inlet of the upper channel while keeping the outlet at a lower height. After the membrane on the bottom channel was covered with cells, the outlet was closed and incubated for 2 h to let the cells attach on the membrane.
Quantification of cell migration
Fluorescent images were processed using a semi-automated program created in ImageJ (NIH). The program was designed to process each grayscale image automatically by subtracting the background to obtain an image of only X4 cells. Cellular migration was quantified by evaluating the location of center of mass of X4 cells.
Statictics
Mean values for cell migration were compared by Mann-Whitney test with Dunn's post-test analysis for multiple comparisons (GraphPad Prism). Significant differences were p < 0.05.
Cell-induced CXCL12 gradient model
The gradient model was constructed in Comsol Multiphysics 3.4 using the 3D diffusion mode. The physical geometry was designed to match the top channel of the cell patterning device which was simplified to a 2 mm W × 20 mm L × 0.2 mm H block. Within this block are 7 subdomain sections each set to have a diffusion coefficient of 1.5 e–10 m2/s for the diffusing species, which is similar to that of CXCL12. The geometry of each subdomain matches the spacing of the cell patterning regions of each experimental condition. For the boundary conditions, all surfaces were set to insulation except for the two regions which contain the CXCL12-producing cells and the CXCR7 cells. The CXCL12-producing surface was set to have a constant flux of 9.45 e−14 mole/s/m2 which is an average value based upon experimentally obtained results in culture dish. The CXCR7 surface was set to a flux that is dependent on the concentration within that region to follow Michaelis-Menten Kinetics: V = Vmax*C/(Km + C). Parameters for the Michaelis–Menten equation were estimated by setting Km value equal to the dissociation constant (Kd = 0.32 nM32) of CXCL12 with CXCR7 and Vmax (6.49 e−19 mole/s) was calculated by solving the above Michaelis–Menten equation using V (6.38 e−20 mole/s) and C (2.18 e−18 mole/m3) values obtained from uptake experiments done in culture dish.
The concentration gradient profiles were taken across the width of the block at a height of 0.01 mm above the bottom surface with a line resolution of 200. The simulated data was taken at the 20 h time-step to approximate the gradient profile (Fig. 4A). The specific gradient ahead of the CXCR4 cells was determined by dividing the concentration difference across 0.05mm by the average concentration over that distance (Fig. 4B).
Specific gradient
where c(x) is the concentration at position x and d is the distance of a cell.
Supplementary Material
Insight, innovation, integration.
A microfluidically engineered tumor microenvironment demonstrates that cells expressing the CXCL12 scavenging receptor, CXCR7, can play a critical role in the chemotaxis of adjacent CXCR4-expressing cancer cells towards CXCL12 producing cells. Efficient chemotaxis of CXCR4 cells required precise spatial relationships of CXCL12-secreting cells and CXCR7 cells, and chemotaxis could be blocked by inhibiting CXCR7 receptors. Simulations show that CXCL12 scavenging by CXCR7 cells is critical for gradient formation, and that chemotaxis in these devices occurs at much shallower CXCL12 gradients than required in conventional mono-culture in vitro systems likely due to integrated signaling among co-cultured cells. The results provide new strategies to study as well as block CXCL12-mediated migration of tumor cells in metastatic cancer and other diseases.
Acknowledgments
Research was supported by NIH grants R01CA136553, R01CA136829, and P50CA093990. We thank ChemoCentryx for CCX733.
Footnotes
Published as part of a themed issue on Mechanisms of Directed Cell Migration: Guest Editors David Beebe and Anna Huttenlocher.
Electronic supplementary information (ESI) available: Supplementary Figure 1. X7 cells accumulate CXCL12-cherry. See DOI: 10.1039/c0ib00041h
Contributor Information
Gary D. Luker, Email: gluker@med.umich.edu.
Shuichi Takayama, Email: takayama@umich.edu.
References
- 1.Gurdon JB, Bourillot PY. Nature. 2001;413:797–803. doi: 10.1038/35101500. [DOI] [PubMed] [Google Scholar]
- 2.Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 3.Kunwar PS, Siekhaus DE, Lehmann R. Annu Rev Cell Dev Biol. 2006;22:237–265. doi: 10.1146/annurev.cellbio.22.010305.103337. [DOI] [PubMed] [Google Scholar]
- 4.Friedl P, Weigelin B. Nat Immunol. 2008;9:960–969. doi: 10.1038/ni.f.212. [DOI] [PubMed] [Google Scholar]
- 5.Keenan TM, Folch A. Lab Chip. 2008;8:34–57. doi: 10.1039/b711887b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toetsch S, Olwell P, Prina-Mello A, Volkov Y. Integr Biol. 2009;1:170–181. doi: 10.1039/b814567a. [DOI] [PubMed] [Google Scholar]
- 7.Jeon NL, Baskaran H, Dertinger SK, Whitesides GM, Van de Water L, Toner M. Nat Biotechnol. 2002;20:826–830. doi: 10.1038/nbt712. [DOI] [PubMed] [Google Scholar]
- 8.Wang SJ, Saadi W, Lin F, Nguyen CMC, Jeon NL. Exp Cell Res. 2004;300:180–189. doi: 10.1016/j.yexcr.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 9.Herzmark P, Campbell K, Wang F, Wong K, El-Samad H, Groisman A, Bourne HR. Proc Natl Acad Sci U S A. 2007;104:13349–13354. doi: 10.1073/pnas.0705889104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mosadegh B, Saadi W, Wang SJ, Jeon NL. Biotechnol Bioeng. 2008;100:1205–1213. doi: 10.1002/bit.21851. [DOI] [PubMed] [Google Scholar]
- 11.Hanahan D, Weinberg RA. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 12.Liotta LA, Kohn EC. Nature. 2001;411:375–379. doi: 10.1038/35077241. [DOI] [PubMed] [Google Scholar]
- 13.Tharp WG, Yadav R, Irimia D, Upadhyaya A, Samadani A, Hurtado O, Liu SY, Munisamy S, Brainard DM, Mahon MJ, Nourshargh S, van Oudenaarden A, Toner MG, Poznansky MC. J Leukocyte Biol. 2006;79:539–554. doi: 10.1189/jlb.0905516. [DOI] [PubMed] [Google Scholar]
- 14.Lee JY, Buzney CD, Poznansky MC, Sackstein R. J Leukocyte Biol. 2009;86:1285–1294. doi: 10.1189/jlb.0309214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elbjeirami WM, West JL. Tissue Eng. 2006;12:381–390. doi: 10.1089/ten.2006.12.381. [DOI] [PubMed] [Google Scholar]
- 16.Chung S, Sudo R, Mack PJ, Wan CR, Vickerman V, Kamm RD. Lab Chip. 2009;9:269–275. doi: 10.1039/b807585a. [DOI] [PubMed] [Google Scholar]
- 17.Torisawa Y, Mosadegh B, Luker GD, Morell M, O'Shea KS, Takayama S. Integr Biol. 2009;1:649–654. doi: 10.1039/b915965g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walker GM, Zeringue HC, Beebe DJ. Lab Chip. 2004;4:91–97. doi: 10.1039/b311214d. [DOI] [PubMed] [Google Scholar]
- 19.Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 20.Burns JM, Summers BC, Wang Y, Melikian A, Berahovich R, Miao Z, Penfold ME, Sunshine MJ, Littman DR, Kuo CJ, Wei K, McMaster BE, Wright K, Howard MC, Schall TJ. J Exp Med. 2006;203:2201–2213. doi: 10.1084/jem.20052144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miao Z, Luker KE, Summers BC, Berahovich R, Bhojani MS, Rehemtulla A, Kleer CG, Essner JJ, Nasevicius A, Luker GD, Howard MC, Schall TJ. Proc Natl Acad Sci U S A. 2007;104:15735–15740. doi: 10.1073/pnas.0610444104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Naumann U, Cameroni E, Pruenster M, Mahabaleshwar H, Raz E, Zerwes HG, Rot A, Thelen M. PLoS One. 2010;5:e9175. doi: 10.1371/journal.pone.0009175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zabel BA, Wang Y, Lewen S, Berahovich RD, Penfold ME, Zhang P, Powers J, Summers BC, Miao Z, Zhao B, Jalili A, Janowska-Wieczorek A, Jaen JC, Schall TJ. J Immunol. 2009;183:3204–3211. doi: 10.4049/jimmunol.0900269. [DOI] [PubMed] [Google Scholar]
- 24.Yu SR, Burkhardt M, Nowak M, Ries J, Petrasek Z, Scholpp S, Schwille P, Brand M. Nature. 2009;461:533–537. doi: 10.1038/nature08391. [DOI] [PubMed] [Google Scholar]
- 25.Rappel WJ, Levine H. Proc Natl Acad Sci U S A. 2008;105:19270–19275. doi: 10.1073/pnas.0804702105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen W, Levine H, Rappel WJA. Phys Biol. 2008;5:046004. doi: 10.1088/1478-3975/5/4/046006. [DOI] [PubMed] [Google Scholar]
- 27.Levoye A, Balabanian K, Baleux F, Bachelerie F, Lagane B. Blood. 2009;113:6085–6093. doi: 10.1182/blood-2008-12-196618. [DOI] [PubMed] [Google Scholar]
- 28.Boldajipour B, Mahabaleshwar H, Kardash E, Reichman-Fried M, Blaser H, Minina S, Wilson D, Xu Q, Raz E. Cell. 2008;132:463–473. doi: 10.1016/j.cell.2007.12.034. [DOI] [PubMed] [Google Scholar]
- 29.Luker KE, Gupta M, Luker GD. BioTechniques. 2009;47:625–632. doi: 10.2144/000113126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luker KE, Steele JM, Mihalko LA, Ray P, Luker GD. Oncogene. 2010;29:4599–4610. doi: 10.1038/onc.2010.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Torisawa Y, Chueh B, Huh D, Ramamurthy P, Roth TM, Baraldab KF, Takayama S. Lab Chip. 2007;7:770–776. doi: 10.1039/b618439a. [DOI] [PubMed] [Google Scholar]
- 32.http://www.millipore.com/catalogue/item/HTS138M
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.