

Abstract
The P2Y12 receptor (P2Y12R), one of eight members of the P2YR family expressed in humans, has been identified as one of the most prominent clinical drug targets for inhibition of platelet aggregation. Consequently, extensive mutagenesis and modeling studies of the P2Y12R have revealed many aspects of agonist/antagonist binding1-4. However, the details of agonist and antagonist recognition and function at the P2Y12R remain poorly understood at the molecular level. Here, we report the structures of the human P2Y12R in complex with a full agonist 2-methylthio-adenosine-5′-diphosphate (2MeSADP, a close analogue of endogenous agonist ADP) at 2.5 Å resolution, and the corresponding ATP derivative 2-methylthio-adenosine-5′-triphosphate (2MeSATP) at 3.1 Å resolution. Analysis of these structures, together with the structure of the P2Y12R with antagonist ethyl 6-(4-((benzylsulfonyl)carbamoyl)piperidin-1-yl)-5-cyano-2-methylnicotinate (AZD1283)5, reveals dramatic conformational changes between nucleotide and non-nucleotide ligand complexes in the extracellular regions, providing the first insight into a different ligand binding landscape in the δ-group of class A G protein-coupled receptors (GPCRs). Agonist and non-nucleotide antagonist adopt different orientations in the P2Y12R, with only partially overlapped binding pockets. The agonist-bound P2Y12R structure answers long-standing ambiguities surrounding P2Y12R-agonist recognition, and reveals interactions with several residues that had not been reported to be involved in agonist binding. As a first example of a GPCR where agonist access to the binding pocket requires large scale rearrangements in the highly malleable extracellular region, the structural studies therefore will provide invaluable insight into the pharmacology and mechanisms of action of agonists and different classes of antagonists for the P2Y12R and potentially for other closely related P2YRs.
After sensing their endogenous extracellular ligands, GPCRs activate associated intracellular signal transduction pathways that subsequently lead to physiological responses6. Structures of five GPCRs (rhodopsin7,8, β1 (β1AR)9,10 and β2 adrenergic receptors (β2AR)11,12, A2A adenosine receptor (A2AAR)13,14 and M2 muscarinic receptor15,16) have now been determined in both antagonist- and agonist-bound states. Each of these five receptors, however, belong to the α-group of class A GPCRs17. Here, we describe a 2.5 Å structure of human P2Y12R bound to the full agonist 2MeSADP, and a 3.1 Å structure of P2Y12R bound to a potential partial agonist 2MeSATP (Extended Data Table 1). Together with the 2.7 Å structure of P2Y12R bound to the antagonist AZD1283 reported in the accompanying paper5, this allows the first crystallographic assessment of a receptor with both agonist- and antagonist-bound structures in the δ-group of class A GPCRs.
Extended Data Table 1.
Data collection and refinement statistics. The highest resolution shell is shown in parentheses.
| Data Collection | ||
|---|---|---|
| P2Y12R-2MeSADP | P2Y12R-2MeSATP | |
| Number of Crystals usded | 17 | 6 |
| Space group | C2221 | C2 |
| Cell dimensions | ||
| a, b, c (Ǻ) | 65.1, 104.2, 169.4 | 75.7, 65.1, 100.7 |
| α, β, γ (º) | 90.0, 90.0, 90.0 | 90.0. 95.5, 90.0 |
| Number of reflections processed | 335,625 | 26,125 |
| Number of unique reflections | 20,345 | 8,273 |
| Resolution (Ǻ) | 50.0-2.50 (2.63-2.50) † | 30.0-3.10 (3.27-3.10) † |
| Rmerge (%) | 19.4 (99.5) | 22.2 (92.2) |
| CC1/2 | 0.996 (0.587) | 0.968 (0.413) |
| Mean I/σ(I) | 12.4 (2.3) | 5.6 (2.2) |
| Completeness (%) | 100.0 (100.0) | 92.2 (91.2) |
| Redundancy | 16.5 (8.1) | 3.2 (2.9) |
| Refinement | ||
| Resolution (Ǻ) | 50.0-2.50 | 50.0-3.10 |
| Number of reflections (test set) | 20,345 (1,041) | 8,273 (398) |
| Rwork/Rfree(%) | 19.9/23.0 | 18.8/25.5 |
| Number of atoms | ||
| Protein | 3,145 | 3,058 |
| Ligand | 29 | 33 |
| Cholesterol | 28 | 0 |
| Lipids, PEG and waters | 105 | 33 |
| Overall B values (Ǻ2) | ||
| P2Y12R | 65.1 | 69.3 |
| BRIL | 59.3 | 142.0 |
| Ligand | 43.9 | 65.2 |
| Cholesterol | 87.0 | n/a |
| Lipids and waters | 69.9 | 89.1 |
| RMSD | ||
| Bond lengths (Ǻ) | 0.010 | 0.010 |
| Bond angles (º) | 1.04 | 1.03 |
| Ramachandran plot statistics (%) ‡ | ||
| Favored regions | 98.7 | 95.8 |
| Allowed regions | 1.3 | 4.2 |
| Disallowed regions | 0.0 | 0.0 |
Footnote:
Values in parentheses are for highest-resolution shell.
As defined in MolProbity.
All three P2Y12R structures were determined using the same thermostabilizing construct5. The receptor conformations of the 2MeSADP and 2MeSATP bound complexes are very similar (Cα R.M.S.D. = 0.6 Å), and both ligands overlay well in the same binding pocket (R.M.S.D. of common atoms = 0.6 Å), with the γ-phosphate group of 2MeSATP extended towards the extracellular surface (Fig. 1, Extended Data Fig. 1). Since the two structures are similar, we will focus our discussions on the higher resolution P2Y12R-2MeSADP structure and will point out the specific differences of the 2MeSATP bound complex as needed.
Fig. 1.

Overall structure of the P2Y12R-2MeSADP and P2Y12R-2MeSATP complexes. (a) Side view of P2Y12R-2MeSADP complex structure. The receptor is colored cyan and shown in cartoon representation. The ligand 2MeSADP is shown in sphere representation with orange carbons. The disulfide bonds are shown as yellow sticks; extracellular and intracellular boundaries are shown as dashed lines. (b) Side view of P2Y12R-2MeSATP complex structure. The receptor is colored violet and shown in cartoon representation. The ligand 2MeSATP is shown in sphere representation with gray carbons. (c) Semitransparent surface presentation of the receptor shows the lid formed by ECL2, ECL3 and N-terminus on top of 2MeSADP. (d) Comparison of P2Y12R-2MeSADP with P2Y12R-2MeSATP complex. The ligands are shown as sticks, and phosphorus in 2MeSATP is colored in green to be distinguished from 2MeSADP.
Extended Data Figure 1.
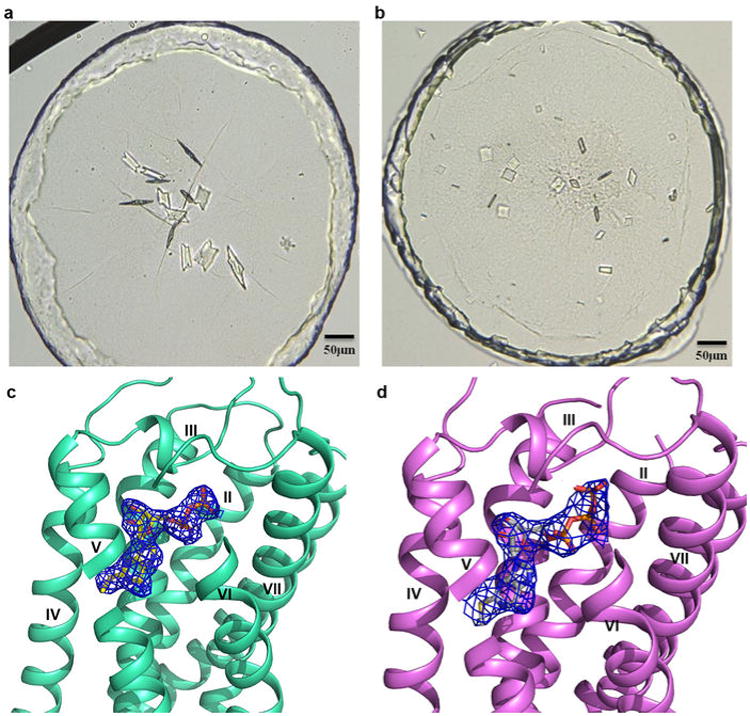
Crystals and electron density of nucleotides for P2Y12R-2MeSADP and P2Y12R-2MeSATP complexes. (a) Crystals of the P2Y12R-2MeSADP complex. The size of the crystals is roughly 80×50×5μm; (b) Crystals of the P2Y12R-2MeSATP complex. The size of the crystals is roughly 30×30×5μm; (c) The 2mFo-DFc map for the 2MeSADP contoured at 1σ; (d) The 2mFo-DFc map for the P2Y12R-2MeSATP contoured at 1σ. The relatively high B-factor of the γ-phosphate group (98 Å2) compared with β-phosphate and surrounding protein atoms (∼75 Å2), and the propensity of 2MeSATP to hydrolyze to 2MeSADP suggest partial occupancy for the β -phosphate group. However, given the differences in crystal forms and packing, as well as the clear density of the γ-phosphate group, the P2Y12R-2MeSATP complex structure should provide relevant information about 2MeSATP binding.
The receptor is folded into a canonical seven transmembrane (7TM) bundle with a partially resolved intracellular helix VIII (Fig. 1). The straight conformation and tilted orientation of helix V observed in the AZD1283-bound structure5, is likely a genuine structural feature inherent to the P2Y12R as it is consistent in all three structures despite the different crystal packing configurations (Extended Data Fig. 2). Both the disulfide bonds, bridging the N-terminus (C17) with helix VII (C2707.25, superscript indicates Ballesteros-Weinstein residue numbering18) and the highly conserved disulfide bond between helix III (C973.25) and extracellular loop 2 (ECL2, C175) are observed.
Extended Data Figure 2.
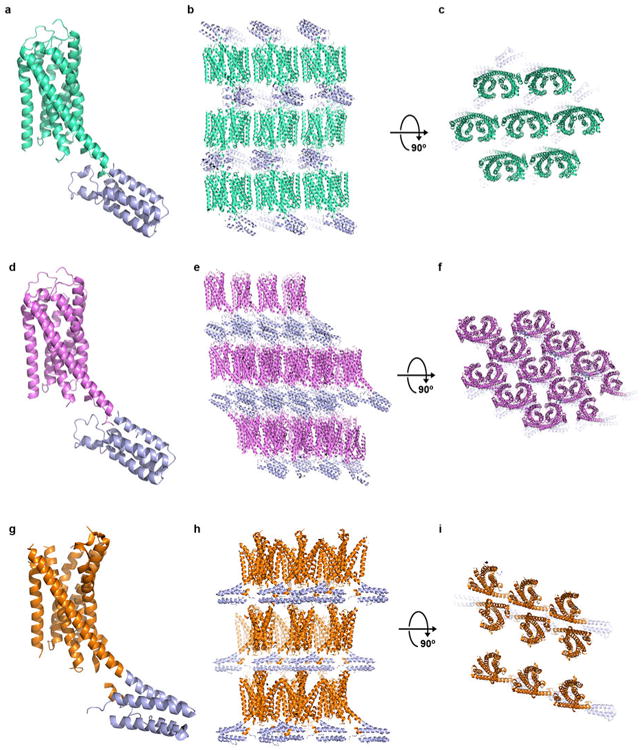
Crystal packing of P2Y12R-2MeSADP, P2Y12R-2MeSATP and P2Y12R-AZD1283 complexes. (a) Overall structure of the P2Y12R-2MeSADP complex, P2Y12R and BRIL are shown in cyan and blue, respectively. (b and c) Crystal packing of P2Y12R-2MeSADP complex at two different views. (d) Overall structure of the P2Y12R-2MeSATP complex, P2Y12R is shown in pink. (e and f) Crystal packing of P2Y12R-2MeSATP complex at two different views. (g) Overall structure of the P2Y12R-AZD1283 complex, P2Y12R is shown in orange. (h and i) Crystal packing of P2Y12R-AZD1283 complex at two different views.
Comparison of the P2Y12R-AZD1283 and P2Y12R-2MeSADP complex structures reveals remarkable differences. The largest conformational change occurs in helices VI and VII. The extracellular part of helix VI in the 2MeSADP-bound structure shifts over 10 Å and helix VII over 5 Å towards the axis of the 7TM helical bundle, as compared to the antagonist-bound structure (Fig. 2). In the AZD1283-bound structure, the phenyl moiety of the antagonist apparently prevents this inward movement of helix VI, which may explain the difference between the two complexes. The inward shift of helices VI and VII in the 2MeSADP complex allows formation of an extensive ionic and polar interaction network with the phosphate groups of 2MeSADP. Connected by a disulfide bond to helix VII, the N-terminus is moved towards the axis of the helical bundle as well; the position of R19, for example, shifts by ∼6.6 Å with agonist bound. Compared with the structure of the antagonist-bound protease-activated receptor 1 (PAR1)19 (the receptor with the closest sequence homology and known structure to P2Y12R), the largest differences are observed in the straight versus bent conformation of helix V, as well as in the positions of the extracellular parts of helix VI, which in PAR1 adopts an intermediate conformation between the agonist- and antagonist-bound P2Y12R structures (Extended Data Fig. 3).
Fig. 2.

Comparison of the P2Y12R-2MeSADP (agonist) and P2Y12R-AZD1283 (antagonist) complexes. (a) The P2Y12R-2MeSADP complex (receptor: cyan cartoon and ligand: sticks of orange carbons) and P2Y12R-AZD1283 complex (receptor: light orange cartoon and ligand: sticks of green carbons) are shown in side view. Movement of the extracellular tips of helices VI and VII toward the center of the 7TM domain is shown by arrows. The extracellular (b) and intracellular (c) views of the comparison are also shown.
Extended Data Figure 3.
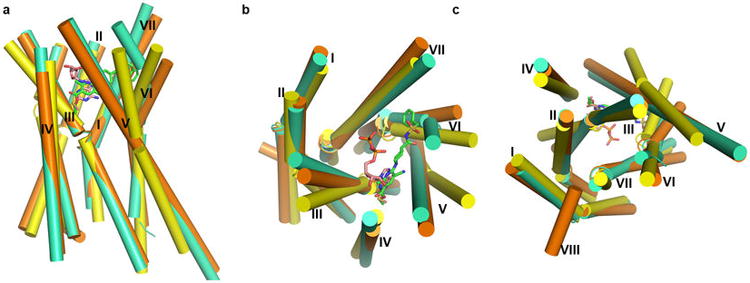
Comparison of antagonist- (orange) and agonist- (greencyan) bound P2Y12R structures with PAR1 structure (yellow). (a) Side view of the three structures. The receptor structures are shown as cylindrical helices, and AZD1283 and 2MeSADP are shown as sticks with green carbons and wheat carbons respectively. (b) Comparison view from the extracellular side. (c) Comparison view from the intracellular side.
All ECLs have clear density and are resolved in the agonist-bound structure, while only ECL3 was resolved in the P2Y12R-AZD1283 complex structure. The highly conserved C973.25- C175ECL2 disulfide bond stabilizing the conformation of ECL2 is clearly observed in the agonist-bound structure, although it was not resolved with antagonist bound5. Formation of this disulfide bond in the P2Y12R-2MeSADP complex requires an unwinding of the helical bulge structure in the extracellular portion of helix III that comprises ∼4.6 residues instead of the ∼3.6 residues in a regular α-helical turn. As a result, there is a Cα rotation of C973.25 along the helical path by over 60°, and a relocation of other residues at this region as compared with the AZD1283 complex (Extended Data Fig. 4). In addition to the structural changes of helix III, the extracellular tip of helix V is also shifted ∼2 Å towards the helical bundle to satisfy formation of this disulfide bond (Fig. 2b).
Extended Data Figure 4.
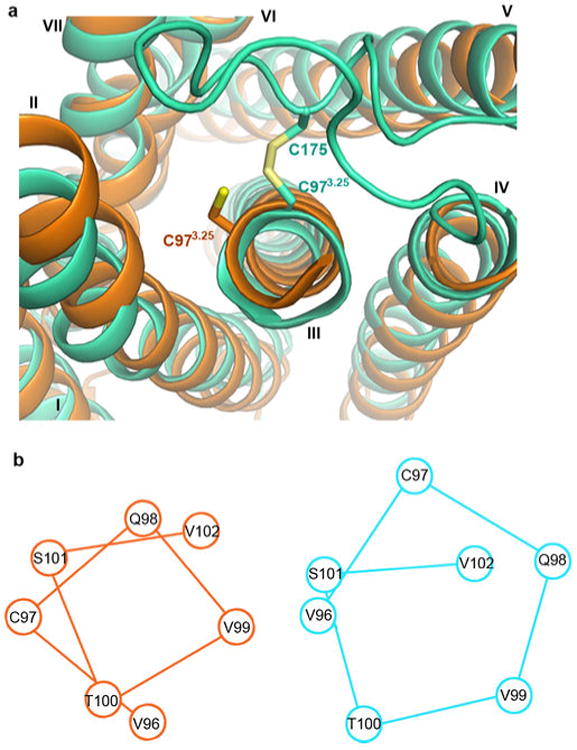
The distortion of helix III by the disulfide bond. (a) Comparison of P2Y12R-AZD1283 (orange) and P2Y12R-2MeSADP (greencyan). (b) Corresponding positions of residues around C973.25 in P2Y12R-AZD1283 (orange) and P2Y12R-2MeSADP (greencyan).
With these substantial rearrangements of the helical bundle, ECLs and the N-terminus, the agonist-bound structure appears to be much tighter compared with the antagonist-bound structure, with 2MeSADP almost completely enclosed in the receptor. A trend for slight contraction of the ligand binding pocket was observed for some α-group GPCRs in agonist-bound as compared to antagonist-bound structures7-16, though these extracellular rearrangements are of unprecedented scale in the P2Y12R. This very high plasticity of the extracellular region in the P2Y12R may help to explain how rather bulky nucleotide ligands gain access into the binding cavity, which is completely covered by loops and N-terminal residues (Fig. 1c). A high plasticity in the ECL region was also proposed for another δ-branch GPCR, PAR1 (ref 19), but the current availability of only an antagonist-bound PAR1 structure precludes crystallographic description of those changes.
Interestingly, the intracellular conformational changes of P2Y12R are less prominent than those at the extracellular side (Fig. 2b,c). In both P2Y12R structures the intracellular domain has a very similar conformation to that observed in the PAR1 structure (Extended Data Fig. 3c). Only minor changes in helices VI and VII were observed in the intracellular part of P2Y12R-2MeSADP. They do not appear consistent, however, with large changes in helical positions observed in the intracellular region in active state agonist-bound A2AAR, or β2AR stabilized by G-protein12,14. It is therefore likely that the P2Y12R-2MeSADP structure represents an “agonist-bound inactive state” with respect to the intracellular region, similar to the one observed in agonist-bound β1AR and β2AR without G-protein or a G-protein mimic stabilizing their active state10,20.
The rearranged 2MeSADP-binding pocket consists of residues from helices III, IV, V, VI and VII as in the P2Y12R-AZD1283 structure5, but also extensively involves ECL2 and the N-terminus. Both pockets described in the P2Y12R-AZD1283 structure are still present, although contracted, especially pocket 1, due to the inward movement of helices VI and VII (Extended Data Fig. 5). In particular, the inward shift of helix VI in the agonist-bound structure shrinks pocket 1 substantially so that it would preclude AZD1283 binding. As a result, although both 2MeSADP and AZD1283 bind to the same pocket, their orientations are completely different, with only partial overlap between them (Fig. 3).
Extended Data Figure 5.
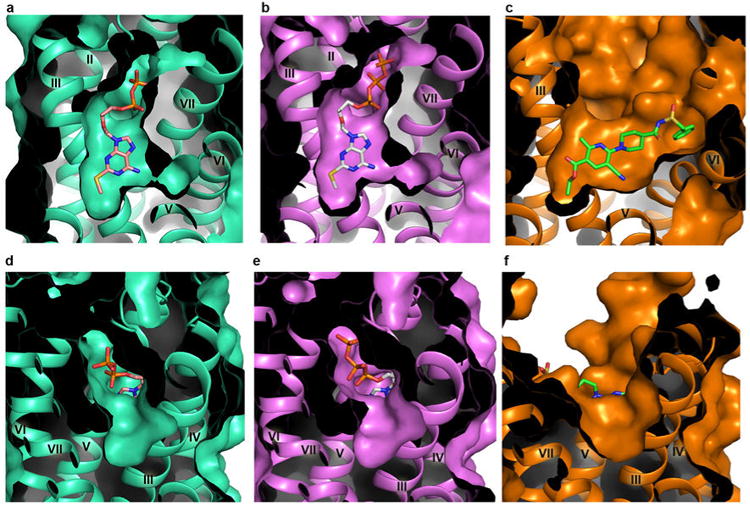
Comparison of pocket 1 (a-c) and pocket 2 (d-f) of P2Y12R structures with different ligands. (a and d) The P2Y12R-2MeSADP structure. (b and e) The P2Y12R-2MeSATP structure. (c and f) The P2Y12R-AZD1283 structure. The 2MeSADP, 2MeSATP and AZD1283 ligands are shown in sticks with wheat, gray and green carbons, respectively.
Fig. 3.
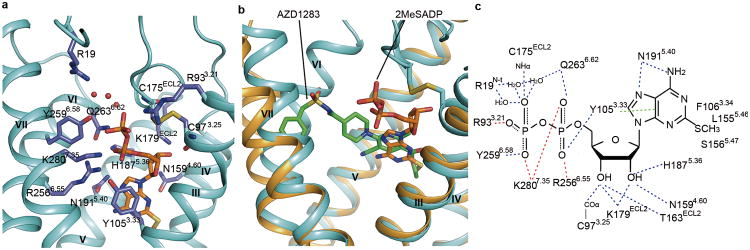
P2Y12R ligand binding pocket for 2MeSADP. (a) The receptor is shown in cyan cartoon representation. The ligand 2MeSADP (orange carbons) and receptor residues (slate carbons) involved in ligand binding are shown in stick representation. Other elements are colored as follows: oxygen, red; nitrogen, dark blue; sulfur, yellow; phosphorus, orange. The water molecules interacting with 2MeSADP are shown as red spheres. (b) Comparison of the 2MeSADP and AZD1283 binding poses in the overlaid P2Y12R complexes. Color scheme as in Fig. 2. (c) Summary of receptor interactions of 2MeSADP. Hydrogen bonds are displayed as blue dashed lines and the salt bridges as red dashed lines. The π-π interaction between 2MeSADP and Y1053.33 is indicated as green dashed lines. The NHα and COα indicate the main chain amine and carbonyl groups of the corresponding residue.
The adenine group of 2MeSADP occupies the same aromatic binding site as the nicotinate group in AZD1283, forming a similar π-π interaction with the Y1053.33 side chain. The 2-thioether inserts into a hydrophobic pocket formed by F1063.34, L1554.56, S1564.57 and N1594.60, and serves as an anchor to maintain the adenine core and the ribose ring in an optimal orientation (Extended Data Fig. 5a). Thus, 2MeSADP binds with greater complementarity, which explains the higher affinity of this ligand compared with ADP. 2-Thioether and amino groups of adenine overlay with the ethyl ester and cyano substituents on the nicotinate group (Fig. 3b). The orientation of the ribose moiety corresponds to that of the methyl group on the nicotinate moiety. The ribose 2′ and 3′ hydroxyl groups interact with K179ECL2 and H1875.36 and with T163ECL2 and K179ECL2, respectively.
The interactions between the receptor and the diphosphate of 2MeSADP involve numerous hydrophilic and positively charged residues (Fig. 3c). As predicted based on the sequence analysis and confirmed through mutagenesis21-23, two essential cationic residues R2566.55 and K2807.35 (Extended Data Table 2) and an aromatic residue Y2596.58 that is conserved in the P2Y12R-like subfamily coordinate phosphate moieties. In addition to these residues, a previously un-implicated in agonist binding third cationic residue R933.21 contact the β-phosphate. Three water molecules also bridge the interaction between β-phosphate with a fourth cationic residue, R19 (N-terminus). Some residues that are thought to participate in agonist binding, however, have no direct contact with the nucleotide ligands. The R265ECL3 side chain, which was previously implicated in activation of the receptor23, is positioned away from 2MeSADP. Interestingly, the conserved K174ECL2, which was previously predicted to interact with 2MeSADP22,24, does not form a direct contact with the agonist, but forms a salt bridge to E2737.28 that apparently stabilizes the agonist-bound conformation.
Extended Data Table 2.
Binding affinities for different P2Y12R constructs. Affinity values of the agonist [3H]2MeSADP determined in saturation binding to WT and mutant P2Y12Rs expressed transiently in COS7 cells (a) and inhibition by antagonist AZD1283 (b).
| a | |
|---|---|
|
| |
| Constructs | [3H]2MeSADP(Kd, nM) |
| WT | 4.9±1.3 |
| S83A | 5.6±1.4 |
| C97A | N.S. |
| R256A | 16.1±6.2 |
| C175A | N.S. |
| K280A | N.S. |
| b | |
|---|---|
|
| |
| Constructs | AZD1283 (Ki, nM) |
| WT | 41.8±16.3 |
| S83A | 36.5±6.8 |
| R256A | 140±39 |
Footnote: Results are expressed as mean ± SEM from 3-6 independent experiments performed in duplicate by methods described in Zhang et al5. N.S., not saturable or negligible specific binding within the radioligand concentrations used (0.4-46 nM).
Cysteine residues forming the conserved disulfide bond are also involved directly in the binding of 2MeSADP. The main chain carbonyl of C973.25 forms a hydrogen bond with the 3′ hydroxyl group of ribose, and the main chain NH group of C175ECL2 interacts with the β-phosphate group. Disruption of this labile disulfide bond, e.g. as possible in the complex with antagonist AZD1283, would also prevent such a bidentate coordination system of the nucleoside. The active metabolites of the thienopyridine drugs are also predicted to destabilize interactions of the P2Y12R with the nucleotide agonist by binding covalently to C973.25. Mutation of either of these cysteine residues to alanine greatly reduces agonist binding (Extended Data Table 2), although it is known that the P2Y12R does not require this disulfide bond for overall structural integrity, i.e. in the AZD1283 complex.
The action of nucleoside 5′-triphosphate derivatives at P2Y12R, i.e. agonism vs. antagonism, has long been ambiguous. Although ATP and 2MeSATP were thought to be antagonists under physiological conditions25, the close conformational similarity between the 2MeSADP and 2MeSATP complexes suggests that ATP derivatives potentially qualify for similar signaling properties at P2Y12R as ADP derivatives, which is also supported by recent pharmacological data3. Moreover, AR-C66096, a non-cleavable triphosphate mimic that could be docked into the P2Y12R in an identical binding mode to 2MeSADP and 2MeSATP, displays characteristics of a partial agonist inhibiting cAMP production in overexpressing P2Y12R/CHO cells (Extended Data Fig. 6). Of course, we emphasize that partial agonist activity of ATP derivatives may be expression level dependent26.
Extended Data Figure 6.
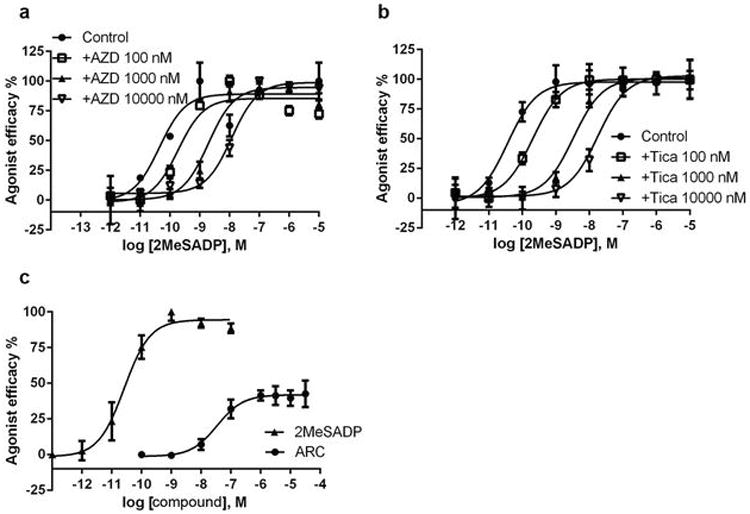
Functional properties of different ligands at P2Y12R. Data (mean ±SEM) were determined in triplicate. (a) Parallel right shifts induced by antagonist AZD1283 (AZD) of the activation curves by agonist 2MeSADP in inhibition of cAMP production in P2Y12R/CHO cells. (b) Parallel right shifts induced by antagonist Ticagrelor (Tica) of the activation curves by agonist 2MeSADP in inhibition of cAMP production in P2Y12R/CHO cells. The pKB values of AZD and Tica are 8.17 ± 0.45 and 7.70 ± 0.18, respectively. (c) Partial agonist effects of AR-C66096 (ARC) in inhibition of cAMP production in P2Y12R/CHO cells. The EC50 value of AR-C66096 was determined to be 34.9 ± 2.9 nM, and its Emax 41.9 ±3.6% compared with 2MeSADP as 100%. A final concentration of 10 μM forskolin was used in the experiment. DMSO was used as a solvent for the stock solution of forskolin, AZD1283 and Ticagrelor. The stock solution of AR-C66096 was made with water.
The 2MeSADP binding cavity appears to accommodate a number of other nucleotide ligands that mimic a triphosphate chain, including AR-C67085 and Cangrelor27 (Extended Data Fig. 7 and 8). However, the nucleoside antagonist Ticagrelor does not dock with a similar conformation to 2MeSADP in the P2Y12R due to the bulky N6 substituent, unless a helical rearrangement occurs, particularly in helix VI. This docking observation suggests that reduction of agonist efficacy of these ligands is likely facilitated by N6 substitutions in the nucleoside scaffold27. Consistently, we have shown that non-nucleotides AZD1283 and Ticagrelor behave as competitive P2Y12R antagonists. Thus, we predict that the ligand-bound receptor conformations and consequently pharmacology of nucleotide-mimetic and non-nucleotide antagonists will be divergent.
Extended Data Figure 7.
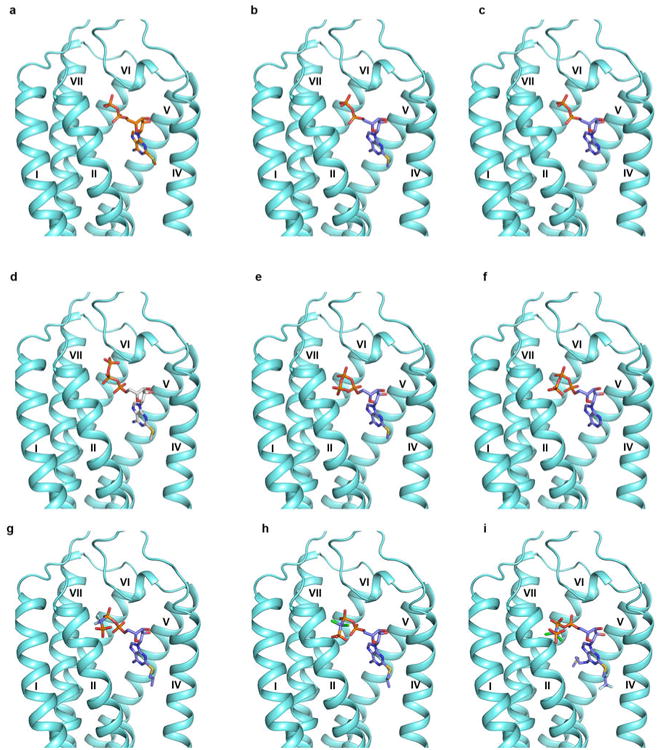
Docking models of different nucleotide analogs to the structure of P2Y12R. (a) The crystal structure of P2Y12R-2MeSADP complex. (b) Docking of 2MeSADP to the P2Y12R structure. (c) Docking of ADP to the P2Y12R structure. (d) The crystal structure of P2Y12R-2MeSATP complex. (e) Docking of 2MeSATP to the P2Y12R structure. (f) Docking of ATP to the P2Y12R structure. (g) Docking of AR-C66096 to the P2Y12R structure. (h) Docking of AR-C67085 to the P2Y12R structure. (i) Docking of AR-C69931MX (Cangrelor) to the P2Y12R structure. 2MeSADP and 2MeSATP poses from corresponding crystal structures are shown in stick with orange and gray carbons respectively. Docking was performed to the conformation of P2Y12R found in the 2MeSADP-bound structure, and the docked ligands are shown in sticks with purple carbons. AR-C66096 and AR-C67085 show the same interactions observed in the 2MeSADP complex. In addition, the C2-propylthio substituent of AR-C66096 and AR-C67085 is located in a hydrophobic pocket in proximity to helix IV surrounded by F1063.34, Y1093.37, M1524.53 and L1554.56. The γ-phosphonate group is directed towards helix III and interacts with K802.60 and R933.21. The C2 substituent and the γ-phosphonate group of AR-C69931MX show similar orientation as observed in the docking pose of AR-C66096 and AR-C67085. The N6 substituent is directed towards helix VI in proximity to Y1093.37, Q1955.44, F2526.51, H2536.52 and R256 6.55.
Extended Data Figure 8.
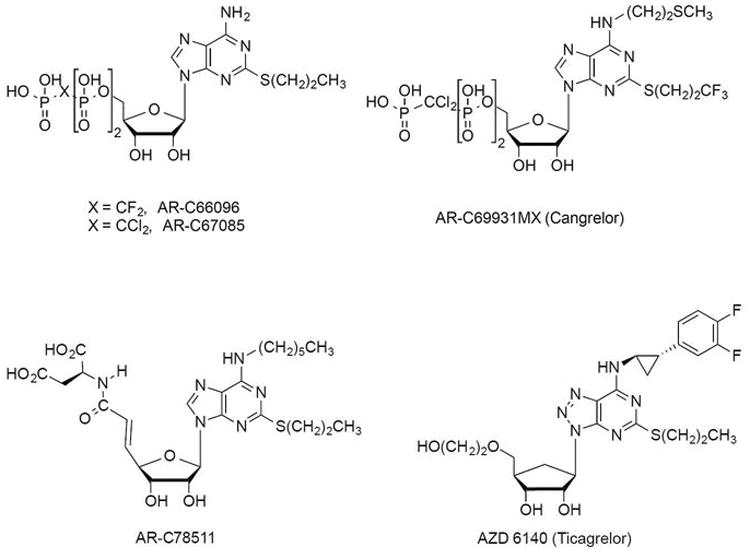
Ligands used in the docking studies. The chemical structures of parts of ligands that are discussed and used in the docking studies are shown. Ticagrelor and AR-C78511 could not be docked in a conformation similar to 2MeSADP because the presence of their bulky N6 substituents would cause a steric clash with helices V and VI. AR-C78511 was previously shown to lack partial agonist properties.
These structural and pharmacological findings provide unexpected insights into the mechanism of agonist and antagonist interactions with P2Y12R, as schematically illustrated in Fig. 4. As the agonist bound pocket is sealed by a “lid” formed by unusually cationic ECLs and the N-terminus, agonist access to the binding pocket of apo P2Y12R would require plasticity of this extracellular region. Closing of the pocket in the absence of charged phosphates would also be disfavored by electrostatic repulsion, with ∼8 arginine and lysine side chains pointing towards the pocket. Binding of a nucleotide agonist like 2MeSADP involves stabilization of an inward position of helices VI and VII and formation of the lid, which is stabilized by numerous electrostatic interactions with the agonist phosphate groups. The additional phosphate group of 2MeSATP is also accommodated by a similar conformation of the lid, though distinct interactions may still impact the 2MeSATP binding and signaling profile. In stark contrast, an open extracellular side conformation is defined by binding of non-nucleotide antagonists like AZD1283, which keeps helices VI and VII away from the pocket and destabilizes the lid.
Fig. 4.

Schematic illustration of conformational changes in P2Y12R extracellular region. (a) Unliganded (apo) state of P2Y12R with open entrance to the pocket and partially disordered lid. The large number of uncompensated electrostatic charges of the side chains forming the pocket (R19Nterm, K802.60, R933.21, K173ECL2, K174 ECL2, H1875.36, R2566.55 and K2807.35) disfavor formation of the closed state. (b) The closed state is stabilized by binding of nucleotide agonist (e.g. ADP, 2MeSADP). (c) A similar conformation with “lid” closure occurs in 2MeSATP structure and for various docked N6 unsubstituted nucleoside triphosphate and triphosphate-mimetic ligands. (d) A helical reorganization is proposed for some N6 substituted nucleoside triphosphate and triphosphate mimetic ligands, especially with bulky N6 substituents. (e) Binding of non-nucleotide antagonist AZD1283 blocks inward movement of helices VI and VII and prevents “lid” closure.
In conclusion, P2Y12R is the first receptor to demonstrate dramatic rearrangements in the extracellular regions, characterized by open and closed access to the ligand binding pocket. Whether such high plasticity of the binding pocket has evolved to enable optimal and specific recognition of negatively charged nucleotide ligands of the P2YR family, or is a more general feature of the δ-group of class A GPCRs is currently unknown. Additional structures for the members of this branch will help to understand similarities and differences in ligand binding and receptor activation.
Methods
Purification of P2Y12R-BRIL protein and crystallization in lipidic cubic phase
P2Y12R-BRIL construction, expression and membrane preparation were performed using the same procedure as described in the companion manuscript5. In brief, the human P2Y12R was subcloned into a modified pFastBac1 vector, with thermostabilized BRIL (PDB 1M6T) inserted at ICL3 (T223-R224) and a D2947.49N mutation. The fusion protein was expressed using Bac-to-Bac Baculovirus Expression System (Invitrogen) in Spodoptera frugiperda (Sf9) cells for 48 h and membrane was washed repeatedly using hypotonic buffer with low and high salt.
Before solubilization, purified membranes were incubated with 20 μM corresponding ligand (2MeSADP or 2MeSATP obtained from Tocris) in the presence of 2 mg/mL iodoacetamide, and EDTA-free protease inhibitor cocktail (Roche) for 30 min. P2Y12R-BRIL was extracted from the membrane by adding n-dodecyl-β-D-maltopyranoside (DDM, Affymetrix) and cholesteryl hemisuccinate (CHS, Sigma) to the membrane solution to a final concentration of 0.5% (w/v) and 0.1% (w/v), respectively, and stirring was continued at 4 °C for 2.5 h. The supernatant was isolated by centrifugation at 160,000 × g for 30 min, followed by incubation in TALON IMAC resin (Clontech) at 4 °C, overnight. The resin was washed with twenty column volumes of 50 mM HEPES, pH 7.5, 1 M NaCl, 10% (v/v) glycerol, 0.05% (w/v) DDM, 0.01% (w/v) CHS, and 30 mM imidazole. The protein was eluted with 5 column volumes of 50 mM HEPES, pH 7.5, 1 M NaCl, 10% (v/v) glycerol, 0.05% (w/v) DDM, 0.01% (w/v) CHS, 270 mM imidazole and 50 μM corresponding ligand. After removing imidazole by using a PD MiniTrap G-25 column (GE Healthcare), ligand concentration was increased to 2 mM. The protein was then treated overnight with His-tagged PreScission protease (20 μg per 500 ml of expressed material) and His-tagged PNGase F (20 μg per 500 ml of expressed material) to remove the C-terminal His-tag and deglycosylate the receptor. PreScission protease, PNGase F and the cleaved 10× His-tag were removed from the sample by passing the sample over Ni-NTA superflow resin (Qiagen). The protein was then concentrated to 30-40 mg/ml with a 100 kDa molecular weight cut-off concentrator (Millipore).
Protein sample was reconstituted into lipidic cubic phase (LCP) by mixing 40% of ∼30 mg/ml protein with 60% lipid (10% (w/w) cholesterol, 90% (w/w) monoolein)28. Crystallization trials were performed using a syringe lipid mixer as previously described29. The protein-lipid mixture was dispensed in 40 nl drops onto glass sandwich plates (Shanghai FAstal BioTechôInc.) and overlaid with 800 nl precipitant solution using a Mosquito LCP robot (TTP LabTech). For P2Y12R-2MeSADP-BRIL complex, crystals appeared after 1 week in 0.30-0.45 M ammonium acetate, 0.1 M sodium citrate, pH 5.0, 30-40% PEG400, 3% v/v 1-Propanol and 500 μM 2MeSADP and reached their full size (80×50×5 μm3) within 2 weeks (Extended Data Fig. 1). For P2Y12R-2MeSATP-BRIL complex, crystals were obtained from precipitant conditions containing 0.15-0.20 M ammonium tartrate, 0.1 M sodium citrate, pH 6.0, 35-40% PEG400, 4% v/v MPD and 500 μM 2MeSATP, reaching their full size (30×30×5 μm3) within 10 days. Crystals were harvested directly from LCP using 50-150 μm micromounts (M2-L19-50/150, MiTeGen) and flash frozen in liquid nitrogen.
Data collection, structure solution and refinement
X-ray diffraction data were collected at the SPring-8 beam line 41XU, Hyogo, Japan, using a Rayonix MX225HE detector (X-ray wavelength 1.0000 Å). The crystals were exposed with a 10 μm minibeam for 1 second and 1° oscillation per frame, and a rastering system was used to find the best diffracting parts of single crystals30. Most crystals of P2Y12R in complex with 2MeSADP diffracted to 3.0 – 2.5 Å resolution. XDS31 was used for integrating and scaling data from 17 best-diffracting crystals for P2Y12R-2MeSADP complexes and 6 crystals for P2Y12R-2MeSATP complexes. The P2Y12R-2MeSADP complex was solved by molecular replacement with Phaser32 using the receptor portion of PAR1 (PDB: 3VW7), converted to polyalanines, and BRIL (PDB: 1M6T) as initial models and refined in Refmac533 and Buster34 The P2Y12R-2MeSATP structure was solved using P2Y12R in complex with 2MeSADP and BRIL (PDB: 1M6T) as starting models and refined under the same procedure.
Ligand binding assays
Wild-type (WT) and mutant human P2Y12R plasmids with single amino acid substitutions were cloned into PCD3.0 vector and transfected into COS7 cells using Lipofectamine 2000 (Life Technologies, Grand Island, NY, USA). Cells were harvested 48 h after transfection. After harvesting, cells were homogenized for 15 sec and then centrifuged for 10 min at 1,000 × g. The suspension was re-centrifuged at 20,000 × g for 60 min. The resulting pellet was re-suspended, homogenized, split into aliquots and maintained at -80 °C in a freezer until use. Protein concentrations were measured using Bio-Rad protein assay reagents.
For saturation experiments, 50 μl [3H]2MeSADP (3.5 Ci/mmol, from 0.4 to 46 nM, Moravek, Brea, CA, USA) was incubated with 100 μl WT and mutant P2Y12R membrane preparations (5 μg/tube) in a total assay volume of 200 μl Tris·HCl buffer containing 10 mM MgCl2. AZD1283 (10 μM) was used to determine the non-specific binding. For displacement experiments, increasing concentrations of AZD1283 were incubated with WT or mutant membrane preparations (5 -10 μg) and [3H]2MeSADP (10 nM) at 25 °C for 30 min. ADP, 2MeSADP and 2MeSATP were obtained from Sigma (St. Louis, MO, USA). AR-C66096 (>98%) was obtained from Tocris (Minneapolis, MN, USA). The reaction was terminated by harvesting with a 24-channnel Brandel cell harvester (Brandel, Gaithersburg, MD, USA) and followed by washing twice with 5 ml cold Tris·HCl buffer containing 10 mM MgCl2. Radioactivity was measured using a scintillation counter (Tri-Carb 2810TR). Data were analyzed using Prism 6 (GraphPad, San Diego, CA, USA).
Cyclic AMP (cAMP) accumulation assay
CHO cells stably expressing the human P2Y12R were plated in 96-well plates in 0.1 ml medium. After overnight incubation, the medium was removed, and cells were washed three times with 0.1 ml Hank's buffer, pH 7.4. Cells were then treated with the antagonists in the presence of rolipram (10 μM) for 20 min before the addition of agonists. Nucleotide derivatives were incubated with cells for 15 min followed by the addition of forskolin (10 μM) and incubation for another 15 min. The reaction was terminated by removing the supernatant, and cells were lysed upon the addition of 100 μL of 0.3% Tween-20. For determination of cAMP production, the ALPHA Screen cAMP assay kit (PerkinElmer, Waltham, MA, USA) was used following the instructions provided with the kit.
Docking
The P2Y12R-2MeSADP structure was prepared using the Protein Preparation Wizard tool implemented in the Schrödinger suite35, adding all the hydrogen atoms and the missing side chains of residues whose backbone coordinates were observed in the structure. The BRIL portion was removed. The orientation of polar hydrogens was optimized, the protein protonation states were adjusted and the overall structure was minimized with harmonic restraints on the heavy atoms, to remove strain. Then, all the hetero groups and water molecules were deleted.
The SiteMap tool of the Schrödinger suite was used to identify potential binding sites in the structure. Molecular docking of selected compounds (ADP, 2MeSADP, ATP, 2MeSATP, AR-C67085, AR-C66096, Cangrelor and Ticagrelor) at the P2Y12R structure was performed by means of the Glide package from the Schr(x000D6)dinger suite. In particular, a Glide Grid was centered on the centroid of residues located within 6 Å from the previously identified cavity. The Glide Grid was built using an inner box (ligand diameter midpoint box) of 14 Å × 14 Å × 14 Å and an outer box that extended 10 Å in each direction from the inner one (so that ligands up to 20 Å could be docked). Docking of ligands was performed in the rigid binding site using the XP (extra precision) procedure. The top scoring docking conformations for each ligand were subjected to visual inspection and analysis of protein-ligand interactions to select the final binding conformations in agreement with the experimental data.
DNA sequence of the crystallization construct
ATGAAGACGATCATCGCCCTGAGCTACATCTTCTGCCTGGTGTTCGCCGACTACAAGGACGATGATGACGGCGCGCCGCAAGCCGTGGACAACCTCACATCAGCCCCTGGCAACACCTCCCTCTGTACCCGCGACTACAAGATCACACAAGTTCTCTTCCCCCTCCTCTACACAGTGTTGTTCTTCGTCGGCCTCATCACCAACGGATTGGCTATGCGTATCTTCTTCCAGATCCGCTCCAAGTCTAACTTCATCATCTTCCTGAAGAACACTGTGATCTCGGACCTGCTCATGATCCTCACATTCCCATTCAAGATCCTGTCAGATGCCAAGCTCGGTACTGGCCCGTTGCGTACATTCGTCTGCCAGGTTACCTCTGTGATCTTCTACTTCACTATGTACATCAGCATCTCATTCCTGGGTCTCATCACCATCGACAGGTACCAAAAGACCACTAGACCCTTCAAGACTAGCAACCCTAAGAACTTGCTGGGCGCTAAGATCCTGAGCGTGGTCATCTGGGCCTTCATGTTCCTCTTGTCACTGCCCAACATGATCCTCACCAACAGGCAGCCTAGAGATAAGAACGTGAAGAAGTGTTCATTCCTCAAGTCGGAGTTCGGATTGGTTTGGCACGAAATCGTGAACTACATCTGCCAAGTCATCTTCTGGATCAACTTCCTGATCGTTATCGTGTGTTACACATTGATCACCAAGGAGCTCTACAGGTCCTACGTCCGTACTGCTGATCTGGAAGACAATTGGGAAACTCTGAACGACAATCTCAAGGTGATCGAGAAGGCTGACAATGCTGCACAAGTCAAAGACGCTCTGACCAAGATGAGGGCAGCAGCCCTGGACGCTCAGAAGGCCACTCCACCTAAGCTCGAGGACAAGAGCCCAGATAGCCCTGAAATGAAAGACTTTCGGCATGGATTCGACATTCTGGTGGGACAGATTGATGATGCACTCAAGCTGGCCAATGAAGGGAAAGTCAAGGAAGCACAAGCAGCCGCTGAGCAGCTGAAGACCACCCGGAATGCATACATTCAGAAGTACCTGCGCGGAGTCGGCAAGGTTCCTAGGAAGAAGGTCAACGTTAAGGTGTTCATCATCATCGCTGTCTTCTTCATCTGCTTCGTTCCATTCCACTTCGCCCGTATCCCGTACACTTTGTCCCAAACACGCGACGTGTTCGATTGTACCGCTGAGAACACTCTGTTCTACGTCAAGGAATCCACATTGTGGCTGACCTCTCTGAACGCCTGCCTCAACCCATTCATCTACTTCTTCCTCTGTAAGTCTTTCCGCAACTCGTTGATCTCCATGCTGAAGTGCCCTAACTCTGCTACCAGCCTGTCCCAAGATAACAGAAAGAAGGAGCAGGACGGAGGCGACCCGAACGAGGAAACCCCGATGGGCCGGCCTCTGGAAGTTCTGTTCCAGGGGCCCCATCATCATCATCATCATCATCATCATCATTAG.
Acknowledgments
This work was supported by the “National Basic Research Program of China” grants 2012CB910400, 2012CB518000 and 2014CB910400 (B.W., Q.Z.), the National Institutes of Health grants R01 AI100604 (B.W., Q.Z., R.C.S.) and U54 GM094618 (V.C., V.K., R.C.S.; Target GPCR-87), the National Science Foundation of China grants 31370729 and National Science and Technology Major Project 2013ZX09507001 and 2012ZX09301001 (B.W., Q.Z.), National Institutes of Health NIDDK Intramural Research Program (K.A.J.) and the National Natural Science Foundation of China 91313000 (H. J.). The authors thank S. Nylander, E. Kiselev and S. Moss for scientific feedback on the manuscript, A. Walker for asistance with manuscript preparation and K. Kadyshevskaya for help with figure preparation.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions: J.Z. optimized the construct, expressed and purified human P2Y12R-BRIL for crystallization, developed the purification procedure, performed crystallization trials, optimized crystallization conditions. K.Z. helped in construct and crystal opimization, and collected diffraction data. Z.G.G. designed, performed and analysed ligand binding and competition assays of wild-type and mutant P2Y12R. S.P. performed and analysed docking assays.D.Z. helped in expression and purification. G.W.H. solved and refined the structure. T.L. helped the expression for crystallization trials. L.M. helped the expression for the activity assays. W.Z. developed the initial expression and purification protocol for P2Y12R. C.E.M. helped to design and analyze pharmacological experiments and wrote the manuscript. H.Y. helped to design and analysed docking assays. H.J. oversaw design and validation of P2Y12R models. V.C. helped to design and optimize LCP crystallization trials, and processed crystallographic data and wrote the manuscript. V.K. performed and analysed molecular modeling simulations, and wrote the manuscript. K.A.J. oversaw, designed and analysed ligand binding assays and docking, and wrote the manuscript. R.C.S. oversaw expression, purification and crystallization, and structure analysis/interpretation of P2Y12R. B.W. and Q.Z. initiated the project, planned and analysed experiments, supervised the research and wrote the manuscript.
Author information: Atomic coordinates and structure factors for the P2Y12R-2MeSADP and P2Y12R-2MeSATP structures have been deposited in the Protein Data Bank with identification codes 4PXZ and 4PY0, respectively.
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of the paper.
References
- 1.Chang H, et al. Modified diadenosine tetraphosphates with dual specificity for P2Y1 and P2Y12 are potent antagonists of ADP-induced platelet activation. J Thromb Haemost. 2012;10:2573–2580. doi: 10.1111/jth.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang H, et al. Agonist and antagonist effects of diadenosine tetraphosphate, a platelet dense granule constituent, on platelet P2Y1, P2Y12 and P2X1 receptors. Thromb Res. 2010;125:159–165. doi: 10.1016/j.thromres.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmidt P, et al. Identification of determinants required for agonistic and inverse agonistic ligand properties at the ADP receptor P2Y12. Mol Pharmacol. 2013;83:256–266. doi: 10.1124/mol.112.082198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Srinivasan S, et al. The P2Y12 antagonists, 2-methylthioadenosine 5′-monophosphate triethylammonium salt and cangrelor (ARC69931MX), can inhibit human platelet aggregation through a Gi-independent increase in cAMP levels. J Biol Chem. 2009;284:16108–16117. doi: 10.1074/jbc.M809780200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang K, et al. Structure of the human P2Y12 receptor in complex with an antithrombotic drug. Nature. 2014 doi: 10.1038/nature13083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou XE, Melcher K, Xu HE. Structure and activation of rhodopsin. Acta Pharmacol Sin. 2012;33:291–299. doi: 10.1038/aps.2011.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palczewski K, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 8.Scheerer P, et al. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 9.Warne T, et al. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Warne T, et al. The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature. 2011;469:241–244. doi: 10.1038/nature09746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cherezov V, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rasmussen SG, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jaakola VP, et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu F, et al. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haga K, et al. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature. 2012;482551:547. doi: 10.1038/nature10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kruse AC, et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 2013;504:101–106. doi: 10.1038/nature12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- 18.Ballesteros JA, Weinstein H. In: Methods in Neurosciences. Stuart C Sealfon., editor. Vol. 25. Academic Press; 1995. pp. 366–428. [Google Scholar]
- 19.Zhang C, et al. High-resolution crystal structure of human protease-activated receptor 1. Nature. 2012;492:387–392. doi: 10.1038/nature11701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenbaum DM, et al. Structure and function of an irreversible agonist-beta(2) adrenoceptor complex. Nature. 2011;469:236–240. doi: 10.1038/nature09665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoffmann K, Sixel U, Di Pasquale F, von Kugelgen I. Involvement of basic amino acid residues in transmembrane regions 6 and 7 in agonist and antagonist recognition of the human platelet P2Y(12)-receptor. Biochem Pharmacol. 2008;76:1201–1213. doi: 10.1016/j.bcp.2008.08.029. [DOI] [PubMed] [Google Scholar]
- 22.Cattaneo M. The platelet P2Y12 receptor for adenosine diphosphate: congenital and drug-induced defects. Blood. 2011;117:2102–2112. doi: 10.1182/blood-2010-08-263111. [DOI] [PubMed] [Google Scholar]
- 23.Ignatovica V, Megnis K, Lapins M, Schioth HB, Klovins J. Identification and analysis of functionally important amino acids in human purinergic 12 receptor using a Saccharomyces cerevisiae expression system. FEBS J. 2012;279:180–191. doi: 10.1111/j.1742-4658.2011.08410.x. [DOI] [PubMed] [Google Scholar]
- 24.Daly ME, et al. Identification and characterization of a novel P2Y 12 variant in a patient diagnosed with type 1 von Willebrand disease in the European MCMDM-1VWD study. Blood. 2009;113:4110–4113. doi: 10.1182/blood-2008-11-190850. [DOI] [PubMed] [Google Scholar]
- 25.Kauffenstein G, Hechler B, Cazenave JP, Gachet C. Adenine triphosphate nucleotides are antagonists at the P2Y receptor. J Thromb Haemost. 2004;2:1980–1988. doi: 10.1111/j.1538-7836.2004.00926.x. [DOI] [PubMed] [Google Scholar]
- 26.Fujioka M, Omori N. Subtleties in GPCR drug discovery: a medicinal chemistry perspective. Drug Discov Today. 2012;17:1133–1138. doi: 10.1016/j.drudis.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 27.Ding Z, Kim S, Kunapuli SP. Identification of a potent inverse agonist at a constitutively active mutant of human P2Y12 receptor. Mol Pharmacol. 2006;69:338–345. doi: 10.1124/mol.105.014654. [DOI] [PubMed] [Google Scholar]
- 28.Zhao Q, Wu BL. Ice breaking in GPCR structural biology. Acta Pharmacol Sin. 2012;33:324–334. doi: 10.1038/aps.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caffrey M, Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat Protoc. 2009;4:706–731. doi: 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cherezov V, et al. Rastering strategy for screening and centring of microcrystal samples of human membrane proteins with a sub-10 microm size X-ray synchrotron beam. J R Soc Interface. 2009;6(Suppl 5):S587–597. doi: 10.1098/rsif.2009.0142.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vagin AA, et al. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr D Biol Crystallogr. 2004;60:2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- 34.Smart OS, et al. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr D Biol Crystallogr. 2012;68:368–380. doi: 10.1107/S0907444911056058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]