

Abstract
We describe a rare primary mixed granular cell astrocytoma and fibrosarcoma neoplasm, occurring in a 52-year-old female, with morphologic, immunohistochemical and molecular genetic features, whose tumor was entirely composed of granular cells and fibrosarcoma competent. This represents, to the best of our knowledge, the first report of the mixed granular cell astrocytoma and fibrosarcoma neoplasm. Moreover, two parts forming a complex arrangement that excluded it being assessed as a coincidental collision tumor. We discuss the relationship of two parts of this rare tumor by fluorescence in situ hybridization (FISH). Sarcomatous components in this tumor had the same aberrations of chromosomes to the gliomatous components of neoplasms, consisting of 1p 19q loss and no evidence of PTEN allele loss and amplification of EGFR. It was suggested that the sarcomatous component may be derived from glioma cells i this case.
Keywords: Granular cell astrocytoma, fibrosarcoma, 1p 19q loss
Introduction
Granular cell astrocytoma (GCA) is considered as a rare variant of an infiltrative astrocytoma containing a remarkable proportion of granular cells. Histological characteristics have been described in previous articles in order to accurately diagnose GCAs. GCAs commonly coexist with conventional infiltrating astrocytoma or glioblastoma. Among the reported cases of GCAs, up to 82% contain components of glial origin [1,2]. While GCAs are rarely accompanied by fibrosarcoma component, we are describing a rare case which was composed of two dissimilar tissues namely two different components--one, of glial origin, essentially like that in GCA; the other, of mesenchymal origin, resembling a spindle cell fibrosarcoma. Tumor of this type hasn’t been described in published literatures yet.
In this article, we report a case of 52-years-old woman who presented with a rare tumor that was composed entirely of GCA cells and fibrosarcoma cells. The tumor was macroscopically totally removed. And she received radiotherapy and chemotherapy during the following months and showed no symptoms or signs of recurrence of tumor. We therefore present the clinical, light microscopic, immunohistochemical and molecular findings of this rare tumor composed of granular cell astrocytoma tissues and fibrosarcoma tissues.
Case report
A 52-year-old female presented with complaints of progressive weakness of left side of the body and mild dizziness for 1 month. A progressive loss of strength in the left lower limb and she presented with history of convulsion in left limb 2 weeks before admission. On examination, the patient was awake. There was slight reduction of muscle strength in the left side. MRI (Figure 1) showed an irregular enhanced lesion in right frontal area with a significant surrounding edema.
Figure 1.
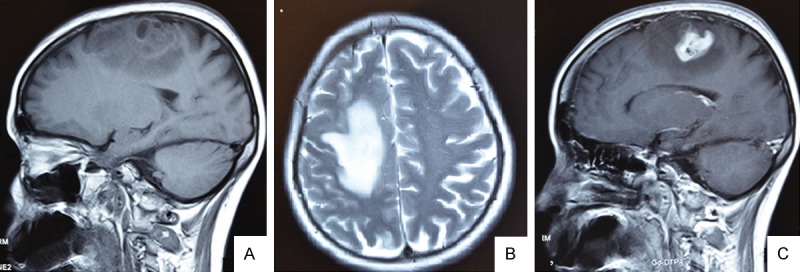
MR Images of this case displaying a bulging lesion with marked peripheral edema localized on the right frontal region. MRI showing a T1-isointensity (A), T2-heterointensity (B) and the central part of lesion was observed after contrast medium injection (C). (A, C, sagittal view; B and A, axial view).
The patient underwent gross excision of the mass after clinical and radiological diagnosis of gliocytoma. The right frontal craniotomy revealed a dark‑red, tough and bloody lesion in the right frontal with a blurry fringe. Thetumor was macroscopically totally removed. After then she received radiotherapy and chemotherapy during following months. Up to now, the patient has showed no symptoms or signs of recurrence of tumor, with a survival time of more than 15 months.
On gross examination, the surgical specimen was a hard, dark- red mass, measured 4 cm × 3 cm × 3 cm in size. The cut surface was dark‑red and heterogeneous, with various firmness. The tumor tissue for morphological examination was fixed in 10% buffered formalin and paraffin embedded. The specimens were stained with H&E and immunohistochemically with the following antibodies: glial fibrillary acid protein (GFAP, 1:500), vimentin (1:50), CD68 (1:100), Ki-67 (1:100), cytokeratin (CK, 1:50), CD34 (1:50), S-100 protein (1:400), and anti-neuronal nuclei (NeuN, Chemicon, 1:100). All antibodies (except NeuN) are purchased from DAKO, Denmark. In addition, positive and negative controls were included and evaluated appropriately for each procedure. PAS staining and reticular fiber staining were also performed. The specimens were also observed by GFAP and Vimentin double-staining.
Microscopically, tumor was characterized by mixed cell proliferation forming gliomatous and sarcomatous areas (Figure 2A).
Figure 2.
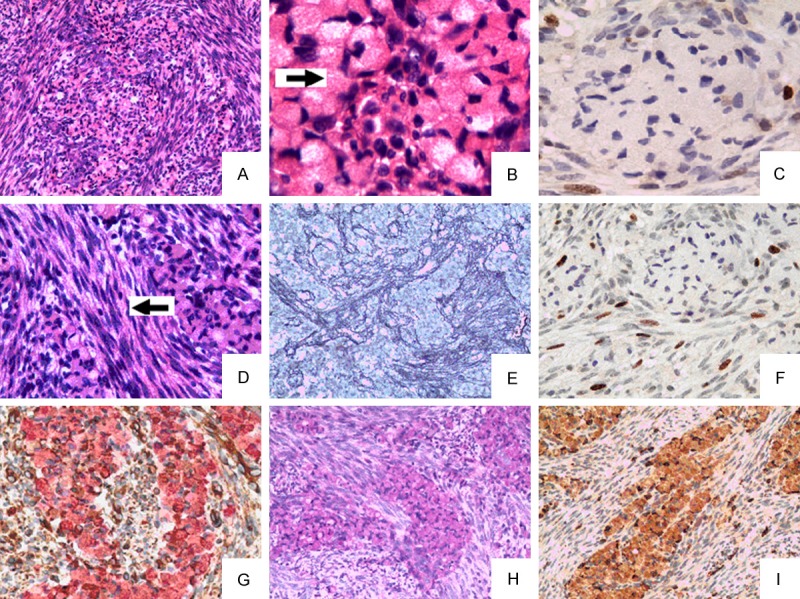
Gliomatous areas are irregularly intermingled with sarcomatous components composed of spindle-shaped cells (HE A, × 200).Gliomatous component demonstrates tumor cells with abundant cytoplasm and eccentrically placed (HE B, × 400, arrow) Ki-67 labelling index low in granular cell astrocytoma tumor cells (C, × 400). Sarcomatous areas were composed predominantly of spindle-shaped cells arranged in intersecting fascicles in herringbone and storiform patterns (HE D, × 400, arrow). Sarcomatous areas were strongly positive for reticular fiber staining (E, × 100). Ki-67 labelling index high in sarcomatous areas tumor cells (F, × 200). Double staining for vimentin and GFAP showed that granular cell astrocytoma tumor cells were all GFAP-positive (pink), and sarcomatous areas cells were all Vimentin-positive (brown). (G, × 400); Granular cell astrocytoma tumor cells were all PAS-positive (H, × 400) and CD68-positive (I × 200).
Gliomatous areas were composed of typical glial cells with generally round or oval cells. The nuclei were mostly round with smooth contours; the tumor cells displayed abundant coarsely granular eosinophilic cytoplasm (Figure 2B). No mitoses, vascular proliferation or necrosis was present. Immunohistochemically, the plump cytoplasm of tumor cells were strongly positive for GFAP (Figure 2G), PAS (Figure 2H) and CD68 (Figure 2I). CK, NeuN and reticular fibers immunostains were negative. Ki-67 Labelling index was low in tumor cells (2-4%) (Figure 2C). This part of tissue was considered to be of glial origin and was considered as granular cell astrocytoma (WHO II).
The other part, sarcomatous areas, was composed predominantly of spindle-shaped cells arranged in intersecting fascicles in herringbone and storiform patterns (Figure 2D). Hyperchromatic nudei and occasional mitoses were noted. In order to evaluate the mesenchymal component, histochemical study to reticulin fibers was performed and the results demonstrated a rich network of fibrils in this area (Figure 2E). Immunohistochemically, the Sarcomatous component cells were strongly positive for Vimentin (Figure 2G). CD68, CD34 and GFAP immunostains were negative. Labelling index of Ki-67 was high in tumor cells (15-20%) (Figure 2F). This part was considered to be of mesenchymal origin and accounted as fibrosarcoma.
In the tumor area, all small islands of the granular cell tumor component were entrapped within reticulin-rich sarcomatous areas. These two tissues were intimately interwoven in many areas.
Fluorescence in situ hybridization (FISH) was performed on patient’s tumor, using the techniques previously described [3]. Briefly, 4-mm slides from the formalin-fixed paraffin-embedded tissue blocks were deparaffinized before hybridization. Dual-color FISH was performed using LSI 1p36/LSI 1q25 and LSI 19q13/19p13 dual-color probe (Vysis/Abbott Molecular) and LSI PTEN/CEP 10 dual color probe (Vysis/Abbott Molecular) for losses of 1p36/19q13 and PTEN. The EGFR gene copy number was determined by FISH using the LSI EGFR spectrum-orange/CEP 7 spectrum-green probe (Vysis/Abbott Molecular). Fluorescent signals were visualized and quantitated under fluorescence microscope. A minimum of 100 non-overlapping intact nuclei were assessed by hybridization. At least 30% or more increase in nuclei numbers is necessary for a signal to be scored as a deletion. Amplification of EGFR was defined as ratio of EGFR signal to CEP7 signal was equal to or greater than two. Percentage of the cells showing deletion and amplification was estimated separately and independently for two component parts of this tumor.
FISH results revealed that the part of granular cell tumor showed deletion of 1p36 (Figure 3A) and 19q13 (Figure 3A), but no loss of PTEN and no amplification of EGFR. The part of fibrosarcoma also showed the same situation as well (deletion of 1p36 (Figure 3B) and 19q13 (Figure 3D). No loss/amplification of PTEN/EGFR). Two component parts of this tumor shared similar genetic alterations.
Figure 3.
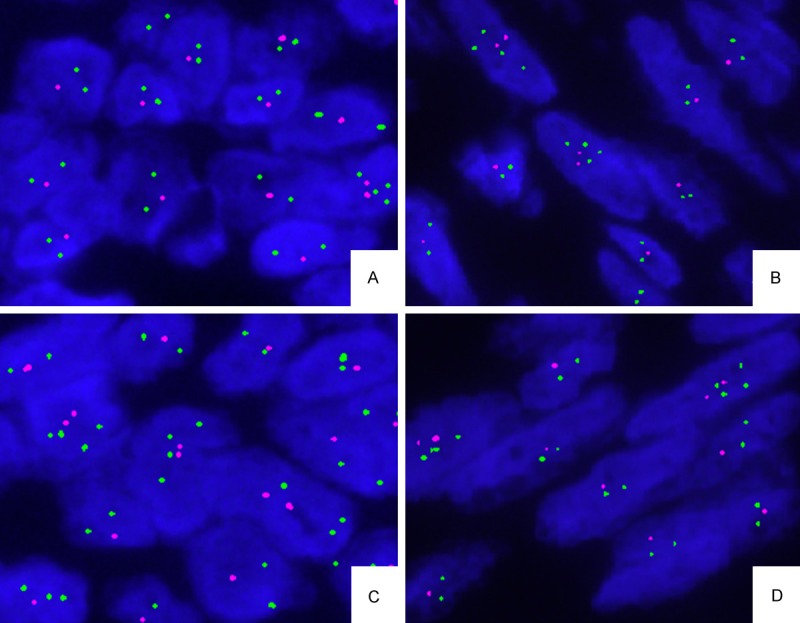
FISH performed on the two components cell of this tumor for chromosome 1p36 and 19q13. The part of granular cell tumor cells showed deletion of 1p36 (A) and 19q13 (C); The part of fibrosarcoma showed deletion of 1p36 (B)and 19q13 (D).
The present case was diagnosed as mixed granular cell astrocytoma (GCA) and fibrosarcoma. As this is the first reported case, it is difficult to determine the grading merely by pathologic findings, a longer follow-up probably could help to understand the real biology of this tumor.
Discussion
GCA is an uncommon morphologic variant of infiltrative glioma that contains a prominent population of atypical granular cells. Morphologically, the tumor cells are plump, round or oval with eosinophilic and periodic acid-Schiff (PAS) positive granular cytoplasm [1]. Normally, GCAs are accompanied by the conventional-type astrocytoma or glioblastoma. However, the occurrence of GCA being accompanied by fibrosarcoma is very rare. In this case, the sarcomatous areas were geographically intermingled with the GCA areas, forming a complex arrangement that excluded it being assessed as a coincidental collision tumor. Moreover, GCAs and sarcomatous areas also could be readily distinguished by reticulin staining; the former was devoid of retiulin fibers, while the latter was rich of reticulin. GCAs cells had no mitoses, no vascular proliferation or necrosis. The sarcomatous components appeared similar to fibrosarcoma, with atypical hyperchromatic nudei and high labeling index of Ki-67.
Microscopically, the histopathology of this tumor was similar to gIiosarcoma (GS) to some extent. Both of them were composed of an admixture of dissimilar tissues. One component resembled the gliomatous part; the other, resembled a spindle cell fibrosarcoma. However, the important differences between the two tumors lay in the gliomatous areas. The gliomatous part for GS resembled the glioblastoma multiforme and was malignant (WHO IV). The gliomatous part for our case resembled GCA (WHO II). The same component, sarcomatous part, for our case resembled fibrosarcoma. The mass in this case was classified as mixed granular cell astrocytoma and fibrosarcoma.To better understand and further investigate the real biology of this tumor, a longer follow-up would be done.
The GCAs showed higher frequencies of loss of heterozygosity (LOH) at 1p and 19q than typical infiltrating astrocytomas of similar grades [4]. To explain the origin of the sarcomatous component of this mixed tumor, we examined aberrations of chromosomes 1p, 19q, PTEN and gene amplification of EGFR using FISH method in GCA component and sarcomatous component. LOH of 1p and 19q were both found in GCA and sarcomatous components. The LOH of PTEN and EGFR gene amplification were not found in both parts of this tumor. Sarcomatous components in this case had the same aberrations of chromosomes to the GCA components. We know Biernat et al. first proposed the monoclonal theory in GS, and his study demonstrated identical p53 mutations in both tumor areas (gliomatous and sarcomatous areas) [5]. Subsequently, many studies found both components in GS shared the similar genetic characteristics by comparative genomic hybridization, FISH, cytogenetic analysis and microsatellite analysis [6]. Many results suggested that the sarcomatous component of GS may be derived from glioma cells of GS [6,7]. Our FISH findings also suggested that the sarcomatous component of this tumor may be derived from glioma cells.
The treatment after surgical resection for this case did not differ from that performed on gliomas that are, consisting of radiotherapy and chemotherapy [8,9]. Perry et al. and Salvati M. et al. reported gliosarcoma patients who received multimodality therapy (surgery, radiotherapy and chemotherapy) had a longer overall median survival time compared to patients submitted to surgery only (single-modality) [10]. After the surgical resection of the tumor, the patient underwent a course of radiotherapy (standard dose ranging from 59.4 Gy at 1.8 Gy/day 5 times/week). Adjuvant temozolomide therapy, 150 mg/m2 per day 5 days per month, was administered in 5 cycles. No recurrence was noted after radiotherapy and chemotherapy until the last follow-up (at the time of manuscripting, already 15 months had passed after surgery) and the long-time follow-up is still in progress and has been cared for. It is worth mentioning that this case had at least already showed a more prolonged survival time than that of some gliosarcoma patients.
To our knowledge, this is a new case of mixed granular cell astrocytoma and fibrosarcoma, which pathologists should be aware of. A longer follow-up and multimodality therapy could be of help to patients with this new type of tumor.
Acknowledgements
This work was supported by the Open Research Fund of the Beijing Key Lab of Epilepsy Research (Grant No. 2013DXBL03), Joint Fund for basic and clinical research cooperation project of Capital Medical University (Grant No. 12JL87), Capital health research and development of special (Grant No. 2011-1020-01), Beijing Municipal Natural Science Foundation (Grant No. 7122088) and Beijing Postdoctoral Research Foundation (Grant No. 2013ZZ-22).
Disclosure of conflict of interest
None.
References
- 1.Sahn EE, Dunlavey ES, Parsons JL. Multiple cutaneous granular cell tumors in a child with possible neurofibromatosis. J Am Acad Dermatol. 1997;36:327–330. doi: 10.1016/s0190-9622(97)80410-0. [DOI] [PubMed] [Google Scholar]
- 2.Wierzba-Bobrowicz T, Lewandowska E, Matyja E, Dziduszko J, Koszewski W, Stępień T, Błażejewska-Hyżorek B. Granular cell astrocytoma: A case report with immunohistochemical and ultrastructural characterization. Folia Neuropathol. 2008;46:286–293. [PubMed] [Google Scholar]
- 3.Scartozzi M, Bearzi I, Mandolesi A, Pierantoni C, Loupakis F, Zaniboni A, Negri F, Quadri A, Zorzi F, Galizia E, Berardi R, Biscotti T, Labianca R, Masi G, Falcone A, Cascinu S. Epidermal growth factor receptor (EGFR) gene copy number (GCN) correlates with clinical activity of irinotecan-cetuximab in K-RAS wild-type colorectal cancer: a fluorescence in situ (FISH) and chromogenic in situ hybridization (CISH) analysis. BMC Cancer. 2009;9:303. doi: 10.1186/1471-2407-9-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castellano-Sanchez AA, Ohgaki H, Yokoo H, Scheithauer BW, Burger PC, Hamilton RL, Finkelstein SD, Brat DJ. Granular cell astrocytomas show a high frequency of allelic loss but are not a genetically defined subset. Brain Pathol. 2003;13:185–194. doi: 10.1111/j.1750-3639.2003.tb00018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biernat W, Aguzzi A, Sure U, Grant JW, Kleihues P, Hegi ME. Identical mutations of the p53 tumor supressor gene in the gliomatous and the sarcomatous components suggests a common origin from glial cells. J Neuropathol Exp Neurol. 1995;54:651–656. doi: 10.1097/00005072-199509000-00006. [DOI] [PubMed] [Google Scholar]
- 6.Actor B, Cobbers JMJL, Buschges R, Wolter M, Knobbe CB, Reifenberger G, Weber RG. Comprehensive analysis of genomic alterations in gliosarcoma and its two tissue components. Genes Chromosomes Cancer. 2002;34:416–427. doi: 10.1002/gcc.10087. [DOI] [PubMed] [Google Scholar]
- 7.Machuca TN, Prevedello DM, Pope LZ, Haratz SS, Araújo JC, Bleggi Torres LF. Gliosarcoma: Report of four cases with immunohistochemical findings. Arq Neuropsiquiatr. 2004;62:608–612. doi: 10.1590/s0004-282x2004000400008. [DOI] [PubMed] [Google Scholar]
- 8.Lutterbacha J, Guttenberger R, Pagenstecher A. Gliosarcoma: a clinical study. Radiother Oncol. 2001;61:57–64. doi: 10.1016/s0167-8140(01)00415-7. [DOI] [PubMed] [Google Scholar]
- 9.Han SJ, Yang I, Tihan T, Prados MD, Parsa AT. Primary gliosarcoma: key clinical and pathologic distinctions from glioblastoma with implications as a unique oncologic entity. J Neurooncol. 2010;96:313–320. doi: 10.1007/s11060-009-9973-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salvati M, Caroli E, Raco A, Giangaspero F, Delfini R, Ferrante L. Gliosarcomas: analysis of 11 cases do two subtypes exist? J Neurooncol. 2005;74:59–63. doi: 10.1007/s11060-004-5949-8. [DOI] [PubMed] [Google Scholar]