

Abstract
Acromelic frontonasal dysostosis (AFND) is a rare disorder characterized by distinct craniofacial, brain, and limb malformations, including frontonasal dysplasia, interhemispheric lipoma, agenesis of the corpus callosum, tibial hemimelia, preaxial polydactyly of the feet, and intellectual disability. Exome sequencing of one trio and two unrelated probands revealed the same heterozygous variant (c.3487C>T [p. Arg1163Trp]) in a highly conserved protein domain of ZSWIM6; this variant has not been seen in the 1000 Genomes data, dbSNP, or the Exome Sequencing Project. Sanger validation of the three trios confirmed that the variant was de novo and was also present in a fourth isolated proband. In situ hybridization of early zebrafish embryos at 24 hr postfertilization (hpf) demonstrated telencephalic expression of zswim6 and onset of midbrain, hindbrain, and retinal expression at 48 hpf. Immunohistochemistry of later-stage mouse embryos demonstrated tissue-specific expression in the derivatives of all three germ layers. qRT-PCR expression analysis of osteoblast and fibroblast cell lines available from two probands was suggestive of Hedgehog pathway activation, indicating that the ZSWIM6 mutation associated with AFND may lead to the craniofacial, brain and limb malformations through the disruption of Hedgehog signaling.
Main Text
Over a 20 year period, four children with classic features of acromelic frontonasal dysostosis (AFND [MIM 603671]) sought care at our center and were enrolled in this study after the institutional review board (Seattle Children’s Hospital) had given approval and all participants had provided written consent, including permission to include images of affected individuals in this manuscript. Each child presented with neurocognitive and motor delays, severe symmetric frontonasal dysplasia associated with median cleft face, carp-shaped mouth, widely spaced nasal alae, hypertelorbitism, variable parietal foramina, interhemispheric lipoma, and unilateral or bilateral tibial hemimelia with preaxial polydactyly. Other variable features included periventricular nodular heterotopia, aplastic or hypoplastic corpus callosum, absent olfactory bulbs, vertical clivus, patellar hypoplasia, hypopituitarism, and cryptorchidism (Figure 1; see also Table S1 in the Supplemental Data available with this article online). The neurocognitive delays were severe, and the affected adults have not been able to live independently. Severe upper airway obstruction and facial malformation required tracheostomy and gastrostomy in three of the four probands, and one proband has a seizure disorder. Although each case has variable structural and functional features, the combination of preaxial polydactyly, tibial hemimelia, and frontonasal dysplasia makes AFND a highly recognizable syndrome.1–3
Figure 1.
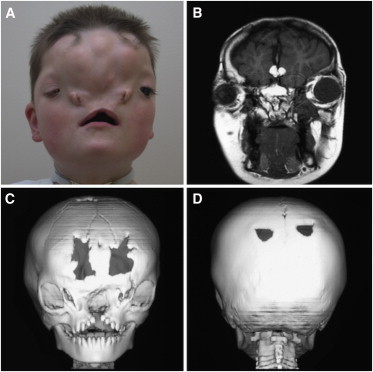
Classic Craniofacial Phenotype Represented by Individual 1
(A) Anteroposterior view of the facial features of individual 1. Note the severe frontonasal dysplasia with ocular hypertelorism, widely spaced nasal alae remnants, and carp-shaped mouth.
(B) Coronal MRI demonstrates a large interhemispheric lipoma.
(C and D) Craniofacial CT demonstrates (C) anterior cranium bifidum, an interfrontal bone, a true median cleft of the maxilla, complete disruption of nasal structures, and (D) symmetric parietal foramina.
The mode of inheritance of AFND was previously unknown; however, there was evidence in the case of individual 1 to support a possible vertical transmission because the proband’s father had a mild phenotype consisting of broad nasal tip, columella, and hypertelorism, suggesting an autosomal-dominant mutation with variable expressivity, dramatically reduced penetrance, and/or possible germline mosaicism.2 For individuals 2 and 3, there was no family history of AFND and no reported consanguinity, suggesting a plausible de novo model of inheritance versus autosomal-recessive inheritance. Individual 4 was adopted, and there is no phenotype information available on the biological parents. For 15 years, it has been thought that the AFND phenotype was consistent with activation of the Hedgehog pathway;1–3 however, candidate-gene screening studies that focused on Sonic Hedgehog (SHH [MIM 600725]) pathway members (i.e., GLI3 [MIM 165240], IHH [MIM 600726], and WNT10a [MIM 606268]) were unsuccessful in determining the causal mutation for AFND.1
In an effort to elucidate the causal mutation, we undertook a broader approach by exome sequencing because this technique has been successful in determining causal variants.4,5 We conducted whole-exome sequencing (WES) on one trio with a mildly affected father and on two unrelated probands (Figure S1). In brief, 1 μg genomic DNA was subjected to a series of shotgun library construction steps, including fragmentation through acoustic sonication (Covaris), end-polishing (NEBNext End Repair Module), A-tailing (NEBNext dA-Tailing Module) and ligation of 8 bp barcoded sequencing adaptors (Enzymatics Ultrapure T4 Ligase). Prior to exome capture, the library was PCR amplified (BioRad iProof). We hybridized 1 μg barcoded shotgun library to capture probes targeting 64 Mb of coding exons (Roche Nimblegen SeqCap EZ Human Exome Library v.3.0) per the manufacturer’s protocol, except that we added custom blockers complementary to the full length of the flanking adaptor and barcodes. Enriched libraries were amplified by PCR before sequencing was performed (BioRad iProof). Library quality was determined from molecular-weight distribution and sample concentration (Agilent Bioanalyzer). Pooled, barcoded libraries were sequenced via paired-end 50 bp reads with an 8 bp barcode read on Illumina HiSeq sequencers. Demultiplexed BAM files were aligned to a human reference (hg19) with the Burrows-Wheeler Aligner (0.6.1). All aligned read data were subjected to (1) removal of duplicate reads (Picard), (2) indel realignment with the GATK IndelRealigner, and (3) base quality recalibration with GATK Recalibration. Variant detection and genotyping were performed with the UnifiedGenotyper (UG) tool from GATK (refv1.6).6,7 Variant data for each sample were formatted as a variant call format (v.4.0)8 and flagged with the filtration walker (GATK) to mark sites that were of lower quality and were potential false positives (e.g., they had quality scores ≤ 50, allelic imbalance ≥ 0.75, long homopolymer runs (> 3), and/or low quality by depth (QD) < 5). Variants were annotated with the SeattleSeq134 Annotation Server.
Our initial analysis plan focused on finding variants that supported a dominant model, the assumption being that the father of individual 1 was affected but with a mild phenotype. We did not find any impactful variants that were present in the two unaffected cases and fit this model of inheritance. We explored other models of inheritance and, by using custom Perl scripts trained to detect de novo variants in probands from exome trio data, we detected one de novo variant in individual 1. Variants present in the 1000 Genomes or the Exome Sequencing Project (ESP) data were excluded, as were intergenic variants and variants that were flagged as low quality or potential false positives (quality scores ≤ 30, long homopolymer runs > 5, low quality by depth < 5, within a cluster of SNPs). The remaining variant at position chr5:60839983 (build hg19) corresponds to ZSWIM6, and an identical mutation was also observed in individuals 2 and 3. Sanger sequencing of trios confirmed a de novo mutation in individuals 1, 2, and 3. Sanger sequencing also confirmed that individual 4 had the variant, albeit at a 60:40 ratio of wild-type to mutant allele, which is suggestive of mosaicism (Figure 2A). We could not establish its de novo status because DNA was not available from the biological parents.
Figure 2.
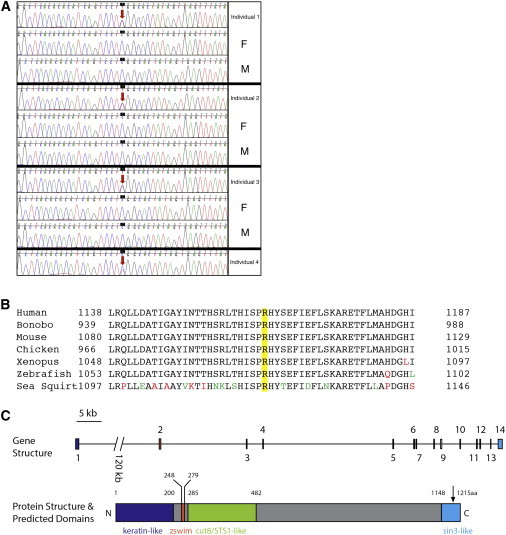
Sanger Validation, Evolutionary Comparative Analysis, Genomic Structure, and Predicted Protein Regions
(A) Sanger sequencing of ZSWIM6 confirms the exon 14 c.3487C>T mutation that leads to the amino acid substitution p.Arg1163Trp in all four cases. The CADD score for this substitution is 22.9, suggesting that it has moderate impact on protein function.
(B) Alignments of amino acids spanning 25 residues on either side of p.Arg1163Trp (yellow) in diverse species, from sea squirts (XP_002126311) to Homo sapiens (NP_065979.1), show that this region is highly conserved. Divergent residues are marked in red, and conservative substitutions are marked in green.
(C) ZSWIM6 is composed of 14 exons that encode the protein, and only one transcript has been detected in vivo. The de novo substitution p.Arg1163Trp is indicated by the black arrow.
The variant c.3487C>T is located within a CpG dinucleotide in ZSWIM6 (RefSeq NM_020928.1) and is predicted to cause a nonsynonymous coding change (p.Arg1163Trp) in this 1216 aa protein. This variant has a GERP score of 4.46 and a CADD score of 22.9, suggesting that it is of moderate impact when mutated. It is in a highly conserved residue among very diverse species, including bonobo (XP_003827517), mouse (NP_663431), chicken (XP_004937320), Xenopus (XP_002934254), zebrafish (NP_001129959), and sea squirt (XP_002126311; Figure 2B). In addition, the substituted arginine residue at position 1163 and 12 neighboring amino acids are conserved among ZSWIM6 family members ZSWIM4 (NP_075560) and ZSWIM5 (NP_065934; Figure S2).
ZSWIM6 encodes a 133.5 kDa protein containing a zinc finger SWIM domain. Proteins with SWIM domains have been identified in a diverse number of species, from bacteria to eukaryotes, and are predicted to have DNA binding and protein-protein interacting properties, but little has been elucidated about their function in vivo.9 In an effort to understand the potential impact of the p.Arg1163Trp substitution, we conducted detailed in silico structural modeling of amino acid substitutions in ZSWIM6. Sequence analysis of ZSWIM6 predicts an all-α-helical structure with novel fold topology and identifies >75% conservation across 97 orthologous proteins from residues 269–1215 (Table S2). One poly-alanine and two poly-glycine repeats occur in the N-terminal 200 residues of the protein, for which a disordered or extended structure is predicted; low-level similarity to the head regions of keratins 24 (E-value = 0.024), 16, 15, 6, and 3 suggest that this region plays a physiologic role in initiating extended polymeric helical structures. The protein’s name is derived from residues 248–279, matching the stereotypical pattern of the SWIM proteins (containing SWI2/SNF2 and MuDR-type zinc fingers), which are thought to carry out protein-protein and protein-DNA interactions.9 ZSWIM6 residues 285–482 align (E-value = 0.74) to cut8/STS1 (Protein Databank identifier 3q5w), which targets proteasomes to the nucleus. Residues 453-1147 show similarity only to paralogs ZSWIM5 (74% identity, 97% alignment) and ZSWIM4 (64% identity, 81% alignment). Residues 1148–1215 show similarity to structurally defined portions of four paired amphipathic helix Sin3 proteins (Protein DataBank identifiers 2czy, 2cr7, 1e91, and 1g1e), enabling atomic resolution modeling (Figure 3A). Sin3 proteins interact with RE1-silencing transcription factor (REST, also known as neuron restrictive silencer factor, or NRSF) proteins, which repress expression of neuron-specific genes in nonneural cells and neuronal progenitors.10–12
Figure 3.
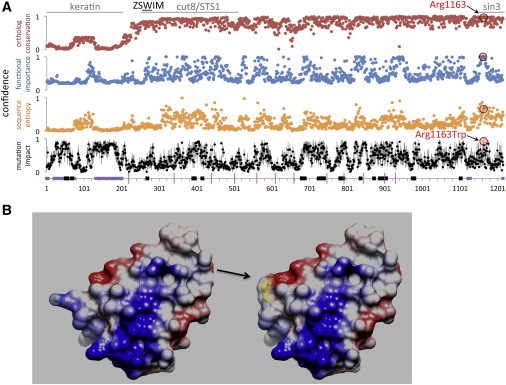
In Silico Protein Modeling
(A) One-dimensional analyses of the predicted functional relevance of protein domains in ZSWIM6. Arginine 1163 occurs in a highly conserved domain from 1148–1215 with similarity to Sin3. Substitution to tryptophan is predicted to have severe effects on protein function. Regions with detectable similarity to non-ZSWIM proteins are denoted at the top. One-dimensional analyses are plotted from the ZSWIM6 N terminus (residue 1, left) to C terminus (1215, right). Plots are shown for percent identity among 97 orthologous proteins (red), predicted functional importance (blue, measured by MFS), relative sequence entropy (orange, measured by HMMRE) and predicted impact of substitution (measured by HUSCY; the mean and standard deviation for all possible single-nucleotide variations are shown in black and gray). At bottom, predicted disordered regions (as measured by DISpro) are shown in purple, domain boundaries (determined by DOMpro) in black, and exon borders as pink lines. Scores for p.Arg1163 and p.Arg1163Trp are highlighted in red and black circles.
(B) Protein modeling of amino acid residues 1148–1215 of the wild-type (left) and mutant (right) shows that the p.Arg1163Trp substitution (arrow) dramatically alters the structure and hydrophobicity of the protein.
Arginine 1163 occurs in the most evolutionarily conserved part of the protein (from mammals to invertebrates)—specifically, in a sequence within residues 1154–1167 (SRLTHISPRHYSEF), which are conserved in ZSWIM paralogs ZSWIM4 and ZSWIM5 and have predicted functional importance (measured by a meta-functional signature, or MFS)13 >5 SD above the mean (z score), suggesting a direct role for this residue in molecular interactions (Figure 3A). Determination of relative sequence entropy (performed with HMMRE),14 disorder analysis (performed with DISpro),15 and domain boundary prediction (performed with DOMpro)16 suggest that this region occurs within a well-defined globular domain from residues 1129–1215 that would maintain biochemical function for in vitro assessment (Figure 3A). A high predicted impact of substitution (as predicted by hereditarily unfit SNV computational yardstick, or HUSCY)17 for p.Arg1163Trp (z score = 4.3) suggests interactions with other functional residues, and the change to tryptophan is the most disruptive substitution that could occur at this position (Figure 3B). The atomic-resolution model of ZSWIM6 residues 1148–1215 predicts that the p.Arg1163 side chain resides on the protein surface between a nonrandom display of negative and positive charges (Figure 3B), most likely patterned for protein-protein interactions that would be severely disrupted by the change to tryptophan.
To gain further insight into the expression and function of zswim6, we utilized zebrafish and mouse models. The zebrafish genome contains a single ortholog of ZSWIM6.18 Zebrafish zswim6 is highly conserved with human ZSWIM6 (Figure 2B). To examine zebrafish zswim6 expression, we cloned a zebrafish zswim6 cDNA product and performed fluorescent RNA in situ hybridization.19 We used Dlx2a and zswim6 cDNA probes for in situ hybridization and dlx2a as a reference for expression pattern.
Zswim6 shows a low level of ubiquitous expression in early embryos, consistent with RT-PCR experiments (data not shown), until 24 hr postfertilization (hpf), when it exhibits increased, localized expression domains in the telencephalon and in the midbrain (Figures 4A and 4B). At 48 hpf, telencephalic expression of zswim6 persisted; however, there was increased expression in the midbrain, hindbrain, and retina (Figures 4C and 4D). This expression pattern is consistent with domains seen in mice and is where zswim6 would be predicted to function on the basis of the human phenotype when this gene is mutated.21
Figure 4.
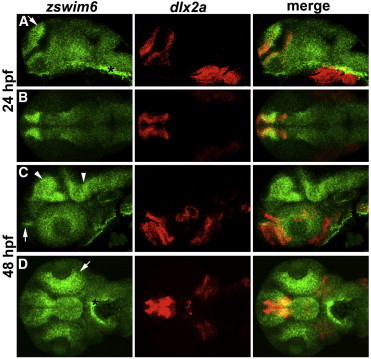
Zswim6 RNA In Situ Staining in Zebrafish
Zebrafish zswim6 expression in the embryonic brain. The NCBI zebrafish zswim6 (RefSeq accession number NM_001136487.1) was used for creation of a zswim6 cDNA clone from 24 hr wild-type cDNA; PCR primers zswim6 F 5′-GCTTTTCCTCTCCGTATCCCG-3′ and zswim6 R 5′-GTATCCGCCCTGAGACAGGA-3′ were used. Zebrafish RNA in situ hybridizations were performed as previously described.19
(A–D) Fluorescent RNA in situ hybridization patterns for zswim6 (green) and dlx2a (red) are shown along with their overlap (merge) at 24 hpf (A and B) and 48 hpf (C and D). Images are shown in lateral view (A and C) and dorsal view (B and D); anterior is to the left. At 24 hpf, zswim6 shows enhanced expression in the dorsal telencephalon (A, arrow). At 48 hpf, zswim6 shows enhanced expression in a telencephalic domain that borders dlx2a expression in the subpallium (C, arrow),20 in the midbrain and hindbrain (C, arrowheads), and in the retina (D, arrow). An asterisk (∗) indicates yolk background stain.
For immunohistochemistry (IHC), we used staged formalin-fixed Swiss Webster mouse embryos and P0 neonatal tissues. IHC of mid-gestation (embryonic day 13.5 [E13.5]) and neonatal mice (P0) demonstrated diffuse protein localization in the central nervous system (data not shown). The most discrete domains of protein localization were at E13.5 in the lens fiber cells (Figures 5A and 5B, arrow) and at P0 in the stereocilia of the outer hair cells of the cochlea (Figure 5C, arrow), a subpopulation of cells in the external root sheath of the hair follicle (Figure 5D, arrow), ameloblasts and odontoblasts (Figure 5E, arrows), and a subset of skeletal-muscle cells (Figure 5F, arrows).
Figure 5.
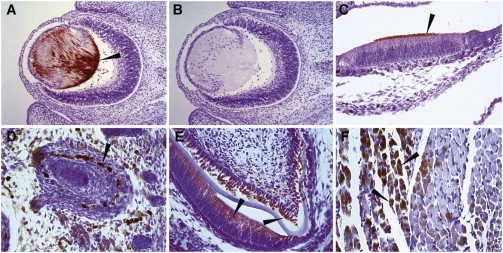
ZSWIM6 Localization during Mouse Development
(A) Immunohistochemistry demonstrating discrete localization of ZSWIM6 in fiber cells of the developing lens of an E13.5 mouse embryo (arrow); polyclonal rabbit anti-human ZSWIM6 antibody (ab122301, Abcam) was used at a dilution of 1:100 and incubated on the sections overnight at 4°C. Immunoreactivity was detected with ImmPACT DAB (SK-4105, Vector Labs), followed by counterstaining with Hematoxylin QS (H-3404, Vector Labs). Slides were dehydrated, mounted in Permount media, and photographed with a Leica DM 4000B digital microscope with Leica Application Suite v 4.3.0 software.
(B) Neighboring-section control of (A) without primary antibody. Postnatal day 0 mouse expression of ZSWIM6 in stereocilia of the outer hair cells of the cochlea (C, arrow), subpopulation of cells in the outer root sheath of the hair follicle (D, arrow), odontoblasts and ameloblasts of the tooth (E, arrows), and variable expression in skeletal muscle (F, arrows) are shown.
Although the molecular underpinnings of AFND are unknown, Vargas et al. (1998)22 speculated that the disorder could be caused by a perturbation of the sonic hedgehog (SHH) pathway members as the clinical manifestations are similar to mouse models. Doublefoot (Dbf) mice have median cleft face, preaxial polydactyly and tibial hemimelia associated with dysregulation of Indian hedghog homolog (IHH) signaling and altered Gli3 processing.23–25 Furthermore, Gli3XT mice exhibit craniofacial defects and preaxial polydactyly, further implicating the SHH pathway because Gli3XT is a zinc-finger DNA-binding transcription factor that mediates downstream SHH signaling.26 Preliminary qRT-PCR experiments performed with a limited number of available cell lines from our cases supports the possibility that activation of the hedgehog pathway leads to the malformations seen in AFND. (Figure S3). In addition, the DECIPHER27 database includes multiple individuals who have heterozygous deletions spanning ZSWIM6 but who do not have a phenotype similar to AFND, suggesting that the c.3487C>T mutation confers gain-of-function.
In summary, we utilized whole-exome sequencing to identify an identical de novo mutation in ZSWIM6 (c.3487C>T) in three unrelated probands (individuals 1–3) and one isolated proband (individual 4), consistent with a molecular cause of AFND. The variable phenotypic expression seen in our four probands (particularly the milder phenotype in individual 4, as described in Table S1) suggests that mosaicism or other gene modifiers are involved in the phenotype. Although preliminary, our qRT-PCR data support the previous hypothesis that AFND is caused by dysregulation of the hedgehog pathway. Identification of an identical alteration in a highly conserved domain suggests a critical role for this domain in embryonic development. The lack of information on the function of ZSWIM6 and other ZSWIM family members suggests that this discovery might introduce more candidates for conditions that include frontonasal dysplasia, preaxial polydactyly, and tibial hemimelia, among other features of AFND. Experimental model systems will be critical for elucidating the function of ZSWIM6 and the developmental pathways impacted by this mutation.
Acknowledgments
We would like to thank the participating families and the following people for their participation and support in developing the manuscript: Colleen Davis, Stephanie Krauter, and Jason Underwood for editorial assistance. Our work is supported in part by grants from National Human Genome Research Institute and National Heart, Lung and Blood Institute of the National Institutes of Health (1U54HG006493 to M.B., D.N., and J.S.; 1RC2HG005608 to M.B., D.N., and J.S.; and 5R000HG004316 to H.K.T.) and the Jean Renny Endowment for Craniofacial Medicine (M.L.C.). Brigham Mecham is founder and CEO of Trialomics.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
BLINK, http://www.ncbi.nlm.nih.gov/sutils/blink.cgi?mode=query
DECIPHER, https://decipher.sanger.ac.uk/
Ensembl, http://www.ensembl.org/
Exome Variant Server, http://evs.gs.washington.edu/EVS/
FASTX-Toolkit: http://hannonlab.cshl.edu/fastx_toolkit/
NHLBI-Exome Sequencing Project, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PCR Data Analysis, http://pcrdataanalysis.sabiosciences.com/pcr/arrayanalysis.php
Picard tools, http://picard.sourceforge.net/
QIAGEN RT,2 http://www.qiagen.com/products/catalog/assay-technologies/real-time-pcr-and-rt-pcr-reagents/rt2-profiler-pcr-arrays?catno=PAHS-078Z
SAMtools, http://samtools.sourceforge.net/
SeattleSeq, http://snp.gs.washington.edu/SeattleSeqAnnotation138/
References
- 1.Slaney S.F., Goodman F.R., Eilers-Walsman B.L., Hall B.D., Williams D.K., Young I.D., Hayward R.D., Jones B.M., Christianson A.L., Winter R.M. Acromelic frontonasal dysostosis. Am. J. Med. Genet. 1999;83:109–116. [PubMed] [Google Scholar]
- 2.Hing A.V., Syed N., Cunningham M.L. Familial acromelic frontonasal dysostosis: Autosomal dominant inheritance with reduced penetrance. Am. J. Med. Genet. A. 2004;128A:374–382. doi: 10.1002/ajmg.a.30091. [DOI] [PubMed] [Google Scholar]
- 3.Toriello H.V., Radecki L.L., Sharda J., Looyenga D., Mann R., Opitz J.M., Reynolds J.F. Frontonasal “dysplasia,” cerebral anomalies, and polydactyly: Report of a new syndrome and discussion from a developmental field perspective. Am. J. Med. Genet. Suppl. 1986;2:89–96. doi: 10.1002/ajmg.1320250613. [DOI] [PubMed] [Google Scholar]
- 4.Ng S.B., Bigham A.W., Buckingham K.J., Hannibal M.C., McMillin M.J., Gildersleeve H.I., Beck A.E., Tabor H.K., Cooper G.M., Mefford H.C. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bamshad M.J., Ng S.B., Bigham A.W., Tabor H.K., Emond M.J., Nickerson D.A., Shendure J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011;12:745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 6.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Danecek P., Auton A., Abecasis G., Albers C.A., Banks E., DePristo M.A., Handsaker R.E., Lunter G., Marth G.T., Sherry S.T., 1000 Genomes Project Analysis Group The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Makarova K.S., Aravind L., Koonin E.V. SWIM, a novel Zn-chelating domain present in bacteria, archaea and eukaryotes. Trends Biochem. Sci. 2002;27:384–386. doi: 10.1016/s0968-0004(02)02140-0. [DOI] [PubMed] [Google Scholar]
- 10.Chen Z.-F., Paquette A.J., Anderson D.J. NRSF/REST is required in vivo for repression of multiple neuronal target genes during embryogenesis. Nat. Genet. 1998;20:136–142. doi: 10.1038/2431. [DOI] [PubMed] [Google Scholar]
- 11.Chong J.A., Tapia-Ramírez J., Kim S., Toledo-Aral J.J., Zheng Y., Boutros M.C., Altshuller Y.M., Frohman M.A., Kraner S.D., Mandel G. REST: A mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell. 1995;80:949–957. doi: 10.1016/0092-8674(95)90298-8. [DOI] [PubMed] [Google Scholar]
- 12.Schoenherr C.J., Anderson D.J. The neuron-restrictive silencer factor (NRSF): A coordinate repressor of multiple neuron-specific genes. Science. 1995;267:1360–1363. doi: 10.1126/science.7871435. [DOI] [PubMed] [Google Scholar]
- 13.Wang K., Horst J.A., Cheng G., Nickle D.C., Samudrala R. Protein meta-functional signatures from combining sequence, structure, evolution, and amino acid property information. PLoS Comput. Biol. 2008;4:e1000181. doi: 10.1371/journal.pcbi.1000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang K., Samudrala R. Incorporating background frequency improves entropy-based residue conservation measures. BMC Bioinformatics. 2006;7:385. doi: 10.1186/1471-2105-7-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng J., Sweredoski M.J., Baldi P. Accurate prediction of protein disordered regions by mining protein structure data. Data Min. Knowl. Discov. 2005;11:213–222. [Google Scholar]
- 16.Cheng J., Sweredoski M.J., Baldi P. DOMpro: Protein domain prediction using profiles, secondary structure, relative solvent accessibility, and recursive neural networks. Data Min. Knowl. Discov. 2006;13:1–10. [Google Scholar]
- 17.Horst J.A., Wang K., Horst O.V., Cunningham M.L., Samudrala R. Disease risk of missense mutations using structural inference from predicted function. Curr. Protein Pept. Sci. 2010;11:573–588. doi: 10.2174/138920310794109139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bradford Y., Conlin T., Dunn N., Fashena D., Frazer K., Howe D.G., Knight J., Mani P., Martin R., Moxon S.A. ZFIN: enhancements and updates to the Zebrafish Model Organism Database. Nucleic Acids Res. 2011;39(Database issue):D822–D829. doi: 10.1093/nar/gkq1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yao Z., Farr G.H., 3rd, Tapscott S.J., Maves L. Pbx and Prdm1a transcription factors differentially regulate subsets of the fast skeletal muscle program in zebrafish. Biol. Open. 2013;2:546–555. doi: 10.1242/bio.20133921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.MacDonald W.A., Menon D., Bartlett N.J., Sperry G.E., Rasheva V., Meller V., Lloyd V.K. The Drosophila homolog of the mammalian imprint regulator, CTCF, maintains the maternal genomic imprint in Drosophila melanogaster. BMC Biol. 2010;8:105. doi: 10.1186/1741-7007-8-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith C.M., Finger J.H., Hayamizu T.F., McCright I.J., Xu J., Berghout J., Campbell J., Corbani L.E., Forthofer K.L., Frost P.J. The mouse Gene Expression Database (GXD): 2014 update. Nucleic Acids Res. 2014;42(Database issue):D818–D824. doi: 10.1093/nar/gkt954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vargas F.R., Roessler E., Gaudenz K., Belloni E., Whitehead A.S., Kirke P.N., Mills J.L., Hooper G., Stevenson R.E., Cordeiro I. Analysis of the human Sonic Hedgehog coding and promoter regions in sacral agenesis, triphalangeal thumb, and mirror polydactyly. Hum. Genet. 1998;102:387–392. doi: 10.1007/s004390050709. [DOI] [PubMed] [Google Scholar]
- 23.Hayes C., Lyon M.F., Morriss-Kay G.M. Morphogenesis of Doublefoot (Dbf), a mouse mutant with polydactyly and craniofacial defects. J. Anat. 1998;193:81–91. doi: 10.1046/j.1469-7580.1998.19310081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Babbs C., Furniss D., Morriss-Kay G.M., Wilkie A.O. Polydactyly in the mouse mutant Doublefoot involves altered Gli3 processing and is caused by a large deletion in cis to Indian hedgehog. Mech. Dev. 2008;125:517–526. doi: 10.1016/j.mod.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Y., Guillot P., Boyd Y., Lyon M.F., McMahon A.P. Evidence that preaxial polydactyly in the Doublefoot mutant is due to ectopic Indian Hedgehog signaling. Development. 1998;125:3123–3132. doi: 10.1242/dev.125.16.3123. [DOI] [PubMed] [Google Scholar]
- 26.Wang C., Rüther U., Wang B. The Shh-independent activator function of the full-length Gli3 protein and its role in vertebrate limb digit patterning. Dev. Biol. 2007;305:460–469. doi: 10.1016/j.ydbio.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Firth H.V., Richards S.M., Bevan A.P., Clayton S., Corpas M., Rajan D., Van Vooren S., Moreau Y., Pettett R.M., Carter N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009;84:524–533. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.