

Abstract
Cerebellar dysplasia with cysts (CDC) is an imaging finding typically seen in combination with cobblestone cortex and congenital muscular dystrophy in individuals with dystroglycanopathies. More recently, CDC was reported in seven children without neuromuscular involvement (Poretti-Boltshauser syndrome). Using a combination of homozygosity mapping and whole-exome sequencing, we identified biallelic mutations in LAMA1 as the cause of CDC in seven affected individuals (from five families) independent from those included in the phenotypic description of Poretti-Boltshauser syndrome. Most of these individuals also have high myopia, and some have retinal dystrophy and patchy increased T2-weighted fluid-attenuated inversion recovery (T2/FLAIR) signal in cortical white matter. In one additional family, we identified two siblings who have truncating LAMA1 mutations in combination with retinal dystrophy and mild cerebellar dysplasia without cysts, indicating that cysts are not an obligate feature associated with loss of LAMA1 function. This work expands the phenotypic spectrum associated with the lamininopathy disorders and highlights the tissue-specific roles played by different laminin-encoding genes.
Main Text
Cerebellar dysplasia is a neuroimaging finding that describes abnormalities of both the cerebellar cortex and white matter and is associated with variable neurodevelopmental outcome.1–4 Cerebellar dysplasia is presumed to arise through genetic or acquired developmental defects that affect cerebellar foliation and fissure formation.5 Although in most cases the etiology of generalized cerebellar hemisphere dysplasia is unknown, this imaging finding has been reported in congenital muscular dystrophies (CMDs), most notably POMGNT1-related CMD (MIM 606822),6 and in Chudley-McCullough syndrome (MIM 604213). Although generally rare, cerebellar cysts are frequently identified in individuals with CMD.6 Recently, a distinctive cerebellar dysplasia with cysts (CDC) phenotype was described in seven children (from five families) with ataxia, intellectual disability, and language impairment but without significant muscle disease (Poretti-Boltshauser syndrome).7 Although the nine genes in which loss-of-function mutations have been shown to cause CMD (POMT1 [MIM 607423], POMT2 [MIM 607439], POMGNT1, FKRP [MIM 606596], FKTN [MIM 607440], LARGE [MIM 603590], ISPD [MIM 614631], B3GALNT2 [MIM 610194], and GPR56 [MIM 604110]) were sequenced in these seven children, the genetic cause of CDC was not identified. Here, we report that mutations in LAMA1 (MIM 150320; RefSeq accession number NM_005559.3), encoding laminin subunit α1, cause CDC, and we expand the phenotype to include milder brain-imaging findings and retinal dystrophy.
Initially, we enrolled a series of three CDC-affected individuals from three families under informed consent through human-subjects-research protocols approved by the institutional review boards at the University of Washington and the University of Calgary. The phenotype of these individuals is indistinguishable from that described in Poretti-Boltshauser syndrome.7 To identify the genetic cause of CDC in these individuals, we combined homozygosity mapping and exome sequencing. Genome-wide SNP genotyping was performed with the Affymetrix Genome-wide Human SNP Array 6.0 on peripheral-blood DNA from CA0035 (II-3 in Figure 1B), whose parents are first cousins. Using HomozygosityMapper,8 we identified ten copy-number-neutral homozygosity regions (Figure 1A) spanning >10 Mb, including a 15.9 Mb stretch in chromosomal region 18p11.31–p11.2 between markers rs1940954 and rs2850205.
Figure 1.
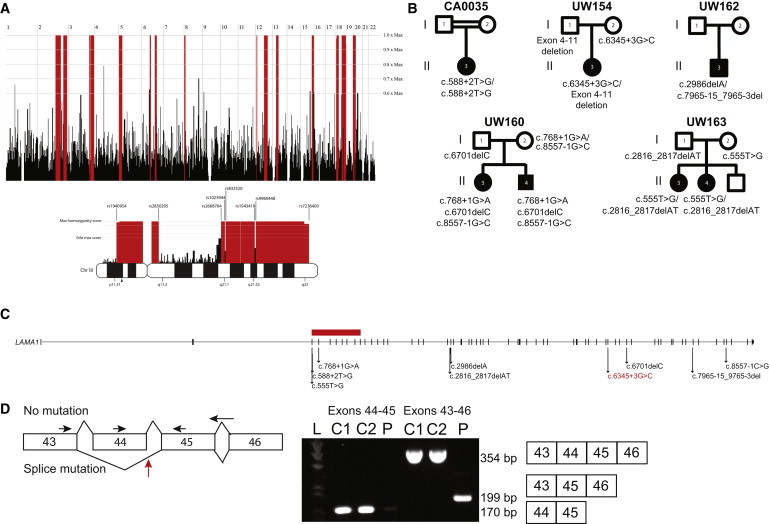
Mutation Identification in Individuals with CDC
(A) Homozygosity mapping in CA0035 identified several regions of homozygosity (red) across the genome. Homozygous regions on chromosome 18 are indicated below the genome-wide mapping. An arrowhead below the chromosome 18 ideogram indicates the location of LAMA1.
(B) Pedigree diagrams showing segregation analysis for five families.
(C) LAMA1 is composed of 63 exons that span chr18: 6,941,743–7,117,813 (UCSC Genome Browser hg19) and encode protein-coding sequence. Arrows indicate the locations of ten different mutations identified in five families. The horizontal red bar represents a deletion spanning LAMA1 exons 4–11.
(D) The splice-site mutation (designated red in C) in UW154-3 results in skipping of exon 45. Total RNA was isolated from fibroblasts with the RNeasy Mini Kit (QIAGEN) and transcribed into cDNA with the SuperScript III First-Strand System and random hexamer primers (LifeTechnologies). RT-PCR was performed under standard conditions with forward primers in exons 43 and 44 and reverse primers in exons 45 and 46 (sequences are available upon request). Primers in exons 44 and 45 generated a 170 bp amplicon in cDNA from two unrelated, unaffected control fibroblasts but minimal product in cDNA from UW154-3. Primers in exons 43 and 46 generated a 354 bp amplicon in control cells and a 199 bp product in UW154-3. Sanger sequencing of this product revealed that exon 43 is spliced to exon 45 in UW154-3 (data not shown). The horizontal arrows indicate primer positions. The red arrow indicates the location of the splice-site mutation in intron 44. “L” indicates the ladder lane, “C1” and “C2” indicate the two control samples, and “P” indicates individual UW154-3. The expected size and exon composition of the respective products are indicated to the right of the gel.
We then performed whole-exome sequencing on peripheral-blood DNA from all three individuals (Table S1, available online), as described previously.9 In brief, genomic DNA was captured with the Agilent SureSelect v.5 exome capture oligonucleotide library and sequenced with paired-end 100 bp reads on Illumina HiSeq 2000. TTN (MIM 188840) and LAMA1 were the only genes found to harbor rare (<5% in 1000 Genomes), deleterious variants in all affected individuals and lie within homozygous regions identified in CA0035 (Table S2 and Figure 1A). Because TTN is a very large gene that frequently harbors variants mainly as a result of its large size, and no individuals with CDC have the dilated or hypertrophic cardiomyopathy that has been associated with TTN mutations, it was excluded from consideration. The LAMA1 variants are not present in 1000 Genomes, the NHLBI Exome Sequencing Project Exome Variant Server (EVS), dbSNP version 137, or an in-house exome database consisting of ∼2,000 samples and are predicted to truncate the protein or result in aberrant splicing by Human Splicing Finder v.3.0.10 CA0035 carries a homozygous canonical splice variant in the donor site for exon 5 of LAMA1 (c.588+2T>G). UW162-3 (II-3 in Figure 1B) carries a heterozygous 13 bp deletion (c.7965−15_7965−3del) that is predicted to alter splicing and a heterozygous 1 bp deletion (c.2988_2989delA) that is predicted to cause a frameshift and premature truncation of the protein (p.Thr996Hisfs28∗). UW154-3 (II-3 in Figure 1B) carries a single heterozygous variant at the third nucleotide of the splice donor site in intron 44 of LAMA1 (c.6345+3G>C). This mutation results in a transcript that is missing exon 45 (Figure 1D) and is predicted to generate a truncated protein (p.Thr2064Ansfs∗2). These variants were confirmed by Sanger sequencing and segregated appropriately in the families (Figure 1B and Figure S1).
Because we identified only a single LAMA1 variant in UW154-3, we hypothesized that a heterozygous deletion in LAMA1 might be present. We used the whole-exome sequencing data to analyze copy-number variants (CNVs) with FishingCNV.11 To identify rare CNVs, we used the same exome sequencing protocol to compare the read depth of the variants in UW154-3 against the distribution of reads in more than 50 control individuals with unrelated genetic disorders. This analysis identified a significant heterozygous LAMA1 deletion variant (p = 4.98 × 10−45) spanning exons 4–11 (chr18: 7,038,808–7,050,935, hg19). There is a long tandem repeat (L1MA9) on the centromeric side, but not on the telomeric side, of this CNV. No deletion at this position has been reported in the Database of Genomic Variants.12 Quantitative PCR confirmed the deletion and found that it was paternally inherited (Figure S2).
In parallel, we performed exome sequencing in a cohort of 40 individuals with a clinical diagnosis of Joubert syndrome (MIM 213300) and in whom targeted sequencing had excluded mutations in all published genes implicated in Joubert and Meckel syndromes (NPHP1 [MIM 607100], AHI1 [MIM 608894], CEP290 [MIM 610142], RPGRIP1L [MIM 610937], TMEM67 [MIM 609884], CC2D2A [MIM 612013], ARL13B [MIM 608922], INPP5E [MIM 613037], OFD1 [MIM 300170], TMEM216 [MIM 613277], CEP41 [MIM 610523], TMEM237 [MIM 614423], TCTN2 [MIM 613846], KIF7 [MIM 611254], TCTN1 [MIM 609863], TMEM138 [MIM 614459], MKS1 [MIM 609883], C5ORF42 [MIM 614571], TMEM231 [MIM 614949], TCTN3 [MIM 613847], CSPP1 [MIM 611654], PDE6D [MIM 602676], IFT172 [MIM 607386], ZNF423 [MIM 604557], TTC21B [MIM 612014], B9D1 [MIM 614144], and B9D2 [MIM 611951]). Joubert syndrome is a recessive neurodevelopmental disorder typically defined by a pathognomonic brain malformation: “the molar-tooth sign,” which consists of cerebellar vermis hypoplasia, thickened, horizontally oriented superior cerebellar peduncles, and sometimes a deep interpeduncular fossa.13 CDC and Joubert syndrome share nonspecific clinical features, including cognitive impairment, hypotonia, ataxia, and abnormal eye movements, but CDC does not manifest with the alternating tachypnea and apnea often seen in Joubert syndrome. Some individuals with Joubert syndrome also develop progressive retinal dystrophy, cystic kidney disease, and/or liver fibrosis that result in significant morbidity and mortality. Surprisingly, exome sequencing in one of the families previously diagnosed with Joubert syndrome revealed in one affected sibling (UW160-3 [II-3 in Figure 1B]) four rare LAMA1 variants, three of which were maternally inherited (c.8557–1G>C [in 0/13,006 EVS chromosomes], c.2657C>T [p.Ala886Val] [in 34/13,006 EVS chromosomes, PolyPhen-2 scores 0.907 and 0.753], and c.768+1G>A [in 0/13,006 EVS chromosomes]) and one of which was paternally inherited (c.6701delC [p.Pro2334Leufs9∗]). Sanger sequencing confirmed that the other affected sibling (UW160-4 [II-4 in Figure 1B]) also carries all four variants. In a second family with two siblings (UW163-3 and UW163-4) previously diagnosed with mild Joubert syndrome, we identified compound-heterozygous mutations (the paternally inherited c.2816_2817delAT [p.Tyr939Leufs27∗] and the maternally inherited c.555T>G [p.Tyr185∗] [in 0/13,006 EVS chromosomes]) in LAMA1 in UW163-3. Sanger sequencing confirmed that the other affected sibling (UW163-4) shares these variants in LAMA1.
We reviewed the clinical and imaging features for the affected individuals from all five families (Table 1). Four individuals showed classic CDC findings, including cerebellar dysplasia with cysts (more severe rostrally and ventrally) and cerebellar vermis hypoplasia. In three, the superior cerebellar peduncles were elevated and splayed, and the fourth ventricle was elongated rostrocaudally and laterally (Figure 2). Minor abnormalities of the brainstem included a short pons (CA0035 and UW154-3), narrow isthmus (UW154-3 and UW160-4), and long midbrain (UW154-3). Although UW160-4 had somewhat long superior cerebellar peduncles, they were not classic for the molar-tooth appearance that defines Joubert syndrome.13 Similarly, UW163-3 and UW163-4 were previously categorized as having mild Joubert syndrome without the classic molar-tooth sign. A few supratentorial abnormalities present on imaging included a single periventricular heterotopia in CA0035 (Figure 2E), partial agenesis of the corpus callosum (a missing rostrum and a markedly thin isthmus and splenium) and partial absence of the septum pellucidum in UW154-3 (Figures 2I and 2J), and patchy increased T2-weighted fluid-attenuated inversion recovery (T2/FLAIR) in CA0035 and UW154-3 (Figures 2E and 2J). Notably, four affected individuals had large eye globes, consistent with high myopia (>−6 diopters). In contrast, UW163-3 and UW163-4 had superior cerebellar dysplasia without cysts (Figure 2L) and mild vermis hypoplasia (Figure 2N). UW163-3 also had patchy increased T2/FLAIR (Figure 2O) and a suprasellar arachnoid cyst, whereas UW163-4 had slightly elevated superior cerebellar peduncles and large globes (data not shown).
Table 1.
Brain MRI Features in Affected Individuals with LAMA1 Mutations
| CA0035 | UW154-3 | UW162-3 | UW160-4 | UW163-3 | UW163-4 | |
|---|---|---|---|---|---|---|
| cDNA mutation 1 | c.588+2T>G | c.6345+3G>C (maternal) | c.7965−15_7965−3del (paternal) | c.6701delC (paternal) | c.2816_2817delAT (paternal) | c.2816_2817delAT (paternal) |
| Protein alteration 1 | canonical splice | splice | splice | p.Pro2334Leufs9∗ | p.Tyr939Leufs27∗ | p.Tyr939Leufs27∗ |
| cDNA mutation 2 | c.588+2T>G | deletion of exons 4–11 (paternal) | c.2988_2989delA (maternal) | c.8557−1G>C, c.768+1G>A (maternal) | c.555T>G (maternal) | c.555T>G (maternal) |
| Protein alteration 2 | canonical splice | NA | p.Pro996Hisfs28∗ | splice, splice | p.Tyr185∗ | p.Thr185∗ |
| Age at MRI | 8 months | 5 and 15 months | 9 months | unknown | 25 months | 5 months |
| Cerebellar dysplasia | yes | yes | yes | yes | superior only | superior only |
| Cerebellar cysts | yes | yes | yes | yes | no | no |
| Vermis hypoplasia | global | global | global | global | inferior only | inferior only |
| SCPs | elevated and splayed | elevated and splayed | normal | elevated and splayed | normal | slightly elevated |
| Fourth ventricle | enlarged | enlarged | enlarged | enlarged | mildly enlarged | normal |
| Brainstem | short pons, thin isthmus | short pons, long midbrain, mildly enlarged tectum | normal | thin isthmus | mass effect from arachnoid cyst | normal |
| Ventricles | normal | moderate VM, partial agenesis of CC and SP | normal | normal | normal | normal |
| Eyes | large | large | large | large | normal | large |
| Increased T2/FLAIR in white matter | patchy increased, periventricular | patchy increased, periventricular | normal | normal | patchy increased, periventricular | normal |
| Other | heterotopia in right lateral ventricle | NA | NA | NA | suprasellar and interpeduncular cyst | NA |
Abbreviations are as follows: CC, corpus callosum; NA, not applicable; SCP, superior cerebellar peduncle; SP, septum pellucidum; and VM, ventricuolmegaly.
Figure 2.
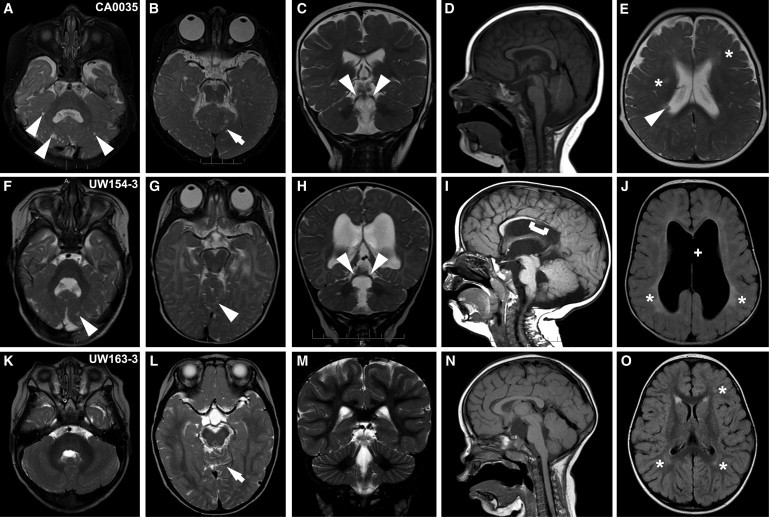
Brain and Eye Imaging Findings in Affected Individuals with LAMA1 Mutations
(A–E) Individual CA0035 has cerebellar cysts (arrowheads in A), superior cerebellar dysplasia (arrow in B) and large globes (B), elevated and splayed superior cerebellar peduncles (arrowheads in C), an enlarged fourth ventricle (A and D), and patchy increased T2/FLAIR (asterisks in E). The periventricular heterotopia (arrowhead in E) is atypical.
(F–J) Similar findings in UW154-3 include cerebellar cysts (arrowhead in F), superior cerebellar dysplasia (arrowhead in G) and large globes (G), elevated and splayed superior cerebellar peduncles (arrowheads in H), an enlarged fourth ventricle (F and I), and patchy increased T2/FLAIR (asterisks in E). In addition, UW154-3 has partial absence of the corpus callosum (bracket in I) and septum pellucidum (plus sign in J).
(K–N) UW163-3 has only superior cerebellar dysplasia (arrow in L) and patchy increased T2/FLAIR (asterisks in O), but no cerebellar cysts (K). The superior cerebellar peduncles (M) and the fourth ventricle (K and N) are normal.
All seven affected individuals had a history of motor delay, three had speech delay, and one was diagnosed with Asperger syndrome, but one was able to graduate from college and live independently as an adult (Table 2). All individuals also had abnormal eye movements, myopia (five with high myopia), and retinal differences, including thinning (UW154-3), atrophy (CA0035 and UW160-4), absent pigment (UW162-3), and overt early-onset retinal dystrophy with visual impairment (UW160-3) and abnormal electroretinography (ERG) (UW163-3 and UW163-4). UW160-3 also had macular heterotopia, and UW160-4 had bilateral cataracts noted at 22 years of age. Creatine kinase levels were not substantially elevated in three individuals, and no other clinical features were observed in more than one individual. The individuals previously diagnosed with Joubert syndrome did not display evidence of cystic kidney disease, liver fibrosis, polydactyly, coloboma, or other clinical features associated with Joubert syndrome.
Table 2.
Clinical Features in Affected Individuals with LAMA1 Mutations
| CA0035 | UW154-3 | UW162-3 | UW160-3 | UW160-4 | UW163-3 | UW163-4 | |
|---|---|---|---|---|---|---|---|
| Ethnicity | Iranian | mixed European | mixed European | mixed European | mixed European | Asian and African American | Asian and African American |
| Sex | female | female | male | female | male | female | female |
| Age | 36 months | 36 months | 25 months | 29 years | 23 years | 5.5 years | 4.5 years |
| OFC (age) | 50th percentile (12 months) | >98th percentile (36 months) | 20th percentile (17 months) | 50th percentile (8 years) | 30th percentile (29 months) | 35th percentile (29 months) | 35th percentile (16 months) |
| Height or length (age) | 90th percentile (12 months) | 4th percentile (36 months) | 60th percentile (17 months) | 155 cm (adult) | 168 cm (adult) | 60th percentile (25 months) | 40th percentile (16 months) |
| Weight (age) | 97th percentile (12 months) | 65th percentile (36 months) | 25th percentile (17 months) | 77 kg (adult) | 93 kg (adult) | 60th percentile (25 months) | 30th percentile (16 months) |
| NDV | moderate motor and speech delay | moderate motor delay (no standing or walking), mild speech delay | motor delay (cruising, but no walking), hypotonia | history of motor delay, normal speech, normal IQ, college graduate, lives independently | history of motor and speech delay, normal IQ, autism spectrum disorder (Asperger), lives with parents | mild motor and speech delay, hypotonia | motor and speech delay, hypotonia |
| Eye movements | strabismus, amblyopia | strabismus, nystagmus | strabismus, nystagmus | strabismus, OMA | strabismus, amblyopia | OMA | OMA, strabismus S/P surgery, nystagmus |
| Retina | atrophic appearing | thinned | absent pigment | lattice and peripheral degeneration, macular heterotopia, increased pigment | atrophic appearing | retinal dysfunction: cones more affected than rods on ERG | chorioretinal atrophy, macular and peripheral involvement, cones worse than rods on ERG |
| Acuity | high myopia (−13.5) | high myopia (−17) | myopia | high myopia (−16.5) | high myopia (−13.5) | Myopia (−3.00) | high myopia (−11.5) |
| CK (ref.) | 177 (0–170 IU/l) | 53 (26–40 IU/l) | 141 (24–204 IU/l) | ND | ND | ND | ND |
| Other | aminoaciduria, consanguinity | one partial seizure (on Keppra), maternally inherited heterozygous 800 kb deletion: 10p12.1–p12.2 | mother on Clomid, normal SNP array | fatty liver on ultrasound, syndactyly in second and third toes | bilateral cataracts at 22 years old, possibly echogenic liver on ultrasound, syndactyly in second and third toes | NA | NA |
Abbreviations are as follows: CK, creatine kinase; ERG, electroretinography; NA, not applicable; ND, not done; NDV, neurodevelopmental features; OMA, oculomotor apraxia; ref., reference; and S/P, status post.
LAMA1-related CDC contributes to the growing list of phenotypes associated with loss of function of laminin-encoding genes. An overlapping spectrum of posterior predominant brain phenotypes are characteristic of mutations in LAMA2 (MIM 156225), LAMB1 (MIM 150240), LAMC1 (MIM 150290), and LAMC3 (MIM 604349) and range from occipital cortical defects to Dandy-Walker malformation.14–17 Recessive LAMA2 mutations are the primary cause of CMD with occasional occipital polymicrogyria and epilepsy as a result of a complete lack of merosin, a component of the skeletal-muscle extracellular matrix protein laminin-2.17 Recessive mutations in LAMB1 and LAMC3 each cause occipital cobblestone malformation, whereas mutations in LAMB1 additionally cause cerebellar vermis and brain stem hypoplasia with variable ocular defects and occasional occipital cephalocele.14,16 Heterozygous LAMC1 mutations cause Dandy-Walker malformation and occipital cephalocele.15 Strikingly, none of these phenotypes includes the cerebellar cysts that are prominent in individuals with LAMA1 mutations.
Laminins are extracellular matrix proteins required for basement membrane assembly and critical for early embryonic development.18 In mice, LAMA1 localizes exclusively in basement membranes, including within the mesenchyme adjacent to the developing cerebellum.19 Mice with homozygous knockout of Lama1 die in the early embryonic period as a result of impaired epiblast differentiation.20 However, Lama1-conditional-knockout mice that overcome this early lethality have motor and coordination deficits and an overproliferation of granule cell precursors and subsequent excessive cell death, leading to mislocalization of granule cells and an overall reduced size of the adult cerebellum.21,22 In addition, the eye phenotypes described in mouse and zebrafish models parallel the findings described in our cohort. Hypomorphic and null Lama1 mutations in mice cause defects in the retinal inner limiting membrane, attenuated ERG, abnormal retinal vasculature, and progressive cell loss from the inner nuclear and ganglion cell layers.23,24 Zebrafish lama1-mutant larvae exhibit attenuated ERG, lens and vasculature abnormalities, shortened and ectopic photoreceptor cells, and axon-guidance defects, whereas adult zebrafish mosaic for lama1 mutations exhibit cataracts, similar to the cataracts in UW160-4.25,26 Further evaluation of the eye findings in individuals with LAMA1 mutations could reveal a distinctive phenotype that will be helpful for diagnosis in the future. Intriguingly, LAMA1 has also been proposed to be the relevant gene in the MYP2 locus associated with high myopia; however, two studies evaluating the association between high myopia and SNPs in LAMA1 yielded conflicting results.27,28
In addition to identifying LAMA1 mutations as the cause of the recently described, distinctive CDC phenotype, this work expands the CDC spectrum to include individuals with retinal dystrophy, mild cerebellar dysplasia, and no cysts. High myopia and large eye globes are also prevalent features. Of note, a homozygous LAMA1 frameshift mutation was identified in siblings (born to consanguineous parents) with a phenotypic description limited to “mild intellectual disability and strabismus,”29 so it is unclear whether these individuals have CDC or whether LAMA1 loss of function can cause isolated intellectual disability. The combination of cerebellar vermis hypoplasia, abnormal superior cerebellar peduncles, and retinal dystrophy could result in misdiagnosis of Joubert syndrome. Although close examination of the retina and brain MRI might appropriately classify most individuals, genetic testing should be the most definitive way to separate the disorders. On the basis of the known mutations, we cannot identify genotype-phenotype correlations, given that the siblings with the mildest brain phenotype (UW163-3 and UW163-4) have a combination of a relatively early stop-gain mutation and a frameshift mutation. Finally, this work expands our understanding of the phenotypic spectrum associated with lamininopathy disorders and highlights the tissue-specific roles played by different laminin-encoding genes.
Acknowledgments
We are grateful to all of the families who elected to participate in this study. This work was selected for study by the Care4Rare (Enhanced Care for Rare Genetic Diseases in Canada) Consortium Gene Discovery Steering Committee: Kym Boycott (lead; University of Ottawa), Alex MacKenzie (co-lead; University of Ottawa), Jacek Majewski (McGill University), Michael Brudno (University of Toronto), Dennis Bulman (University of Ottawa), and David Dyment (University of Ottawa). This work was funded in part by Genome Canada, the Canadian Institutes of Health Research, the Ontario Genomics Institute, the Ontario Research Fund, Genome Quebec, the Children’s Hospital of Eastern Ontario Foundation, Alberta Children’s Hospital Research Institute, and Alberta Innovates Health Solutions. The authors wish to acknowledge the contribution of the high-throughput sequencing platform of the McGill University and Genome Quebec Innovation Centre in Montreal. This work was also supported by the University of Washington Center for Mendelian Genomics (National Human Genome Research Institute award number U54HG006493), the Alberta Children’s Hospital Foundation, University of Washington School of Medicine Department of Pediatrics funds, private donations from families to the University of Washington Hindbrain Malformation Research Program, and the NIH National Institute for Neurological Disorders and Stroke under award number R01NS050375. The opinions and assertions contained herein are the views of the authors and are not to be construed as official or as reflecting the views of the United States Department of Defense.
Contributor Information
A. Micheil Innes, Email: micheil.innes@albertahealthservices.ca.
Dan Doherty, Email: ddoher@uw.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
Database of Genomic Variants (DGV), http://dgv.tcag.ca/dgv/app/home
HomozygosityMapper, http://www.homozygositymapper.org
Human Splicing Finder, http://www.umd.be/HSF3/index.html
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Demaerel P. Abnormalities of cerebellar foliation and fissuration: classification, neurogenetics and clinicoradiological correlations. Neuroradiology. 2002;44:639–646. doi: 10.1007/s00234-002-0783-1. [DOI] [PubMed] [Google Scholar]
- 2.Jissendi-Tchofo P., Pandit F., Soto-Ares G., Vallee L. Neuropsychological evaluation and follow-up of children with cerebellar cortical dysplasia. Dev. Med. Child Neurol. 2011;53:1119–1127. doi: 10.1111/j.1469-8749.2011.04117.x. [DOI] [PubMed] [Google Scholar]
- 3.Poretti A., Boltshauser E. Cerebellar dysplasia. In: Boltshauser E., Schmahmann J., editors. Cerebellar disorders in children. Mac Keith Press; London: 2012. pp. 172–176. [Google Scholar]
- 4.Soto-Ares G., Delmaire C., Deries B., Vallee L., Pruvo J.P. Cerebellar cortical dysplasia: MR findings in a complex entity. AJNR Am. J. Neuroradiol. 2000;21:1511–1519. [PMC free article] [PubMed] [Google Scholar]
- 5.Boddaert N., Desguerre I., Bahi-Buisson N., Romano S., Valayannopoulos V., Saillour Y., Seidenwurm D., Grevent D., Berteloot L., Lebre A.S. Posterior fossa imaging in 158 children with ataxia. J. Neuroradiol. 2010;37:220–230. doi: 10.1016/j.neurad.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 6.Poretti A., Klein A., Boltshauser E. Cerebellar cysts and neuroimaging in congenital muscular dystrophies. In: Boltshauser E., Schmahmann J., editors. Cerebellar disorders in children. Mac Keith Press; London: 2012. pp. 177–183. [Google Scholar]
- 7.Poretti A., Häusler M., von Moers A., Baumgartner B., Zerres K., Klein A., Aiello C., Moro F., Zanni G., Santorelli F.M. Ataxia, intellectual disability, and ocular apraxia with cerebellar cysts: a new disease? Cerebellum. 2014;13:79–88. doi: 10.1007/s12311-013-0521-8. [DOI] [PubMed] [Google Scholar]
- 8.Seelow D., Schuelke M., Hildebrandt F., Nürnberg P. HomozygosityMapper—an interactive approach to homozygosity mapping. Nucleic Acids Res. 2009;37(Web Server issue):W593–W599. doi: 10.1093/nar/gkp369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Srour M., Putorti M.L., Schwartzentruber J., Bolduc V., Shevell M.I., Poulin C., O’Ferrall E., Buhas D., Majewski J., Brais B., FORGE consortium Mutations in riboflavin transporter present with severe sensory loss and deafness in childhood. Muscle Nerve. 2014 doi: 10.1002/mus.24224. Published online February 24, 2014. [DOI] [PubMed] [Google Scholar]
- 10.Desmet F.O., Hamroun D., Lalande M., Collod-Béroud G., Claustres M., Béroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi Y., Majewski J. FishingCNV: a graphical software package for detecting rare copy number variations in exome-sequencing data. Bioinformatics. 2013;29:1461–1462. doi: 10.1093/bioinformatics/btt151. [DOI] [PubMed] [Google Scholar]
- 12.MacDonald J.R., Ziman R., Yuen R.K., Feuk L., Scherer S.W. The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2014;42(Database issue):D986–D992. doi: 10.1093/nar/gkt958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maria B.L., Hoang K.B., Tusa R.J., Mancuso A.A., Hamed L.M., Quisling R.G., Hove M.T., Fennell E.B., Booth-Jones M., Ringdahl D.M. “Joubert syndrome” revisited: key ocular motor signs with magnetic resonance imaging correlation. J. Child Neurol. 1997;12:423–430. doi: 10.1177/088307389701200703. [DOI] [PubMed] [Google Scholar]
- 14.Barak T., Kwan K.Y., Louvi A., Demirbilek V., Saygı S., Tüysüz B., Choi M., Boyacı H., Doerschner K., Zhu Y. Recessive LAMC3 mutations cause malformations of occipital cortical development. Nat. Genet. 2011;43:590–594. doi: 10.1038/ng.836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Darbro B.W., Mahajan V.B., Gakhar L., Skeie J.M., Campbell E., Wu S., Bing X., Millen K.J., Dobyns W.B., Kessler J.A. Mutations in extracellular matrix genes NID1 and LAMC1 cause autosomal dominant Dandy-Walker malformation and occipital cephaloceles. Hum. Mutat. 2013;34:1075–1079. doi: 10.1002/humu.22351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Radmanesh F., Caglayan A.O., Silhavy J.L., Yilmaz C., Cantagrel V., Omar T., Rosti B., Kaymakcalan H., Gabriel S., Li M. Mutations in LAMB1 cause cobblestone brain malformation without muscular or ocular abnormalities. Am. J. Hum. Genet. 2013;92:468–474. doi: 10.1016/j.ajhg.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vigliano P., Dassi P., Di Blasi C., Mora M., Jarre L. LAMA2 stop-codon mutation: merosin-deficient congenital muscular dystrophy with occipital polymicrogyria, epilepsy and psychomotor regression. Eur. J. Paediatr. Neurol. 2009;13:72–76. doi: 10.1016/j.ejpn.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 18.Domogatskaya A., Rodin S., Tryggvason K. Functional diversity of laminins. Annu. Rev. Cell Dev. Biol. 2012;28:523–553. doi: 10.1146/annurev-cellbio-101011-155750. [DOI] [PubMed] [Google Scholar]
- 19.Sasaki T., Giltay R., Talts U., Timpl R., Talts J.F. Expression and distribution of laminin alpha1 and alpha2 chains in embryonic and adult mouse tissues: an immunochemical approach. Exp. Cell Res. 2002;275:185–199. doi: 10.1006/excr.2002.5499. [DOI] [PubMed] [Google Scholar]
- 20.Schéele S., Falk M., Franzén A., Ellin F., Ferletta M., Lonai P., Andersson B., Timpl R., Forsberg E., Ekblom P. Laminin alpha1 globular domains 4-5 induce fetal development but are not vital for embryonic basement membrane assembly. Proc. Natl. Acad. Sci. USA. 2005;102:1502–1506. doi: 10.1073/pnas.0405095102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heng C., Lefebvre O., Klein A., Edwards M.M., Simon-Assmann P., Orend G., Bagnard D. Functional role of laminin α1 chain during cerebellum development. Cell Adhes. Migr. 2011;5:480–489. doi: 10.4161/cam.5.6.19191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ichikawa-Tomikawa N., Ogawa J., Douet V., Xu Z., Kamikubo Y., Sakurai T., Kohsaka S., Chiba H., Hattori N., Yamada Y., Arikawa-Hirasawa E. Laminin α1 is essential for mouse cerebellar development. Matrix Biol. 2012;31:17–28. doi: 10.1016/j.matbio.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edwards M.M., Mammadova-Bach E., Alpy F., Klein A., Hicks W.L., Roux M., Simon-Assmann P., Smith R.S., Orend G., Wu J. Mutations in Lama1 disrupt retinal vascular development and inner limiting membrane formation. J. Biol. Chem. 2010;285:7697–7711. doi: 10.1074/jbc.M109.069575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edwards M.M., McLeod D.S., Grebe R., Heng C., Lefebvre O., Lutty G.A. Lama1 mutations lead to vitreoretinal blood vessel formation, persistence of fetal vasculature, and epiretinal membrane formation in mice. BMC Dev. Biol. 2011;11:60. doi: 10.1186/1471-213X-11-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biehlmaier O., Makhankov Y., Neuhauss S.C. Impaired retinal differentiation and maintenance in zebrafish laminin mutants. Invest. Ophthalmol. Vis. Sci. 2007;48:2887–2894. doi: 10.1167/iovs.06-1212. [DOI] [PubMed] [Google Scholar]
- 26.Semina E.V., Bosenko D.V., Zinkevich N.C., Soules K.A., Hyde D.R., Vihtelic T.S., Willer G.B., Gregg R.G., Link B.A. Mutations in laminin alpha 1 result in complex, lens-independent ocular phenotypes in zebrafish. Dev. Biol. 2006;299:63–77. doi: 10.1016/j.ydbio.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 27.Sasaki S., Ota M., Meguro A., Nishizaki R., Okada E., Mok J., Kimura T., Oka A., Katsuyama Y., Ohno S. A single nucleotide polymorphism analysis of the LAMA1 gene in Japanese patients with high myopia. Clin. Ophthalmol. 2007;1:289–295. [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao Y.Y., Zhang F.J., Zhu S.Q., Duan H., Li Y., Zhou Z.J., Ma W.X., Li Wang N. The association of a single nucleotide polymorphism in the promoter region of the LAMA1 gene with susceptibility to Chinese high myopia. Mol. Vis. 2011;17:1003–1010. [PMC free article] [PubMed] [Google Scholar]
- 29.Najmabadi H., Hu H., Garshasbi M., Zemojtel T., Abedini S.S., Chen W., Hosseini M., Behjati F., Haas S., Jamali P. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478:57–63. doi: 10.1038/nature10423. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.