

Abstract
Centronuclear myopathies (CNMs) are characterized by muscle weakness and increased numbers of central nuclei within myofibers. X-linked myotubular myopathy, the most common severe form of CNM, is caused by mutations in MTM1, encoding myotubularin (MTM1), a lipid phosphatase. To increase our understanding of MTM1 function, we conducted a yeast two-hybrid screen to identify MTM1-interacting proteins. Striated muscle preferentially expressed protein kinase (SPEG), the product of SPEG complex locus (SPEG), was identified as an MTM1-interacting protein, confirmed by immunoprecipitation and immunofluorescence studies. SPEG knockout has been previously associated with severe dilated cardiomyopathy in a mouse model. Using whole-exome sequencing, we identified three unrelated CNM-affected probands, including two with documented dilated cardiomyopathy, carrying homozygous or compound-heterozygous SPEG mutations. SPEG was markedly reduced or absent in two individuals whose muscle was available for immunofluorescence and immunoblot studies. Examination of muscle samples from Speg-knockout mice revealed an increased frequency of central nuclei, as seen in human subjects. SPEG localizes in a double line, flanking desmin over the Z lines, and is apparently in alignment with the terminal cisternae of the sarcoplasmic reticulum. Examination of human and murine MTM1-deficient muscles revealed similar abnormalities in staining patterns for both desmin and SPEG. Our results suggest that mutations in SPEG, encoding SPEG, cause a CNM phenotype as a result of its interaction with MTM1. SPEG is present in cardiac muscle, where it plays a critical role; therefore, individuals with SPEG mutations additionally present with dilated cardiomyopathy.
Main Text
Congenital myopathies manifest early in life with varying degrees of skeletal-muscle dysfunction and hypotonia and are subclassified on the basis of histopathological findings.1 Centronuclear myopathies (CNMs) are a common subtype characterized by a large number of myofibers with central nuclei in the absence of other diagnostic features.1–3 Mutations in MTM1 (MIM 300415; encoding myotubularin [MTM1]), DNM2 (MIM 602378; encoding dynamin 2), BIN1 (MIM 601248; encoding bridging integrator 1), RYR1 (MIM 180901; encoding ryanodine receptor 1), and TTN (MIM 188840; encoding titin) are associated with 60%–80% of CNM cases; the rest have an unknown genetic basis.4–8 Alterations in proteins encoded by these genes, except for TTN, are postulated to affect the assembly or function of triads, the specialized membrane structures responsible for excitation-contraction (E-C) coupling.6,8–12 X-linked centronuclear myopathy (MIM 310400), a severe and the most common type of CNM, is characterized by congenital onset in boys and is associated with MTM1 mutations. MTM1 encodes MTM1, a lipid phosphatase predominantly located at the junctional sarcoplasmic reticulum (SR) of the muscle triads. This protein has a critical function in SR remodeling and promotion of SR membrane curvature and is hence essential for calcium homeostasis and E-C coupling.9,11,13
To better understand the molecular mechanisms of MTM1 function, we used a yeast two-hybrid (Y2H) screen, performed by Hybrigenics Services (Paris), to identify MTM1-interacting proteins in a human adult and fetal skeletal muscle library. Using full-length MTM1 as the bait, we detected striated muscle preferentially expressed protein kinase (SPEG) as an MTM1-interacting partner by isolating six different overlapping clones encoding portions of SPEG (Figure 1A). Aligning the sequences revealed that MTM1 interacts with a region encompassing SPEG amino acids 2,530–2,674, which includes one entire Ig-like domain and ends just proximal to the second fibronectin type III domain. To identify MTM1 regions that interact with SPEG, we tested a peptide encompassing residues 2,530–2,674 of human SPEG for interaction with various MTM1 fragments by using an interaction-domain-mapping assay. Both full-length MTM1 and a fragment containing residues 155–603, including the phosphatase and coiled-coil domains, were confirmed to interact with SPEG (Figure 1B). Interestingly, subfragments including either the phosphatase or the coiled-coil domains alone did not interact with SPEG, suggesting that the critical binding region either overlaps the junction or might include cooperative binding sites in both regions of MTM1. In support of the Y2H data, full-length SPEG and MTM1 coimmunoprecipitated with each other from differentiated C2C12 myotube lysates with the use of either anti-SPEG or anti-MTM1 antibodies (Figure 1C). In addition, double-label immunofluorescence experiments using anti-SPEG and anti-MTM1 antibodies revealed apparent colocalization of SPEG and MTM1 (Figures 1D–1F).16 Together, these data show that SPEG most likely interacts with MTM1, suggesting that it might play a critical role in E-C coupling,9,11 cytoskeletal organization,16 or other functional roles17 of MTM1. Given these potential functional relationships, we hypothesized that SPEG complex locus (SPEG) is a candidate disease-associated gene for CNM subjects without a known genetic basis.
Figure 1.
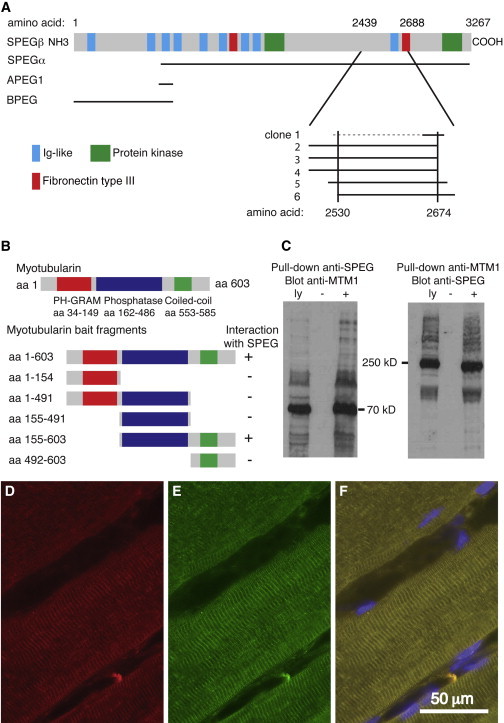
SPEG Interacts with MTM1
(A) Schematic of the six different SPEG Y2H prey clones that interacted with full-length MTM1 bait; they are lined up under the four alternative SPEG transcripts (indicated on the left). The broken line indicates that the 5′ end of clone 1 was unknown.
(B) A schematic of MTM1 illustrates the location of three major domains—PH-GRAM (amino acids 34–149), phosphatase (amino acids 162–486), and coiled coil (amino acids 553–585)—above the map of fragments used for deletion mapping of the region responsible for interactions with SPEG. Deletion analysis of MTM1 showed that the phosphatase and coiled-coil domains together were necessary to mediate the interaction with SPEG.
(C) SPEG and MTM1 coimmunoprecipitated from C2C12 myotube lysates with the use of rabbit anti-SPEG generated against a FLAG-tagged APEG-1 fusion protein14 and anti-MTM1 antibodies (r1947).15 Abbreviations are as follows: ly, total cell lysates; −, no precipitating antibody; and +, immunoprecipitated with “pull-down” antisera (indicated at the top) prior to gel electrophoresis and immunoblotting with indicated antisera.
(D–F) Indirect immunofluorescence analysis of SPEG (red, D), MTM1 (green, E), and a merged image, including blue DAPI-stained nuclei (F), with the use of rabbit anti-SPEG14 and mouse anti-MTM1 (1:80, HPA010008, Sigma-Aldrich) antibodies revealed their colocalization in human skeletal muscle. The scale bar represents 50 μm.
In a parallel project, to identify additional genes involved in CNM, we performed whole-exome sequencing on DNAs from a cohort of 29 unrelated CNM individuals without known gene mutations. These individuals were enrolled under appropriate procedures followed in accordance with the ethical standards of the responsible committees on human experimentation (institutional and national) and provided proper informed consent.8 DNA samples were enriched with exomic sequences with the Illumina Exome Enrichment protocol, and captured libraries were sequenced with the Illumina HiSeq 2000 platform. The reads were mapped to the human reference genome (UCSC Genome Browser, hg19 assembly) with the Burrows-Wheeler Aligner (version 0.5.8). SNPs and small indels were called with SAMtools (version 0.1.7) or ANNOVAR. The annotated variants were filtered against variants reported in 1000 Genomes and the NHLBI Exome Sequencing Project (ESP) Exome Variant Server.
Remarkably, two CNM individuals carried potentially pathogenic recessive variants in SPEG, located in chromosomal region 2q35 in humans and an excellent candidate CNM-associated gene given our interaction studies with MTM1. We reached out to our collaborators around the world to find additional subjects carrying SPEG mutations. The Australian group, led by Drs. Ravenscroft and Laing, used whole-exome sequencing to identify a third subject (enrolled after informed consent was approved by their human experimentation institutional review board) carrying recessive SPEG mutations. All these variants were confirmed via Sanger sequencing. None of the SPEG mutations were present in the NHLBI ESP Exome Variant Server or 1000 Genomes.
One of the three affected families was consanguineous; pedigrees are shown in Figure 2A, and clinical and molecular findings are summarized in Table 1. A newborn (II:4 or P1) from family 1 (Figure 2A) was born at term gestation to consanguineous parents in Turkey and died at 3 weeks of age of apparently severe muscle weakness. She had insufficient respiratory efforts, needed tube feeding, and was severely hypotonic with bilateral hip contractures. Other relevant clinical findings included a narrow and high-arched palate, microstomia, and retromicrognathia. She did not undergo a cardiac evaluation. Her serum creatine kinase level was 280 IU/l (normal range is 0–300 IU/l). Family history was significant for the prior death of two female siblings, each at 2 days of age, but one male sibling is alive and well. In exon 30 of SPEG, she carried a homozygous nonsense variant (c.6697C>T [p.Gln2233∗]; RefSeq accession number NM_005876.4, hg19) predicted to truncate SPEG. The father and the only surviving sibling were heterozygous for the change, whereas the mother and a first cousin of the father declined to enroll in the study.
Figure 2.
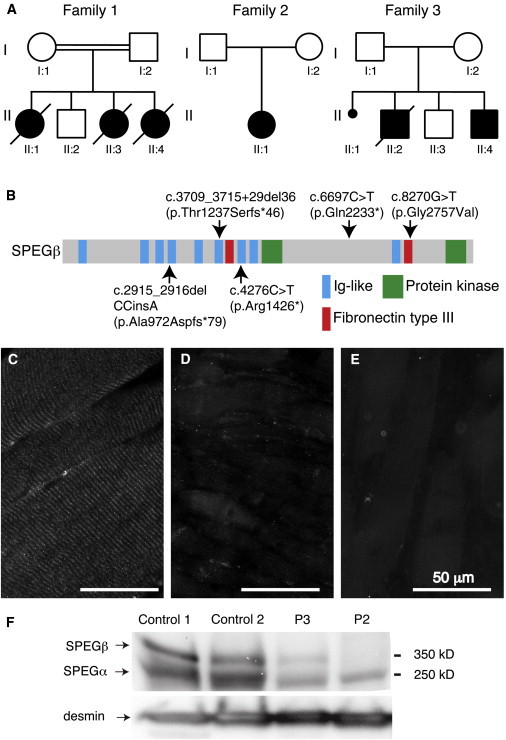
Genetic and Molecular Findings in Three Families Affected by SPEG Mutations
(A) Pedigree of the three families carrying SPEG mutations. Family 1 was consanguineous, whereas families 2 and 3 were not. The probands were II:4 (P1) for family 1, II:1 (P2) for family 2, and II:4 (P3) for family 3.
(B) Distribution of alterations across the schematic of SPEG. Domains are also indicated.
(C–E) Indirect immunofluorescence analysis using rabbit anti-SPEG antibody (NBP1-90134, Novus Biologicals) in muscle-biopsy specimens from a representative age-matched human control individual (C) and probands P2 (II:4 from family 2, D) and P3 (II:4 from family 3, E) showed a loss of the striated pattern and a marked and reproducible reduction of overall SPEG staining in muscle from both subjects.
(F) Immunoblot analysis using rabbit anti-SPEG in muscle-biopsy specimens from two unaffected control individuals and subjects 2 and 3.14 Restaining the filter with anti-desmin antibodies (1466-1, Epitomics) confirmed adequate loading of lanes and demonstrated robust levels of desmin in these muscles.
Table 1.
Molecular and Clinicopathological Findings in Individuals Carrying SPEG Mutations
|
Subject |
|||
|---|---|---|---|
| P1 | P2 | P3 | |
| Sex | female | female | male |
| Current age | died at 3 weeks of life | 6 years | 1.5 years |
| SPEG exons | exon 30 | exons 18 and 13 | exons 10 and 35 |
| Allele 1 (maternal) | c.6697C>T (p.Gln2233∗) | c.4276C>T (p.Arg1426∗) | c.2915_2916delCCinsA (p.Ala972Aspfs∗79) |
| Allele 2 (paternal) | c.6697C>T (p.Gln2233∗) | c.3709_3715+29del36 (p.Thr1237Serfs∗46) | c.8270G>T (p.Gly2757Val) |
| Clinical information | full-term, breech delivery; consanguineous parents; died of severe muscle weakness; family history of two female siblings who died at 2 days old | severe hypotonia since birth; needed tracheostomy; 24 hr mechanical ventilation; gastrostomy; dilated cardiomyopathy; congestive cardiac failure in neonatal age was treated with digoxin and captopril | hypotonia since birth; dilated cardiomyopathy and nephrolithiasis; orogastric feeds; no assisted ventilation |
| Biopsy findings | marked increase in myofibers with central nuclei; few necklace fibers | hypotrophic myofibers; marked increase in central nuclei | myopathic changes; variation in fiber size that was more prominent in some fascicles; increased central nuclei in hypotrophic fibers; predominant type 1 fibers |
Individual II:1 (P2) from family 2 (Figure 2A) was compound heterozygous for two SPEG variants, a frameshift (c.3709_3715+29del36 [p.Thr1237Serfs∗46]) and a nonsense change (c.4276C>T [p.Arg1426∗]); both parents carried one variant each. A tracheostomy was performed when she was 1 year old, and a gastrostomy tube was placed early in life. Now alive at 6 years of age, she continues to need ventilatory and feeding support via tracheostomy and gastrostomy tubes. She has proximal muscle weakness, ophthalmoplegia, and facial weakness. Her motor milestones are grossly delayed, given that she sat unsupported at 2.5 years of age and is currently unable to walk unsupported. She wears bilateral ankle foot orthotics. Other findings include bifid uvula, a high-arched palate, and retrognathia. Her serum creatine kinase level was 22 IU/l. An initial echocardiogram at 6 days of age was normal, but a subsequent study at 2 months of age demonstrated a dilated left ventricle with moderate to severe depression of systolic function, diastolic dysfunction of both left and right ventricles, and mild mitral and tricuspid valve insufficiency. In the neonatal period, she was started on digoxin, furosemide, carvedilol, and captopril to support her cardiac function, and over the next few months, she required increased dosages of several of these drugs and close monitoring by the cardiology team with serial echocardiograms. Remarkably, her echo findings improved over time and by 1 year of age revealed normal ventricular function. Her cardiac medications were discontinued at 2.5 years of age.
Individual II:4 (P3) from family 3 (Figure 2A) is a 19-month-old Turkish boy with no known consanguinity and was born at 36 weeks of gestation by cesarean section for fetal bradycardia. He stayed in the neonatal intensive-care unit for 2 weeks for cyanosis and respiratory difficulty but was not placed on assisted ventilation. He was noted to be very hypotonic, and echocardiography at 1 month of age showed dilated cardiomyopathy. Family history was significant for the previous demise of a 40-day-old sibling who had respiratory insufficiency (no cardiac evaluation was performed) and a very similar presentation according to the parents. He has delayed motor milestones: head control was achieved at 16 months, and unsupported sitting was reached at 18 months of age. He has antigravity movements of both the upper and lower extremities. He breathes independently but continues to require nasogastric feeds. He has a history of frequent pulmonary and urinary-tract infections. He has mild facial weakness, a high-arched palate, axial hypotonia, and absent deep tendon reflexes. His serum creatine kinase level was within normal limits at 106 IU/l. He is currently being treated with captopril, digoxin, furosemide, and carnitine for decreased left ventricular function, dilated cardiomyopathy, and mitral insufficiency. He is compound heterozygous for a frameshift variant (c.2915_2916delCCinsA [p.Ala972Aspfs∗79]) and a missense mutation (c.8270G>T [p.Gly2757Val]) in SPEG, and each segregated appropriately from the parents. The missense mutation was predicted to be pathogenic by several software programs, including SIFT (deleterious, 0.002), PROVEAN (deleterious, −6.23), PolyPhen-2 (probably damaging, 1.00), and MutationTaster (disease causing, 1.0).
Histopathologically, all three probands were diagnosed with CNM prior to enrollment in this study. Their muscle biopsies revealed numerous small myofibers with central nuclei (Figures 3A, 3C, and 3D). In P1 (II:4 from family 1 in Figure 2A), subsarcolemmal ringed and central dense areas, so-called “necklace fibers,” were identified on oxidative staining (NADH tetrazolium reductase) (Figure 3B).
Figure 3.
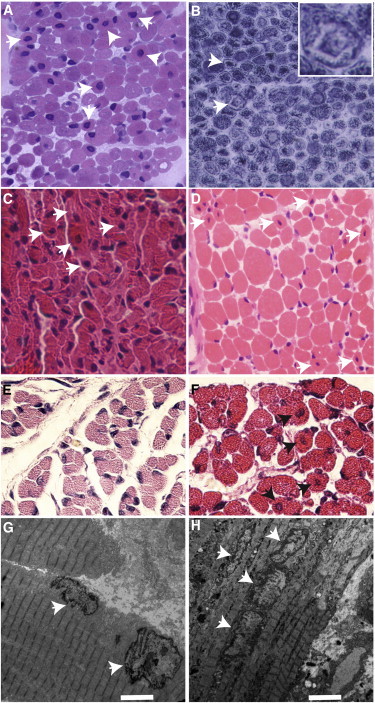
Histopathological Findings in Human Subjects and a Mouse Model of SPEG Deficiency
(A–D) Light microscopic findings in muscle-biopsy specimens from human probands P1 (A and B), P2 (II:4 from family 2 in Figure 2, C), and P3 (II:4 from family 3 in Figure 2, D) included increased central nuclei on hematoxylin and eosin (H&E) staining (arrows in A, C, and D) and subsarcolemmal ringed and central dense areas, also called necklace fibers (arrows and inset, with NADH tetrazolium reductase staining, in B).
(E and F) Histopathological and ultrastructural findings in skeletal muscles from SPEG-deficient and wild-type (WT) littermate mice. H&E staining of paraspinal muscles revealed higher numbers of central nuclei (arrows) in 1-day-old SPEG-deficient mice (F) than in WT controls (E).
(G and H) Transmission electron microscopic findings in skeletal-muscle (quadriceps) specimens obtained from Speg-KO (H) and WT littermate (G) mice. Several centrally placed nuclei were present in the specimen from a Speg-KO mouse (arrows, H), and in comparison, peripherally located nuclei were seen in the control mouse (arrows, G). Scale bars represent 4 μm.
To evaluate whether the SPEG mutations were associated with altered SPEG abundance and/or localization, we performed immunofluorescence analyses on skeletal-muscle specimens available from P2 (II:4 from family 2 in Figure 2) and P3 (II:4 from family 3 in Figure 2A) and an age-matched control individual by using anti-SPEG antibody. As predicted from the nature of the mutations, compared to muscle from the unaffected control individual (Figure 2C), muscle fibers from both P2 (II:1 from family 2 in Figure 2A) and P3 (II:4 from family 3 in Figure 2A) showed reduced or absent SPEG staining (Figures 2D and 2E). To further confirm this finding, we performed immunoblotting experiments by using anti-SPEG antibody and anti-desmin as a control. Compared to muscle from the age-matched control individual (Figure 2F), muscle from individuals P2 (II:1 from family 2 in Figure 2) and P3 (II:4 from family 3 in Figure 2) showed a marked reduction or absence of both major isoforms, SPEGα and SPEGβ.
The murine SPEG has four isoforms (SPEGα, SPEGβ, aortic peg [APEG], and brain peg [BPEG]) derived from two transcription start sites and alternative splicing.14 SPEGα and SPEGβ are both found in striated muscles, APEG is predominantly present in vascular tissues, and BPEG is found in the brain and aorta.18 The two largest isoforms, SPEGα and SPEGβ, share homology with myosin light-chain kinase (MLCK) family members, which include titin, twitchin, obscurin, kalirin, trio, and death-associated protein kinases. SPEG has unique homology with obscurin given that both contain two tandemly arranged serine/threonine kinase (MLCK) domains.18,19 They also contain immunoglobulin and fibronectin domains that are characteristic of the MLCK family. SPEGα is predominantly present during skeletal-muscle differentiation, whereas both SPEGα and SPEGβ are present in neonatal cardiomyocytes during maturation and differentiation.14 All the mutations identified in our three CNM kindreds are predicted to disrupt both SPEGα and SPEGβ but are distal to the 3′ terminus of both APEG1 and BPEG (Figure 2B).
Deletion of Speg common exons 8–10 in mice (i.e., Speg knockout [KO]) led to dilation of both atria and ventricles by embryonic day 18.5, and cardiac myofibril degeneration and marked reduction of cardiac function led to death by postnatal day 2.18 At least two of our three affected individuals exhibited significant cardiac dysfunction. Although a respiratory cause for this cannot be ruled out, the early onset in P2 (II:1 from family 2 in Figure 2) and P3 (II:4 from family 3 in Figure 2) and the improving cardiac function in P2 (II:1 from family 2 in Figure 2) in the context of continued respiratory compromise, in addition to our knowledge of the severe mouse cardiac phenotype and SPEG expression in the heart, but not the lungs, suggest that these individuals suffered from a primary cardiomyopathy.
Because of the early lethality, the previous murine studies focused on cardiac manifestations of the newborn Speg-KO mice. Given the significant degree of skeletal-muscle weakness in our affected individuals, we have now more carefully evaluated the effects of SPEG deficiency on skeletal muscles in these animals. Hematoxylin and eosin staining of multiple muscle groups revealed a significantly higher mean number of myofibers with central nuclei in six Speg-KO mice (8.5 ± 1.3%, range 5.32%–12.6%, Figure 3F) than in three wild-type (WT) littermate controls (2.2 ± 0.8%, range 0.95%–3.09%, Figure 3E) (p = 0.015). Electron microscopy revealed similar findings—a higher number of central nuclei lined up within myofibers of Speg-KO mice (Figure 3H) than in those of WT littermate controls (Figure 3G). Overall, the mouse model of SPEG deficiency resembles human SPEG-mutation-positive subjects who present with increased central nuclei within the skeletal myofibers and both skeletal and dilated cardiac myopathies.
Our observation that SPEG and MTM1 colocalize on longitudinal sections of myofibers (Figures 1D–1F) is consistent with previously published data that murine SPEG appears to localize over Z lines with desmin, another MTM1-interacting protein.14,16 Because MTM1 is known to be a component of the junctional SR at the triads, immediately flanking the Z lines, where it plays a role in E-C coupling through promotion of proper SR and T-tubule morphology,9,11,15 we performed double-label immunolocalization experiments for SPEG in murine and human skeletal muscle by using additional antibodies against proteins representative of various parts of the myofibers. Ultrathin transverse sections of quadriceps muscles exhibited SPEG immunofluorescence in a reticular staining pattern around the periphery of the myofibrils; this immunofluorescence colocalized with dihydropyridine receptors and the SR Ca2+ATPase (SERCA) (Figure 4A), both of which are present in the SR. Examination in glancing sections showed that it did not colocalize with myosin and only partially overlapped with the Z line protein α-actinin-2 (Figure 4A), consistent with localization around, but not within, the myofibrils. Upon examination of longitudinal sections, SPEG staining appeared as a double band flanking desmin, which appeared as a series of dots between the Z lines, and significantly overlapping staining for triadin, a protein found in the terminal cisternae of the SR (Figure 4B).
Figure 4.
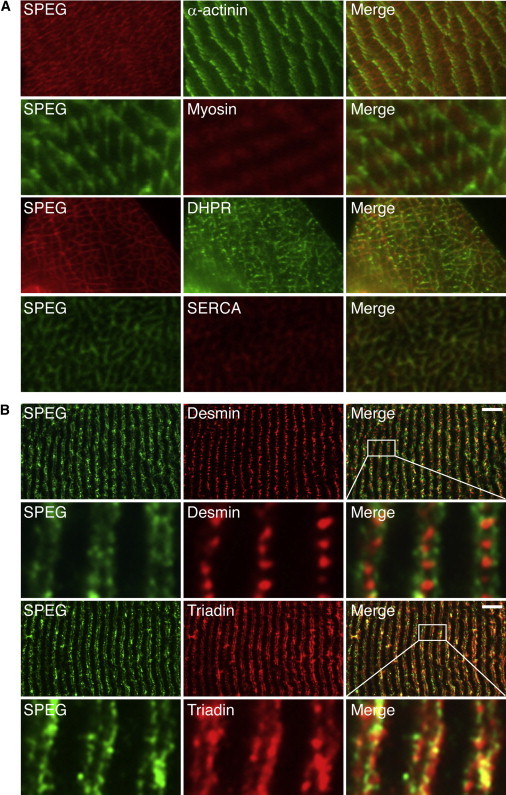
SPEG Localization in Normal Skeletal Muscle
(A) Ultrathin sections from frozen WT mouse quadriceps muscle were coimmunostained with mouse monoclonal anti-α-actinin-2 (clone EA-53, Sigma), mouse monoclonal anti-myosin heavy chain fast (clone MY-32, Sigma), mouse monoclonal anti-dihydropyridine receptor (DHPR, clone D218, Sigma), mouse monoclonal anti-sarcoplasmic reticulum Ca2+ATPase (SERCA, clone IIH11, Sigma), and rabbit anti-SPEG14 (indicated). As seen in the merged images, SPEG staining partially overlapped with α-actinin depending on the plane of section and did not colocalize with myosin but did colocalize with both DHPR and SERCA.
(B) Longitudinal sections from frozen human quadriceps muscles were coimmunostained with monoclonal mouse anti-human desmin (clone D33, Dako), mouse monoclonal anti-rabbit triadin (clone GE 4.90, Abcam), and rabbit anti-SPEG14 (indicated). SPEG appeared as a doublet over the myofibrils and flanking desmin, which is found between Z lines. SPEG largely colocalized with triadin, which is found at the junctional SR. Scale bars represent 7 μm.
A major role of MTM1 is most likely related to the regulation of phosphatidylinositol 3-monophosphate levels and control of membrane curvature of the SR.11 However, MTM1 also has significant non-phosphatase-dependent activities, including desmin binding,16 given that the phosphatase-dead MTM1 p.Cys375Ser mutant in Mtm1-null mice improves muscle function and restores localization of nuclei, triad alignment, and organization of the desmin intermediate filament network.20 MTM1 deficiency leads to disruption of the MTM1-desmin complex, causing abnormal organization of desmin-containing intermediate filaments. We examined SPEG localization and organization in MTM1-deficient muscles from human subjects with myotubular myopathy and mice with an Mtm1-KO mutation.21 Remarkably, both SPEG and desmin exhibited identical-appearing abnormalities in the MTM1-deficient muscles, suggesting that MTM1 exerts similar activities on both of these proteins (Figure 5). It is interesting to note that the human desmin- and SPEG-encoding genes are tandemly arrayed within 8.3 kb of each other under the control of a common locus control region,22 suggesting evolutionary pressure to coordinately regulate these two proteins. Further, obscurin, the only other MLCK protein with two tandem MLCK domains, is thought to link the SR to sarcomeres, and obscurin-deficient mice display increased centralized nuclei in skeletal muscles as a sign of myopathy.23 Together with our present observations of a SPEG-MTM1 interaction in the region of the junctional SR, the known roles of desmin and obscurin in linking or aligning the triads and SR, respectively, to the sarcomere suggest a central role for this complex in SR organization, as well as nuclear and organelle positioning in skeletal muscle. In summary, SPEG is a striated muscle protein that interacts with MTM1 and is localized to the SR region in myofibers, and SPEG dysfunction is associated with CNM and cardiomyopathy in at least two of three affected individuals with SPEG mutations.
Figure 5.

Analysis of SPEG and Desmin Distribution in MTM1-Deficient Skeletal Muscles
Representative indirect immunofluorescence images for SPEG (rabbit anti-SPEG,14 green) and desmin (clone D33, red) in skeletal-muscle specimens from an unaffected control individual (top row), a boy with X-linked myotubular myopathy due to an MTM1 mutation (second row), a WT mouse (third row), and an Mtm1-KO mouse (bottom row). Merged images, including nuclei stained blue, are shown on the right. Note the abnormal clumping and accumulations of colocalized SPEG and desmin in the MTM1-deficient muscles. The scale bar represents 100 μm.
Acknowledgments
We are grateful to the several centronuclear-myopathy-affected individuals, their families, and their caregivers. We would like to thank Vandana Gupta and Ozge Ceyhan-Birsoy for helpful discussions during this work, Melissa Mercier for sequencing SPEG, and Louise Trakimas of the electron-microscope facility at Harvard Medical School for help with mouse muscle histology. P.B.A. was supported by K08 AR055072 from the NIH National Institute of Arthritis and Musculoskeletal and Skeletal Diseases (NIAMS). This work was also supported by the Muscular Dystrophy Association (MDA201302), NIH NIAMS grant R01 AR044345, the AUism Charitable Foundation and A Foundation Building Strength (to A.H.B.), the National Health and Medical Research Council of Australia (Early Career Researcher Fellowship 1035955 to G.R. and Research Fellowship APP1002147 and Project Grant APP1022707 to N.G.L.), an American Heart Association Scientist Development Grant (to X.L.), and NIH NHLBI grants R01 HL102897 and HL 108801 (to M.A.P.). DNA sequencing was performed by the Boston Children’s Hospital Genomics Program Molecular Genetics Core, supported by NIH grant P30 HD18655. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Contributor Information
Pankaj B. Agrawal, Email: pagrawal@enders.tch.harvard.edu.
Alan H. Beggs, Email: beggs@enders.tch.harvard.edu.
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/
Burrows-Wheeler Aligner, http://bio-bwa.sourceforge.net/
MutationTaster, http://mutationtaster.org
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www/omim.org/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
PROVEAN, http://provean.jcvi.org/
SAMtools, http://sourceforge.net/projects/samtools/files/samtools/0.1.7/
References
- 1.Nance J.R., Dowling J.J., Gibbs E.M., Bönnemann C.G. Congenital myopathies: an update. Curr. Neurol. Neurosci. Rep. 2012;12:165–174. doi: 10.1007/s11910-012-0255-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pierson C.R., Tomczak K., Agrawal P., Moghadaszadeh B., Beggs A.H. X-linked myotubular and centronuclear myopathies. J. Neuropathol. Exp. Neurol. 2005;64:555–564. doi: 10.1097/01.jnen.0000171653.17213.2e. [DOI] [PubMed] [Google Scholar]
- 3.Romero N.B., Bitoun M. Centronuclear myopathies. Semin. Pediatr. Neurol. 2011;18:250–256. doi: 10.1016/j.spen.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 4.Laporte J., Hu L.J., Kretz C., Mandel J.L., Kioschis P., Coy J.F., Klauck S.M., Poustka A., Dahl N. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat. Genet. 1996;13:175–182. doi: 10.1038/ng0696-175. [DOI] [PubMed] [Google Scholar]
- 5.Bitoun M., Maugenre S., Jeannet P.Y., Lacène E., Ferrer X., Laforêt P., Martin J.J., Laporte J., Lochmüller H., Beggs A.H. Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat. Genet. 2005;37:1207–1209. doi: 10.1038/ng1657. [DOI] [PubMed] [Google Scholar]
- 6.Nicot A.S., Toussaint A., Tosch V., Kretz C., Wallgren-Pettersson C., Iwarsson E., Kingston H., Garnier J.M., Biancalana V., Oldfors A. Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat. Genet. 2007;39:1134–1139. doi: 10.1038/ng2086. [DOI] [PubMed] [Google Scholar]
- 7.Bevilacqua J.A., Monnier N., Bitoun M., Eymard B., Ferreiro A., Monges S., Lubieniecki F., Taratuto A.L., Laquerrière A., Claeys K.G. Recessive RYR1 mutations cause unusual congenital myopathy with prominent nuclear internalization and large areas of myofibrillar disorganization. Neuropathol. Appl. Neurobiol. 2011;37:271–284. doi: 10.1111/j.1365-2990.2010.01149.x. [DOI] [PubMed] [Google Scholar]
- 8.Ceyhan-Birsoy O., Agrawal P.B., Hidalgo C., Schmitz-Abe K., DeChene E.T., Swanson L.C., Soemedi R., Vasli N., Iannaccone S.T., Shieh P.B. Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy. Neurology. 2013;81:1205–1214. doi: 10.1212/WNL.0b013e3182a6ca62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Al-Qusairi L., Weiss N., Toussaint A., Berbey C., Messaddeq N., Kretz C., Sanoudou D., Beggs A.H., Allard B., Mandel J.L. T-tubule disorganization and defective excitation-contraction coupling in muscle fibers lacking myotubularin lipid phosphatase. Proc. Natl. Acad. Sci. USA. 2009;106:18763–18768. doi: 10.1073/pnas.0900705106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zvaritch E., Kraeva N., Bombardier E., McCloy R.A., Depreux F., Holmyard D., Kraev A., Seidman C.E., Seidman J.G., Tupling A.R., MacLennan D.H. Ca2+ dysregulation in Ryr1(I4895T/wt) mice causes congenital myopathy with progressive formation of minicores, cores, and nemaline rods. Proc. Natl. Acad. Sci. USA. 2009;106:21813–21818. doi: 10.1073/pnas.0912126106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amoasii L., Hnia K., Chicanne G., Brech A., Cowling B.S., Müller M.M., Schwab Y., Koebel P., Ferry A., Payrastre B., Laporte J. Myotubularin and PtdIns3P remodel the sarcoplasmic reticulum in muscle in vivo. J. Cell Sci. 2013;126:1806–1819. doi: 10.1242/jcs.118505. [DOI] [PubMed] [Google Scholar]
- 12.Gibbs E.M., Davidson A.E., Telfer W.R., Feldman E.L., Dowling J.J. The myopathy-causing mutation DNM2-S619L leads to defective tubulation in vitro and in developing zebrafish. Dis. Model. Mech. 2014;7:157–161. doi: 10.1242/dmm.012286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dowling J.J., Vreede A.P., Low S.E., Gibbs E.M., Kuwada J.Y., Bonnemann C.G., Feldman E.L. Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy. PLoS Genet. 2009;5:e1000372. doi: 10.1371/journal.pgen.1000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsieh C.M., Fukumoto S., Layne M.D., Maemura K., Charles H., Patel A., Perrella M.A., Lee M.E. Striated muscle preferentially expressed genes alpha and beta are two serine/threonine protein kinases derived from the same gene as the aortic preferentially expressed gene-1. J. Biol. Chem. 2000;275:36966–36973. doi: 10.1074/jbc.M006028200. [DOI] [PubMed] [Google Scholar]
- 15.Buj-Bello A., Fougerousse F., Schwab Y., Messaddeq N., Spehner D., Pierson C.R., Durand M., Kretz C., Danos O., Douar A.M. AAV-mediated intramuscular delivery of myotubularin corrects the myotubular myopathy phenotype in targeted murine muscle and suggests a function in plasma membrane homeostasis. Hum. Mol. Genet. 2008;17:2132–2143. doi: 10.1093/hmg/ddn112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hnia K., Tronchère H., Tomczak K.K., Amoasii L., Schultz P., Beggs A.H., Payrastre B., Mandel J.L., Laporte J. Myotubularin controls desmin intermediate filament architecture and mitochondrial dynamics in human and mouse skeletal muscle. J. Clin. Invest. 2011;121:70–85. doi: 10.1172/JCI44021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dowling J.J., Joubert R., Low S.E., Durban A.N., Messaddeq N., Li X., Dulin-Smith A.N., Snyder A.D., Marshall M.L., Marshall J.T. Myotubular myopathy and the neuromuscular junction: a novel therapeutic approach from mouse models. Dis. Model. Mech. 2012;5:852–859. doi: 10.1242/dmm.009746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu X., Ramjiganesh T., Chen Y.H., Chung S.W., Hall S.R., Schissel S.L., Padera R.F., Jr., Liao R., Ackerman K.G., Kajstura J. Disruption of striated preferentially expressed gene locus leads to dilated cardiomyopathy in mice. Circulation. 2009;119:261–268. doi: 10.1161/CIRCULATIONAHA.108.799536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gautel M. Cytoskeletal protein kinases: titin and its relations in mechanosensing. Pflugers Arch. 2011;462:119–134. doi: 10.1007/s00424-011-0946-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amoasii L., Bertazzi D.L., Tronchère H., Hnia K., Chicanne G., Rinaldi B., Cowling B.S., Ferry A., Klaholz B., Payrastre B. Phosphatase-dead myotubularin ameliorates X-linked centronuclear myopathy phenotypes in mice. PLoS Genet. 2012;8:e1002965. doi: 10.1371/journal.pgen.1002965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buj-Bello A., Laugel V., Messaddeq N., Zahreddine H., Laporte J., Pellissier J.F., Mandel J.L. The lipid phosphatase myotubularin is essential for skeletal muscle maintenance but not for myogenesis in mice. Proc. Natl. Acad. Sci. USA. 2002;99:15060–15065. doi: 10.1073/pnas.212498399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tam J.L., Triantaphyllopoulos K., Todd H., Raguz S., de Wit T., Morgan J.E., Partridge T.A., Makrinou E., Grosveld F., Antoniou M. The human desmin locus: gene organization and LCR-mediated transcriptional control. Genomics. 2006;87:733–746. doi: 10.1016/j.ygeno.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 23.Lange S., Ouyang K., Meyer G., Cui L., Cheng H., Lieber R.L., Chen J. Obscurin determines the architecture of the longitudinal sarcoplasmic reticulum. J. Cell Sci. 2009;122:2640–2650. doi: 10.1242/jcs.046193. [DOI] [PMC free article] [PubMed] [Google Scholar]