

Abstract
Psoriasis vulgaris (PsV) risk is strongly associated with variation within the major histocompatibility complex (MHC) region, but its genetic architecture has yet to be fully elucidated. Here, we conducted a large-scale fine-mapping study of PsV risk in the MHC region in 9,247 PsV-affected individuals and 13,589 controls of European descent by imputing class I and II human leukocyte antigen (HLA) genes from SNP genotype data. In addition, we imputed sequence variants for MICA, an MHC HLA-like gene that has been associated with PsV, to evaluate association at that locus as well. We observed that HLA-C∗06:02 demonstrated the lowest p value for overall PsV risk (p = 1.7 × 10−364). Stepwise analysis revealed multiple HLA-C∗06:02-independent risk variants in both class I and class II HLA genes for PsV susceptibility (HLA-C∗12:03, HLA-B amino acid positions 67 and 9, HLA-A amino acid position 95, and HLA-DQα1 amino acid position 53; p < 5.0 × 10−8), but no apparent risk conferred by MICA. We further evaluated risk of two major clinical subtypes of PsV, psoriatic arthritis (PsA; n = 3,038) and cutaneous psoriasis (PsC; n = 3,098). We found that risk heterogeneity between PsA and PsC might be driven by HLA-B amino acid position 45 (pomnibus = 2.2 × 10−11), indicating that different genetic factors underlie the overall risk of PsV and the risk of specific PsV subphenotypes. Our study illustrates the value of high-resolution HLA and MICA imputation for fine mapping causal variants in the MHC.
Introduction
Psoriasis vulgaris (PsV [MIM 177900]) is a common immune-mediated skin disease characterized by epidermal hyperplasia, inflammatory cell infiltration, and vascular remodeling.1,2 Approximately one-third of PsV-affected individuals develop a related inflammatory musculoskeletal condition called psoriatic arthritis (PsA), which is considered more severe than the other more common PsV subtype, cutaneous psoriasis (PsC).3 Previous linkage and association studies have mapped PsV risk to a critical region spanning ∼300 kb within the major histocompatibility complex (MHC) class I region on 6p21 (this region is termed PSORS1).4 Subsequent analyses have identified HLA-Cw6 (MIM 142840) as the risk allele of PSORS1.5,6 Although recent large-scale genome-wide association studies (GWASs) have identified more than 35 genetic risk loci for PsV outside the MHC region, HLA-C∗06:02 (the most frequent four-digit allele equivalent to HLA-Cw6) has consistently demonstrated the strongest association with PsV risk.7–12
However, the effects of the genetic architecture of the MHC region on PsV risk have yet to be fully elucidated. Previous studies have suggested the existence of other risk variants in addition to HLA-C∗06:02 in class I human leukocyte antigen (HLA) genes (HLA-A [MIM 142800] or HLA-B [MIM 142830]) and class II HLA genes (HLA-DRB1 [MIM 142857], HLA-DQA1 [MIM 146880], HLA-DQB1 [MIM 604305], HLA-DPA1 [MIM 142880], or HLA-DPB1 [MIM 142858]).13–18 Investigators have also studied polymorphisms of MHC class I polypeptide-related sequence A (MICA [MIM 600169]), an HLA-like gene that does not present antigen.17,19,20 However, strong and complex linkage-disequilibrium (LD) patterns in the MHC region21,22 have thus far challenged the identification of independent risk signals. Moreover, analyses focusing on the two major subsets of PsV have identified different effect sizes of associated risk alleles (including HLA-C∗06:02) between PsA and PsC, suggesting heterogeneous effects of HLA alleles on the two major subphenotypes.13–16,23
Recently, we devised an analytical approach to fine map risk of the variants in the MHC region by imputing genotypes of amino acid polymorphisms in the classical HLA genes and classical two- and four-digit alleles.24–26 Each classical four-digit HLA allele corresponds to a unique amino acid sequence encoded by the HLA gene, and thus polymorphic residues at each amino acid position could also be targets of disease-risk association studies. This approach has been used for successfully fine mapping HLA alleles of several autoimmune or immune-mediated diseases, including durable host control of HIV infection (MIM 609423), seropositive and seronegative rheumatoid arthritis (MIM 180300), myasthenia gravis (MIM 254200), and follicular lymphoma (MIM 613024).25–29 In certain instances, our approach has been able to pinpoint individual amino acid sites that might account for disease risk within HLA molecules.25–27
In this study, we aimed to fully characterize the genetic architecture of the MHC region for PsV. Our goals were (1) to define the set of risk alleles for PsV at the four-digit HLA allele and amino acid resolutions, (2) to examine how the role of MICA in PsV compares to that of other HLA genes, and (3) to identify a genetic marker that distinguishes the risk of two subtypes, PsA and PsC. To this end, we applied our HLA-variant imputation approach to large-scale PsV GWASs and Immunochip studies comprising 9,247 affected individuals and 13,589 control individuals of European ancestry. We also expanded our approach to impute MICA alleles and MICA amino acid polymorphisms by constructing a MICA imputation reference panel. With the imputed MHC sequence variations, including classical HLA genes and MICA, we fine mapped the MHC associations with overall PsV risk and specifically focused on risk comparisons between the PsA and PsC subphenotypes.
Material and Methods
Samples
We used data from 9,247 PsV-affected individuals and 13,589 control individuals obtained from six case-control PsV data sets, including four GWASs (the Collaborative Association Study of Psoriasis [CASP] and the Genizon, Kiel, and PsA GWASs), a targeted deep follow-up study of CASP (the CASP-DFU), and one Immunochip-based data set of 3,723 affected and 7,595 control subjects (the Psoriasis Association Genetics Extension [PAGE] study), for a total of 9,247 affected and 13,589 control individuals (Table S1, available online).7,9,12 Genotype data of the studies were generated and stringently quality-control (QC) filtered as described elsewhere,7,9,12 and all samples were confirmed to be unrelated individuals of European ancestry according to self-reported ethnicity and results of principal-component (PC) analysis. All participating individuals provided written informed consent and were recruited according to the protocols approved by the institutional review board of each institution.
Phenotype Classification
All PsV-affected individuals were diagnosed by a dermatologist. Diagnosis of PsA was confirmed by a rheumatologist according to the Classification Criteria for Psoriatic Arthritis.30 Individuals who had had PsV for 10 or more years but no signs of PsA were classified as having PsC. Our data set included 3,038 PsA subjects, 3,098 PsC subjects, and 3,111 subjects of unknown PsA or PsC status (Table S1).
Statistical Analysis
HLA Imputation
For each data set, we used SNP2HLA24 to extract SNP genotypes located in the MHC region to impute classical two- and four-digit HLA alleles of and amino acid polymorphisms encoded by the eight class I and class II HLA genes (HLA-A, HLA-B, HLA-C, HLA-DRB1, HLA-DQA1, HLA-DQB1, HLA-DPA1, and HLA-DPB1). We conducted HLA imputation for each data set separately by using HLA and SNP genotypes from the Type 1 Diabetes Genetics Consortium (T1DGC; n = 5,225), which has demonstrated a high imputation accuracy for classical HLA alleles,24,26,31 as a reference panel. We obtained information on HLA-gene polymorphisms from the IMGT/HLA Database.32 Amino acid sequences encoded by the imputed HLA genes are indicated in Figure S1. For HLA amino acid positions, we indicate the start codon of the mature HLA protein as position 1, and we label the codon 5′ to this site as −1.24 SNP2HLA checks concordance of allele strands of the A/T or G/C SNPs between the data set and the reference panel on the basis of allele-frequency comparison.24 We applied postimputation QC criteria of MAF > 0.1% for the association analysis.
MICA Imputation
To expand our HLA imputation protocol into HLA-like genes, we constructed a reference panel for imputation of MICA variants. We obtained classical four-digit MICA alleles for the subjects from a subset of the PsA data set (n = 1,046). These samples were not selected in any particular way. We obtained MICA amino acid sequences from the IMGT/HLA Database32 and the encoded MICA amino acid polymorphisms of the subjects, as well as the genotypes of MICA classical alleles and the genotyped SNPs in the MHC region. Using the constructed MICA reference panel and SNP2HLA,24 we imputed MICA variants for the other data-set collections. Imputed genotypes of the MICA alleles and MICA amino acid polymorphisms were extracted and merged into those obtained from HLA imputation mentioned in the previous section. We empirically assessed the accuracy of imputing MICA variants by additionally genotyping MICA in a subset of the subjects from the PAGE Immunochip data set (n = 104) and comparing concordances of the imputed and genotyped classical MICA variants as described elsewhere.24,26
Statistical Framework for Association Analysis
We used the following analyses to test associations between HLA variants and risk of four binary phenotypes: (1) overall analysis of PsV susceptibility (PsV-affected versus control individuals), (2) stratified analysis of PsA susceptibility (PsA-affected versus control individuals), (3) stratified analysis of PsC susceptibility (PsC-affected versus control individuals), and (4) intra-PsV analysis directly comparing PsA to PsC (PsA-affected versus PsC-affected individuals). For each phenotype, we assessed variant risk with a logistic-regression model assuming additive effects of the allele dosages in the log-odds scale and their fixed effects among the data-set collections. We defined HLA variants to include biallelic SNPs in the MHC region, two- and four-digit biallelic classical HLA or MICA alleles, biallelic HLA or MICA amino acid polymorphisms for respective residues, and multiallelic HLA or MICA amino acid polymorphisms for respective positions. To account for potential population-based and data-set-specific confounding factors, we included the top ten PCs and an indicator variable for each data set as covariates. For HLA variants with m alleles (m = 2 for biallelic variants and m > 2 for multiallelic variants), we included m − 1 alleles, excluding the most frequent allele as a reference, as independent variables in the regression model. This resulted in the following logistic-regression model:
where β0 is the logistic-regression intercept and β1,j is the additive effect of the dosage of allele j for the variant xj. K and L are numbers of the collections and PCs enrolled in the analysis. yk,l is the lth PC for the kth collection, and zk is the indicator variable for the collection-specific intercept. β2,k,l and β3,k parameters are the effects of yk,l and zk, respectively. An omnibus p value of the variant (pomnibus) was obtained by a log-likelihood ratio test comparing the likelihood of the null model against the likelihood of the fitted model. We assessed the significance of the improvement in fit by calculating the deviance (−2 × the log likelihood ratio), which follows a χ2 distribution with m − 1 degree(s) of freedom.
Conditional Association Analysis
For conditional association analysis, we considered the regression model including the additional HLA variants as covariates. When conditioning on specific HLA amino acid position(s), we included multiallelic variants of the amino acid residues as covariates. When conditioning on specific HLA gene(s), we included all two- and four-digit classical alleles of the HLA gene(s) (but not alleles with strong correlations [R2 > 0.97]). We consecutively selected the HLA variants to be included as covariates for each HLA gene separately in a forward-type stepwise fashion until no variant satisfied the genome-wide significant threshold (p < 5.0 × 10−8). We tested a multivariate full regression model by including the HLA-C, HLA-B, HLA-A, and HLA-DQA1 risk variants identified by the stepwise regression analysis as covariates and excluding the most frequent allele (or residue) from each locus (or amino acid position) as a reference allele (Table 1). Assuming a PsV prevalence of 2.0%, we estimated phenotypic variance explained by the risk HLA alleles and amino acid polymorphisms on the basis of the effect sizes obtained from the multivariate regression analysis and a liability threshold model.2
Table 1.
Association Results of the HLA Variants for Overall PsV Risk in Multivariate Regression Model
| HLA Variant |
Frequency |
PsV-Affected versus Control Individuals |
||
|---|---|---|---|---|
| PsV | Control | OR (95% CI)a | pa | |
| Classical HLA-C Alleles | ||||
| HLA-C∗06:02 | 0.23 | 0.093 | 3.26 (3.02–3.52) | 2.1 × 10−201 |
| HLA-C∗12:03 | 0.073 | 0.056 | 1.38 (1.26–1.52) | 6.5 × 10−12 |
| Other four-digit HLA-C alleles | 0.70 | 0.85 | reference | reference |
| HLA-B Amino Acid Position 67 | ||||
| Cys | 0.16 | 0.12 | 1.56 (1.45–1.67) | 6.0 × 10−35 |
| Met | 0.12 | 0.046 | 1.44 (1.30–1.58) | 2.6 × 10−13 |
| Tyr | 0.12 | 0.16 | 1.00 (0.93–1.07) | 0.93 |
| Phe | 0.21 | 0.26 | 1.00 (0.93–1.08) | 0.99 |
| Ser | 0.39 | 0.42 | reference | reference |
| HLA-B Amino Acid Position 9 | ||||
| Asp | 0.096 | 0.11 | 1.33 (1.21–1.45) | 1.6 × 10−9 |
| Tyr | 0.70 | 0.67 | reference | reference |
| His | 0.20 | 0.22 | 0.87 (0.82–0.92) | 1.6 × 10−6 |
| HLA-A Amino Acid Position 95 | ||||
| Val | 0.34 | 0.29 | 1.31 (1.25–1.38) | 4.7 × 10−28 |
| Ile | 0.56 | 0.59 | reference | reference |
| Leu | 0.099 | 0.12 | 0.89 (0.83–0.95) | 7.0 × 10−4 |
| HLA-DQα1 Amino Acid Position 53 | ||||
| Arg | 0.37 | 0.29 | 1.07 (1.01–1.13) | 0.016 |
| Lys | 0.39 | 0.43 | reference | reference |
| Gln | 0.25 | 0.29 | 0.91 (0.86–0.96) | 9.3 × 10−4 |
Abbreviations are as follows: CI, confidence interval; OR, odds ratio; and PsV, psoriasis vulgaris.
Obtained from the multivariate full regression model including the HLA-C, HLA-B, HLA-A, and HLA-DQA1 risk variants identified by the stepwise regression analysis.
Testing for Discordant Effect Sizes on PsA and PsC
We tested whether the effect sizes of m classical four-digit alleles of the HLA gene had concordant risks between PsA and PsC, as described elsewhere.26 For each of the two compared phenotypes (PsA-affected versus control individuals and PsC-affected versus control individuals), we calculated multivariate odds ratios (ORs) of m − 1 alleles by including them as binary independent variables in the regression model, where the most frequent allele was excluded as a reference. Let βPsA,1, …, βPsA,m−1 and vPsA,1, …, vPsA,m−1 be the multivariate log ORs and their variances, respectively, in PsA-affected versus control individuals, and let βPsC,1, …, βPsC,m−1 and vPsC,1, …, vPsC,m−1 be those in PsC-affected versus control individuals. We evaluated discordance of the effect sizes between the compared phenotypes (pheterogeneity) by testing the statistic
which follows a χ2 distribution with m − 1 degrees of freedom under the null hypothesis of concordant effects.
Results
HLA and MICA Imputation
After imputation of HLA and MICA, we obtained genotypes for 7,078 SNPs in the MHC region (29.6–33.2 Mb at chromosome 6, UCSC Genome Browser hg18), 105 two-digit HLA or MICA alleles, 176 four-digit HLA or MICA alleles, and 438 amino acid polymorphisms encoded by HLA or MICA genes. Imputation of MICA demonstrated high concordance between genotyped and imputed genotypes (88.9% for both two- and four-digit alleles), which was comparable to that reported for imputation of other HLA genes.24–26
HLA-C∗06:02 Has the Strongest Association with PsV Risk
Unsurprisingly, when we tested the imputed variants in the MHC region for overall PsV risk (PsV-affected versus control individuals), the top association signal mapped to HLA-C (Figures 1A and 2A; Table S2). The most strongly associated variant was the classical HLA-C∗06:02 allele (p = 1.7 × 10−364), highly consistent with previous reports that HLA-C∗06:02 has the strongest association with PsV risk.7–12,17,18 We observed that no HLA-C amino acid polymorphism was more strongly associated than HLA-C∗06:02 (the smallest p value, pomnibus = 4.5 × 10−250, was at HLA-C amino acid position 156; Figure 3A). When conditioning on HLA-C∗06:02, we observed the top association signal at the four-digit classical allele of HLA-C∗12:03 (p = 2.5 × 10−25). After conditioning on HLA-C∗06:02 and HLA-C∗12:03, we found that no association exceeded the genome-wide significance threshold for the HLA-C variants (p > 5.0 × 10−8). These results suggest that multiple classical HLA-C alleles, exemplified by HLA-C∗06:02, explain the influence of HLA-C on PsV risk.
Figure 1.
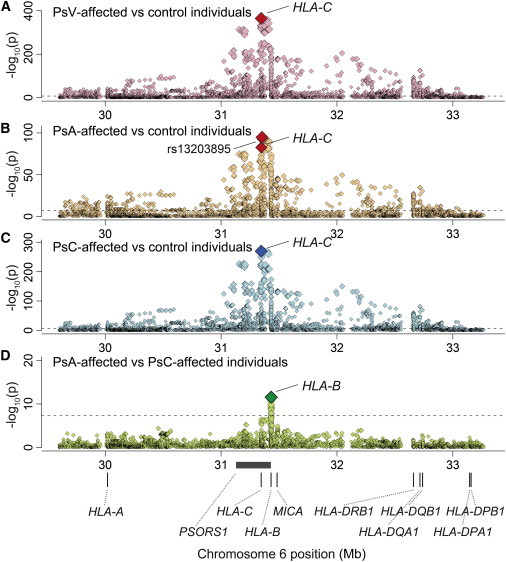
Plots of Nominal Association between the MHC Variants and PsV and Its Subphenotypes of PsA and PsC
Each diamond represents the –log10(p) of the variants, including SNPs, classical HLA or MICA alleles, and amino acid polymorphisms encoded by the HLA genes or MICA. The dotted horizontal line represents the significance threshold of p = 5.0 × 10−8. The bottom panel shows the physical positions of the HLA genes, MICA, and PSORS1 on chromosome 6 (UCSC Genome Browser hg18). We tested four binary phenotypes: (A) PsV-affected versus control individuals, (B) PsA-affected versus control individuals, (C) PsC-affected versus control individuals, and (D) PsA-affected versus PsC-affected individuals.
Figure 2.

Plots of Stepwise Conditional Association of the Variants in the MHC Region
(A–E) Stepwise analysis of HLA-C, HLA-B, HLA-A, and HLA-DQA1 in PsV-affected versus control individuals.
(F and G) Stepwise analysis of HLA-B in PsA versus PsC individuals. Each diamond represents the –log10(p) of the variants, including SNPs, classical HLA or MICA alleles, and amino acid polymorphisms encoded by the HLA genes or MICA. The dotted horizontal line represents the significance threshold of p = 5.0 × 10−8. The most strongly associated amino acid polymorphisms and HLA classical alleles are labeled when their associations satisfied p < 5.0 × 10−8.
Figure 3.
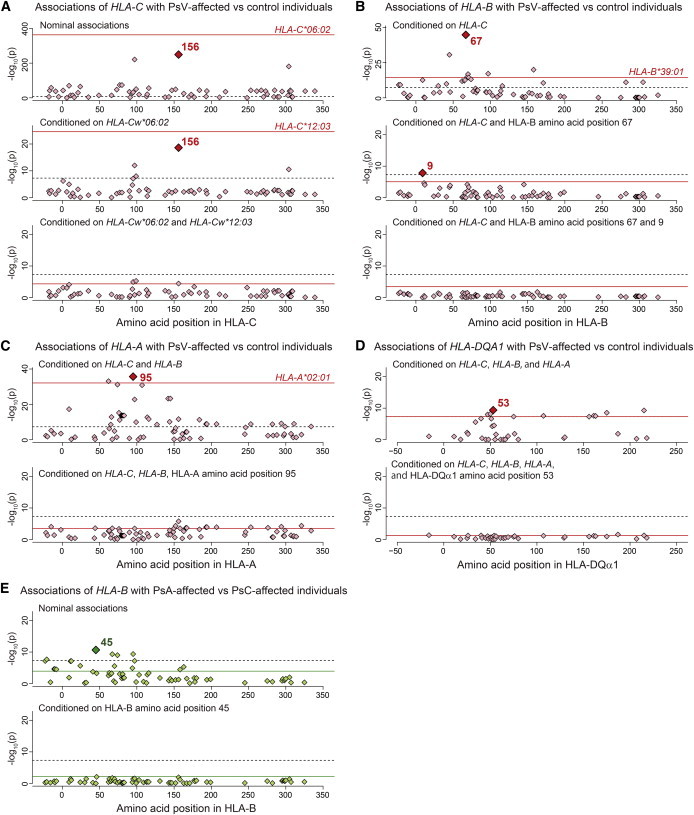
Plots of Stepwise Conditional Association of the HLA Amino Acid Polymorphisms and Classical HLA Alleles
(A–D) Stepwise analysis of HLA-C, HLA-B, HLA-A, and HLA-DQA1 in PsV-affected versus control individuals.
(E) Stepwise analysis of HLA-B in PsA-affected versus PsC-affected individuals. Each diamond represents the –log10(pomnibus) of the amino acid polymorphism encoded by the HLA gene. The colored horizontal line represents the –log10(pomnibus) of the most strongly associated classical allele of the HLA gene. The dotted horizontal line represents the significance threshold of p = 5.0 × 10−8. The most strongly associated amino acid polymorphisms and HLA classical alleles are labeled when their associations satisfied p < 5.0 × 10−8.
PsV Risk Is Associated with Multiple Class I and Class II HLA Genes
We then investigated additional HLA-variant PsV risk independent of HLA-C. When we conditioned on all classical HLA-C alleles, we observed a significant independent association at HLA-B amino acid position 67 (pomnibus = 1.8 × 10−45; Figures 2B and 3B). Stepwise regression analysis of HLA-B variants further identified an independent association at position 9 (pomnibus = 1.5 × 10−8). When we conditioned on HLA-C and HLA-B, we observed a significant independent association at HLA-A amino acid position 95 (pomnibus = 2.1 × 10−36; Figures 2C and 3C). Stepwise analysis did not identify additional association within HLA-A. When we conditioned on the effects of HLA-C, HLA-B, and HLA-A, we observed a significant independent association at HLA-DQα1 amino acid position 53 (pomnibus = 4.2 × 10−10; Figures 2D and 3D). Stepwise analysis did not identify an additional association within HLA-DQA1. When conditioning on HLA-C, HLA-B, HLA-A, and HLA-DQA1, we observed no other significant associations (p > 5.0 × 10−8; Figure 2E). These results demonstrate that PsV risk within the MHC region can be explained by combinations of multiple class I and class II HLA genes.
To highlight specific HLA alleles or amino acid residues that confer overall PsV risk, we defined a multivariate full regression model including all the identified risk variants of HLA-C, HLA-B, HLA-A, and HLA-DQA1 (Table 1; Table S3). For HLA-C, we observed increased risk associated with HLA-C∗06:02 (OR = 3.26, 95% confidence interval [CI] = 3.02–3.52, p = 2.1 × 10−201) and HLA-C∗12:03 (OR = 1.38, 95% CI = 1.26–1.52, p = 6.5 × 10−12). For HLA-B, we observed increased risk associated with Cys67 (OR = 1.56, 95% CI = 1.45–1.67, p = 6.0 × 10−35), Met67 (OR = 1.44, 95% CI = 1.30–1.58, p = 2.6 × 10−13), and Asp9 (OR = 1.33, 95% CI = 1.21–1.45, p = 1.6 × 10−9). HLA-A Val95 (OR = 1.31, 95% CI = 1.25–1.38, p = 4.7 × 10−28) and HLA-DQα1 Arg53 (OR = 1.07, 95% CI = 1.01–1.13, p = 0.016) also demonstrated increased risk. In combination, these risk variants explained 6.7% of phenotype variance of PsV under the assumption of 2.0% of disease prevalence,2 whereas HLA-C∗06:02 alone explained only 4.9% of the variance.
Risk Heterogeneity between PsA and PsC Is Explained by HLA-B
Next, we conducted an analysis focusing on subphenotypes of PsA and PsC. Overall, the association results of the PsA case-control analysis and of the PsC case-control analysis were similar to those of the PsV case-control analysis (Figures 1A–1C). The classical HLA-C∗06:02 allele demonstrated the lowest p value among the HLA variants in the MHC region. Stepwise association analyses, separately conducted for the PsA case-control analysis and the PsC case-control analysis, both revealed independent contributions of other class I HLA genes (HLA-B and HLA-C; Figure S2; Table S2). We did not observe independent signals for HLA class II genes in this stratified analysis, perhaps as a result of reduced statistical power.
Surprisingly, when we directly assessed comparative risk between PsA and PsC subjects, we found the lowest p value of the nominal association signal at HLA-B amino acid position 45 (pomnibus = 2.2 × 10−11; Figures 1D and 2F) rather than at HLA-C alleles. After conditioning on HLA-B amino acid position 45, or all classical HLA-B alleles, we observed no significant association in the MHC region (p > 5.0 × 10−8; Figures 2G and 3E). Of the HLA-B amino acid residues at position 45, HLA-B Glu45 increased PsA susceptibility in comparison to PsC susceptibility (OR = 1.46, 95% CI = 1.31–1.62, p = 2.9 × 10−12; Table 2; Table S2). Examining the classical alleles, we noted that HLA-B∗27, a risk allele for another arthritic disease, ankylosing spondylitis (MIM 106300),33 demonstrated the lowest p value for PsA versus PsC association (p = 1.2 × 10−4; Figure 4; Table S4) but much less significantly than HLA-B Glu45. We note that HLA-B∗27, along with HLA-B∗38, HLA-B∗39, and a number of other alleles, carries Glu at position 45.
Table 2.
Association Results of HLA Variants for Subphenotype Risk Comparisons of PsA and PsC
| HLA Variant |
Frequency |
PsA-Affected versus PsC-Affected Individuals |
||
|---|---|---|---|---|
| PsA | PsC | OR (95% CI) | p | |
| HLA-B Amino Acid Position 45 (Nominal) | ||||
| Glu | 0.43 | 0.33 | 1.46 (1.31–1.62) | 2.9 × 10−12 |
| Thr, Lys, Met | 0.57 | 0.67 | reference | reference |
Abbreviations are as follows: CI, confidence interval; OR, odds ratio; PsA, psoriatic arthritis; and PsC, cutaneous psoriasis.
Figure 4.
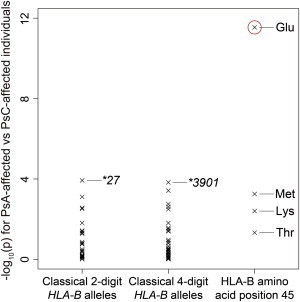
Associations of Classical HLA-B Alleles and HLA-B Amino Acid Position 45 for PsA versus PsC Individuals
The –log10(p) of HLA-B classical two- and four-digit HLA-B alleles and HLA-B amino acid residues at position 45 in PsA versus PsC individuals. HLA-B Glu45 demonstrated the strongest association (p = 2.9 × 10−12; highlighted with a red circle), whereas the HLA-B classical alleles showed much smaller effects (p > 1.0 × 10−4).
Previous studies comparing PsA and PsC have suggested that HLA-C∗06:02 shows increased PsC risk but decreased PsA risk,13,14 and our study replicated this finding in a concordant directional effect (p = 9.4 × 10−6 for PsA versus PsC individuals). However, the differential impact of HLA-C∗06:02 on PsA and PsC risk disappeared after we conditioned on HLA-B amino acid position 45 (p = 0.12), suggesting that this reduced effect was the result of linkage to HLA-B. In contrast, the effect of HLA-B amino acid position 45 with respect to PsA versus PsC risk retained significance even after we conditioned on HLA-C∗06:02 (pomnibus = 2.2 × 10−7).
We evaluated effect-size (OR) heterogeneity in classical four-digit alleles of the HLA genes between the two association analyses of PsA-affected versus control individuals and PsC-affected versus control individuals. Among the eight class I and class II HLA genes that we evaluated in the overall PsV case-control analysis, the HLA-B and HLA-C alleles showed significant risk heterogeneity (pheterogeneity = 5.8 × 10−14 and pheterogeneity = 2.9 × 10−6, respectively, with a significance threshold of p < 0.05/8 = 0.0073). When we conditioned on HLA-B amino acid position 45, risk heterogeneity diminished in both HLA-B and HLA-C classical alleles (pheterogeneity > 0.01). In contrast, when we conditioned on HLA-C∗06:02, risk heterogeneity diminished for the classical HLA-C alleles (pheterogeneity = 0.018), but not in the classical HLA-B alleles (pheterogeneity = 6.5 × 10−5). These results demonstrate that the risk heterogeneity between PsA and PsC primarily derives from HLA-B, but not HLA-C (or other), genes.
No Apparent Contribution of MICA to Psoriasis Risk
MICA variants demonstrated nominally significant associations with PsV, PsA, and PsC risk (the lowest p value, pomnibus = 2.2 × 10−180, was at MICA amino acid position 298 for PsV-affected versus control individuals; Table S2). However, after we conditioned on HLA-C and HLA-B, no MICA variants showed independent association signals (p > 5.0 × 10−8 for any of the four tested phenotype comparisons). Previous studies have suggested independent effects of several MICA variants, such as MICA∗016, MICA∗008:01, and MICA amino acid position 129, on PsV or its subphenotypes,19,20 but our study did not observe apparent associations of these variants when we conditioned on HLA-C∗06:02 or HLA-B amino acid position 67 (p > 0.017). We note that no significant association was observed in the MICA variants in direct comparison between PsA subjects and PsC subjects (p > 5.0 × 10−8). Therefore, our study could not identify an independent contribution of MICA on risk of psoriasis and its clinical subtypes.
Discussion
In this study, we fine mapped PsV risk within the MHC region. In addition to imputing variants of HLA genes, we evaluated risk of the HLA-like gene MICA by creating a MICA reference panel that empirically demonstrated high imputation accuracy. Our study identified multiple HLA-C∗06:02-independent risk variants of both class I and class II HLA genes for PsV susceptibility (HLA-B, HLA-A, and HLA-DQA1), but no apparent risk attributable to MICA. We also observed that risk heterogeneity between PsA and PsC could be explained by polymorphisms of a single amino acid site encoded by HLA-B, suggesting that different genetic architectures underlie the overall risk of PsV and that of its subphenotypes. To our knowledge, ours is the largest HLA fine-mapping study of PsV associations in the MHC to date and defines genetic heterogeneity between PsA and PsC subphenotypes.
HLA-C∗06:02 has the strongest association with PsV risk, as reported previously.7–12,17,18 We demonstrated that no single HLA-C amino acid polymorphism was more strongly associated than the HLA-C∗06:02 classical allele, suggesting that the haplotype sequence including HLA-C∗06:02 itself should be the origin of PsV risk. Several hypotheses might explain this. Clop et al. reported noncoding regulatory variants that are located in enhancer motifs and that are unique to the HLA-C∗06:02 haplotype.34 A combination of the polymorphisms in multiple HLA-C amino acid sites could effectively tag HLA-C∗06:02.35 We did observe a more modest independent effect at HLA-C∗12:03 in addition to the large HLA-C∗06:02 effect; Helms et al. reported that HLA-C∗06:02 and HLA-C∗12:03 share several functional domains and peptide-binding pockets of HLA-C.5 We note that HLA-C∗12:03 did not show an independent association signal after we conditioned on HLA-C∗06:02 and every classical HLA-B allele (p = 0.12), suggesting the possibility that the observed HLA-C∗12:03 association might reflect risk at other HLA-B alleles in LD. Further functional studies will be necessary for elucidating the role of HLA-C in PsV risk.
In contrast, outside of HLA-C, amino acid polymorphisms (HLA-B amino acid positions 67 and 9, HLA-A position 95, and HLA-DQα1 position 53) demonstrated stronger associations than did classical alleles HLA-B, HLA-A, and HLA-DQA1. All of these amino acid sites were located within the HLA antigen binding (Figure 5 and Figure S3). Positions 67 and 9 in HLA-B have been identified in HLA fine-mapping studies for other immune-related diseases.25–27 We note that LD structures between the amino acid positions could yield potential ambiguity in fine mapping of the causal amino acid position, and this might be clarified with larger studies.
Figure 5.
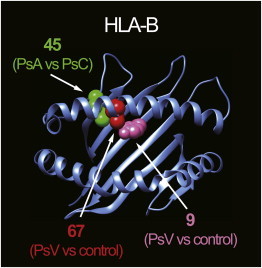
3D Ribbon Models for HLA-B
HLA-B structures are based on Protein Data Bank entry 2bvp36 and were prepared with UCSF Chimera version 1.7.37 Amino acid position 67 and 9 residues associated with overall PsV risk are highlighted as red and pink spheres, respectively. An amino acid position 45 residue associated with subphenotype risk heterogeneity (PsA versus PsC) is highlighted as a green sphere.
The contribution of HLA-like genes to immune-related disease risk has long been a topic of discussion.38,39 Although our imputation of MICA alleles was highly accurate, our study did not observe independent MICA risk of PsV after we conditioned on the neighboring risk HLA genes HLA-C and HLA-B. Previous studies focusing on MICA risk did not apply robust conditioning on all classical HLA-C and HLA-B alleles and thus could have potentially reflected the associations of HLA-C and HLA-B via LD with them.19,20
Here, we were able to successfully decompose the genetic architecture of PsA and PsC to a shared component and a subphenotype-specific component. Our study demonstrates that the HLA gene associated with the risk heterogeneity between PsA and PsC (HLA-B) is distinct from the HLA gene most associated with overall PsV risk (HLA-C). Previous studies have reported that HLA-C∗06:02 has different effect sizes for PsA and PsC, naturally leading to a hypothesis that the heterogeneity is driven by the difference in HLA-C, the major risk factor.13–16,23 However, our observation is more concordant with a model where the two PsV subtypes generally share the same risk alleles, including HLA-C∗06:02, but differ at a specific locus that contributes to subtype differences.
HLA-B amino acid position 45 is the driving MHC position that modulates differential risk of PsA and PsV. The effect of Glu at HLA-B position 45 confers substantial risk of psoriatic arthritis and explains previously reported associations at HLA-B∗27. This site is located within the binding groove of HLA-B and is classified as one of the functional pockets influencing receptor cell-surface expression or antigen peptide binding or presentation.40 Being able to clinically distinguish those individuals who have isolated skin disease (PsC) from those who develop joint disease (PsA) has clinical importance. PsA often occurs in addition to psoriatic skin disease and almost always requires systemic therapy, for example, with anti-TNF or other biologic medications, for the prevention of destructive joint disease. In contrast, PsC can in many instances be managed with topical treatments alone.3 Our findings might contribute to utilizing information on HLA variants to improve diagnostic approaches for clinical subphenotypes, as suggested for other complex diseases.26 There is a possibility that the HLA-B45 amino acid residue tags other HLA-B driving risk variants at other amino acid sites, although we observed only limited LD between these sites (Figure S4). We also note that our method of evaluating OR heterogeneity (i.e., pheterogeneity) might be conservative because of shared control subjects in the PsA and PsC case-control analyses. Further studies will be required for elucidating the functional mechanisms that result in differential PsA and PsC risk.
In summary, our study fine mapped risk of multiple class I and class II HLA genes in PsV and its subphenotypes through large-scale HLA and MICA imputation. Our study should contribute to our understanding of HLA variants in the etiology of PsV.
Acknowledgments
This work was supported by the NIH (1R01AR062886, R01AR042742, R01AR050511, R01AR062382, 5U01GM092691-04, 1R01AR063759-01A1, and 2R01AR050266), the Arthritis Foundation, a Clinical Scientist Development Award to S.R. from the Doris Duke Foundation, the Japan Society of the Promotion of Science, the Japan Science and Technology Agency, the Ann Arbor Veterans Affairs Hospital, the Canadian Institute of Health Research, the Krembil Foundation, the Arthritis Society, a Vernieuwingsimpuls VIDI Award (project 016.126.354) from the Netherlands Organization for Scientific Research, and the Babcock Memorial Trust.
Footnotes
This is an open access article under the CC BY license (http://creativecommons.org/licenses/by/3.0/).
Contributor Information
Paul I.W. de Bakker, Email: pdebakker@umcutrecht.nl.
Soumya Raychaudhuri, Email: soumya@broadinstitute.org.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
IMGT/HLA Database, http://www.ebi.ac.uk/ipd/imgt/hla/
Online Mendelian Inheritance in Man (OMIM), http://omim.org/
Type 1 Diabetes Genetics Consortium (T1DGC), https://www.t1dgc.org/home.cfm
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Nestle F.O., Kaplan D.H., Barker J. Psoriasis. N. Engl. J. Med. 2009;361:496–509. doi: 10.1056/NEJMra0804595. [DOI] [PubMed] [Google Scholar]
- 2.Christophers E. Psoriasis—epidemiology and clinical spectrum. Clin. Exp. Dermatol. 2001;26:314–320. doi: 10.1046/j.1365-2230.2001.00832.x. [DOI] [PubMed] [Google Scholar]
- 3.Fitzgerald O., Winchester R. Psoriatic arthritis: from pathogenesis to therapy. Arthritis Res. Ther. 2009;11:214. doi: 10.1186/ar2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Veal C.D., Capon F., Allen M.H., Heath E.K., Evans J.C., Jones A., Patel S., Burden D., Tillman D., Barker J.N., Trembath R.C. Family-based analysis using a dense single-nucleotide polymorphism-based map defines genetic variation at PSORS1, the major psoriasis-susceptibility locus. Am. J. Hum. Genet. 2002;71:554–564. doi: 10.1086/342289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Helms C., Saccone N.L., Cao L., Daw J.A., Cao K., Hsu T.M., Taillon-Miller P., Duan S., Gordon D., Pierce B. Localization of PSORS1 to a haplotype block harboring HLA-C and distinct from corneodesmosin and HCR. Hum. Genet. 2005;118:466–476. doi: 10.1007/s00439-005-0048-2. [DOI] [PubMed] [Google Scholar]
- 6.Nair R.P., Stuart P.E., Nistor I., Hiremagalore R., Chia N.V., Jenisch S., Weichenthal M., Abecasis G.R., Lim H.W., Christophers E. Sequence and haplotype analysis supports HLA-C as the psoriasis susceptibility 1 gene. Am. J. Hum. Genet. 2006;78:827–851. doi: 10.1086/503821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nair R.P., Duffin K.C., Helms C., Ding J., Stuart P.E., Goldgar D., Gudjonsson J.E., Li Y., Tejasvi T., Feng B.J., Collaborative Association Study of Psoriasis Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat. Genet. 2009;41:199–204. doi: 10.1038/ng.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Strange A., Capon F., Spencer C.C., Knight J., Weale M.E., Allen M.H., Barton A., Band G., Bellenguez C., Bergboer J.G., Genetic Analysis of Psoriasis Consortium & the Wellcome Trust Case Control Consortium 2 A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat. Genet. 2010;42:985–990. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ellinghaus E., Ellinghaus D., Stuart P.E., Nair R.P., Debrus S., Raelson J.V., Belouchi M., Fournier H., Reinhard C., Ding J. Genome-wide association study identifies a psoriasis susceptibility locus at TRAF3IP2. Nat. Genet. 2010;42:991–995. doi: 10.1038/ng.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hüffmeier U., Uebe S., Ekici A.B., Bowes J., Giardina E., Korendowych E., Juneblad K., Apel M., McManus R., Ho P. Common variants at TRAF3IP2 are associated with susceptibility to psoriatic arthritis and psoriasis. Nat. Genet. 2010;42:996–999. doi: 10.1038/ng.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stuart P.E., Nair R.P., Ellinghaus E., Ding J., Tejasvi T., Gudjonsson J.E., Li Y., Weidinger S., Eberlein B., Gieger C. Genome-wide association analysis identifies three psoriasis susceptibility loci. Nat. Genet. 2010;42:1000–1004. doi: 10.1038/ng.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsoi L.C., Spain S.L., Knight J., Ellinghaus E., Stuart P.E., Capon F., Ding J., Li Y., Tejasvi T., Gudjonsson J.E., Collaborative Association Study of Psoriasis (CASP) Genetic Analysis of Psoriasis Consortium. Psoriasis Association Genetics Extension. Wellcome Trust Case Control Consortium 2 Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat. Genet. 2012;44:1341–1348. doi: 10.1038/ng.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eder L., Chandran V., Pellet F., Shanmugarajah S., Rosen C.F., Bull S.B., Gladman D.D. Human leucocyte antigen risk alleles for psoriatic arthritis among patients with psoriasis. Ann. Rheum. Dis. 2012;71:50–55. doi: 10.1136/ard.2011.155044. [DOI] [PubMed] [Google Scholar]
- 14.Eder L., Chandran V., Pellett F., Shanmugarajah S., Rosen C.F., Bull S.B., Gladman D.D. Differential human leucocyte allele association between psoriasis and psoriatic arthritis: a family-based association study. Ann. Rheum. Dis. 2012;71:1361–1365. doi: 10.1136/annrheumdis-2012-201308. [DOI] [PubMed] [Google Scholar]
- 15.Winchester R., Minevich G., Steshenko V., Kirby B., Kane D., Greenberg D.A., FitzGerald O. HLA associations reveal genetic heterogeneity in psoriatic arthritis and in the psoriasis phenotype. Arthritis Rheum. 2012;64:1134–1144. doi: 10.1002/art.33415. [DOI] [PubMed] [Google Scholar]
- 16.Chandran V., Bull S.B., Pellett F.J., Ayearst R., Rahman P., Gladman D.D. Human leukocyte antigen alleles and susceptibility to psoriatic arthritis. Hum. Immunol. 2013;74:1333–1338. doi: 10.1016/j.humimm.2013.07.014. [DOI] [PubMed] [Google Scholar]
- 17.Feng B.J., Sun L.D., Soltani-Arabshahi R., Bowcock A.M., Nair R.P., Stuart P., Elder J.T., Schrodi S.J., Begovich A.B., Abecasis G.R. Multiple Loci within the major histocompatibility complex confer risk of psoriasis. PLoS Genet. 2009;5:e1000606. doi: 10.1371/journal.pgen.1000606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knight J., Spain S.L., Capon F., Hayday A., Nestle F.O., Clop A., Barker J.N., Weale M.E., Trembath R.C., Wellcome Trust Case Control Consortium. Genetic Analysis of Psoriasis Consortium. I-chip for Psoriasis Consortium Conditional analysis identifies three novel major histocompatibility complex loci associated with psoriasis. Hum. Mol. Genet. 2012;21:5185–5192. doi: 10.1093/hmg/dds344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pollock R., Chandran V., Barrett J., Eder L., Pellett F., Yao C., Lino M., Shanmugarajah S., Farewell V.T., Gladman D.D. Differential major histocompatibility complex class I chain-related A allele associations with skin and joint manifestations of psoriatic disease. Tissue Antigens. 2011;77:554–561. doi: 10.1111/j.1399-0039.2011.01670.x. [DOI] [PubMed] [Google Scholar]
- 20.Pollock R.A., Chandran V., Pellett F.J., Thavaneswaran A., Eder L., Barrett J., Rahman P., Farewell V., Gladman D.D. The functional MICA-129 polymorphism is associated with skin but not joint manifestations of psoriatic disease independently of HLA-B and HLA-C. Tissue Antigens. 2013;82:43–47. doi: 10.1111/tan.12126. [DOI] [PubMed] [Google Scholar]
- 21.de Bakker P.I., McVean G., Sabeti P.C., Miretti M.M., Green T., Marchini J., Ke X., Monsuur A.J., Whittaker P., Delgado M. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat. Genet. 2006;38:1166–1172. doi: 10.1038/ng1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okada Y., Yamazaki K., Umeno J., Takahashi A., Kumasaka N., Ashikawa K., Aoi T., Takazoe M., Matsui T., Hirano A. HLA-Cw∗1202-B∗5201-DRB1∗1502 haplotype increases risk for ulcerative colitis but reduces risk for Crohn’s disease. Gastroenterology. 2011;141:864–871. doi: 10.1053/j.gastro.2011.05.048. e1–e5. [DOI] [PubMed] [Google Scholar]
- 23.Ho P.Y., Barton A., Worthington J., Plant D., Griffiths C.E., Young H.S., Bradburn P., Thomson W., Silman A.J., Bruce I.N. Investigating the role of the HLA-Cw∗06 and HLA-DRB1 genes in susceptibility to psoriatic arthritis: comparison with psoriasis and undifferentiated inflammatory arthritis. Ann. Rheum. Dis. 2008;67:677–682. doi: 10.1136/ard.2007.071399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jia X., Han B., Onengut-Gumuscu S., Chen W.M., Concannon P.J., Rich S.S., Raychaudhuri S., de Bakker P.I. Imputing amino acid polymorphisms in human leukocyte antigens. PLoS ONE. 2013;8:e64683. doi: 10.1371/journal.pone.0064683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raychaudhuri S., Sandor C., Stahl E.A., Freudenberg J., Lee H.S., Jia X., Alfredsson L., Padyukov L., Klareskog L., Worthington J. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat. Genet. 2012;44:291–296. doi: 10.1038/ng.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han B., Diogo D., Eyre S., Kallberg H., Zhernakova A., Bowes J., Padyukov L., Okada Y., González-Gay M.A., Rantapää-Dahlqvist S. Fine mapping seronegative and seropositive rheumatoid arthritis to shared and distinct HLA alleles by adjusting for the effects of heterogeneity. Am. J. Hum. Genet. 2014;94:522–532. doi: 10.1016/j.ajhg.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pereyra F., Jia X., McLaren P.J., Telenti A., de Bakker P.I., Walker B.D., Ripke S., Brumme C.J., Pulit S.L., Carrington M., International HIV Controllers Study The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science. 2010;330:1551–1557. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foo J.N., Smedby K.E., Akers N.K., Berglund M., Irwan I.D., Jia X., Li Y., Conde L., Darabi H., Bracci P.M. Coding variants at hexa-allelic amino acid 13 of HLA-DRB1 explain independent SNP associations with follicular lymphoma risk. Am. J. Hum. Genet. 2013;93:167–172. doi: 10.1016/j.ajhg.2013.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gregersen P.K., Kosoy R., Lee A.T., Lamb J., Sussman J., McKee D., Simpfendorfer K.R., Pirskanen-Matell R., Piehl F., Pan-Hammarstrom Q. Risk for myasthenia gravis maps to a (151) Pro→Ala change in TNIP1 and to human leukocyte antigen-B∗08. Ann. Neurol. 2012;72:927–935. doi: 10.1002/ana.23691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taylor W., Gladman D., Helliwell P., Marchesoni A., Mease P., Mielants H., CASPAR Study Group Classification criteria for psoriatic arthritis: development of new criteria from a large international study. Arthritis Rheum. 2006;54:2665–2673. doi: 10.1002/art.21972. [DOI] [PubMed] [Google Scholar]
- 31.Rich S.S., Concannon P., Erlich H., Julier C., Morahan G., Nerup J., Pociot F., Todd J.A. The Type 1 Diabetes Genetics Consortium. Ann. N Y Acad. Sci. 2006;1079:1–8. doi: 10.1196/annals.1375.001. [DOI] [PubMed] [Google Scholar]
- 32.Robinson J., Halliwell J.A., McWilliam H., Lopez R., Parham P., Marsh S.G. The IMGT/HLA database. Nucleic Acids Res. 2013;41(Database issue):D1222–D1227. doi: 10.1093/nar/gks949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cortes A., Hadler J., Pointon J.P., Robinson P.C., Karaderi T., Leo P., Cremin K., Pryce K., Harris J., Lee S., International Genetics of Ankylosing Spondylitis Consortium (IGAS) Australo-Anglo-American Spondyloarthritis Consortium (TASC) Groupe Française d’Etude Génétique des Spondylarthrites (GFEGS) Nord-Trøndelag Health Study (HUNT) Spondyloarthritis Research Consortium of Canada (SPARCC) Wellcome Trust Case Control Consortium 2 (WTCCC2) Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat. Genet. 2013;45:730–738. doi: 10.1038/ng.2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clop A., Bertoni A., Spain S.L., Simpson M.A., Pullabhatla V., Tonda R., Hundhausen C., Di Meglio P., De Jong P., Hayday A.C. An in-depth characterization of the major psoriasis susceptibility locus identifies candidate susceptibility alleles within an HLA-C enhancer element. PLoS ONE. 2013;8:e71690. doi: 10.1371/journal.pone.0071690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Asahina A., Kuwata S., Tokunaga K., Juji T., Nakagawa H. Study of aspartate at residue 9 of HLA-C molecules in Japanese patients with psoriasis vulgaris. J. Dermatol. Sci. 1996;13:125–133. doi: 10.1016/s0923-1811(96)00518-x. [DOI] [PubMed] [Google Scholar]
- 36.Stewart-Jones G.B., Gillespie G., Overton I.M., Kaul R., Roche P., McMichael A.J., Rowland-Jones S., Jones E.Y. Structures of three HIV-1 HLA-B∗5703-peptide complexes and identification of related HLAs potentially associated with long-term nonprogression. J. Immunol. 2005;175:2459–2468. doi: 10.4049/jimmunol.175.4.2459. [DOI] [PubMed] [Google Scholar]
- 37.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 38.Choy M.K., Phipps M.E. MICA polymorphism: biology and importance in immunity and disease. Trends Mol. Med. 2010;16:97–106. doi: 10.1016/j.molmed.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 39.Lo P.H., Urabe Y., Kumar V., Tanikawa C., Koike K., Kato N., Miki D., Chayama K., Kubo M., Nakamura Y., Matsuda K. Identification of a functional variant in the MICA promoter which regulates MICA expression and increases HCV-related hepatocellular carcinoma risk. PLoS ONE. 2013;8:e61279. doi: 10.1371/journal.pone.0061279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buxton S.E., Benjamin R.J., Clayberger C., Parham P., Krensky A.M. Anchoring pockets in human histocompatibility complex leukocyte antigen (HLA) class I molecules: analysis of the conserved B (“45”) pocket of HLA-B27. J. Exp. Med. 1992;175:809–820. doi: 10.1084/jem.175.3.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.